
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A chemical reaction controlled by light-activated molecular switches based on hetero-cyclopentanediyls†
Jonas
Bresien
 *a,
Thomas
Kröger-Badge
a,
Stefan
Lochbrunner
bc,
Dirk
Michalik
ad,
Henrik
Müller
a,
Axel
Schulz
*acd and
Edgar
Zander
a
*a,
Thomas
Kröger-Badge
a,
Stefan
Lochbrunner
bc,
Dirk
Michalik
ad,
Henrik
Müller
a,
Axel
Schulz
*acd and
Edgar
Zander
a
aInstitute of Chemistry, University of Rostock, Albert-Einstein-Str. 3a, D-18059 Rostock, Germany. E-mail: jonas.bresien@uni-rostock.de; axel.schulz@uni-rostock.de
bInstitute of Physics, University of Rostock, Albert-Einstein-Str. 23-24, D-18059 Rostock, Germany
cDepartment of Life, Light & Matter, University of Rostock, D-18051 Rostock, Germany
dLeibniz Institute for Catalysis at the University of Rostock e.V., Albert-Einstein-Straße 29a, D-18059 Rostock, Germany
First published on 18th February 2019
Abstract
Molecular switches are molecules that can reversibly be shifted between at least two stable states with different physical and chemical properties, making them interesting for application as chemical sensors or molecular machines. We recently discovered that five-membered, cyclic biradicals based on group 15 elements are efficient and robust photochemical switches that can be activated by red light. The quantum yield of the photo-isomerization is as high as 24.6%, and the thermal equilibration of the photo-activation product proceeds rapidly at ambient temperature. The fully reversible process was studied by experimental and high-level ab initio techniques. We could further demonstrate that the biradical character could be completely turned on and off, so the system could be applied to control chemical equilibria that involve activation products of the cyclic biradicals.
Introduction
The term “molecular switch” originated in the early 1980s and was first used in conjunction with biological signal transmission.1 Since then, it has found widespread use throughout chemistry, and generally refers to any kind of molecule that can exist in two or more (meta-)stable states. Interconversion can be induced by external stimuli such as temperature, chemical modifications, redox reactions, electric fields, or irradiation with photons.2–5 The most commonly known type of molecular switches are pH indicators, which are based on acid–base equilibria and therefore belong to the group of chemically activated compounds.6,7 Other chemically switchable systems may, for example, find application in the field of gas or ion sensing,5,8 or be used as redox indicators.7Especially light-activated molecular switches promise diverse applications, e.g. as photoactuators9–11 or photo-regulated catalysts.12,13 Therefore, a growing research interest is aimed both at the synthesis of new systems as well as the exploration of new fields of applications.14,15 Most light-activated molecular switches function by conformational changes.3 One of the most prominent examples is probably retinal, the chromophore of the receptor protein rhodopsin, which is found in the rods of the retina in mammals' eyes. Upon irradiation with visible light, retinal undergoes a conformational change resulting in a signalling cascade, which eventually leads to a visual stimulus.16–18 Therefore, it plays a crucial role in the functioning of the eye. Mechanistically, the isomerization of retinal can be traced back to an E/Z isomerization along a C–C double bond. Thus, it is not surprising that many synthetic, light activated molecular switches are based on a similar principle:19 For example, azobenzene may be isomerized from its E to its Z conformer by light in the UV region, while the Z → E isomerization is triggered by light in the blue region of the visible spectrum (Fig. 1a).20,21 The absorption maxima can be shifted by derivatization of azobenzene with electron donating or electron withdrawing groups.4 Another popular type of photo-switching is represented by photo-cyclization reactions,22,23 as observed, for example, in case of diarylethenes (Fig. 1a).24 This class of molecular switches is based on the formation or breaking of chemical bonds. In fact, most (light-activated) molecular switches can be divided into these two categories, namely stereoisomerism (change of structure without modification of the chemical bonding pattern) and structural isomerism (including a redistribution of bonds).3 Other mechanisms are discussed in the literature, such as dipole, charge, or spin switching,3 but these processes often include some kind of structural change, too.
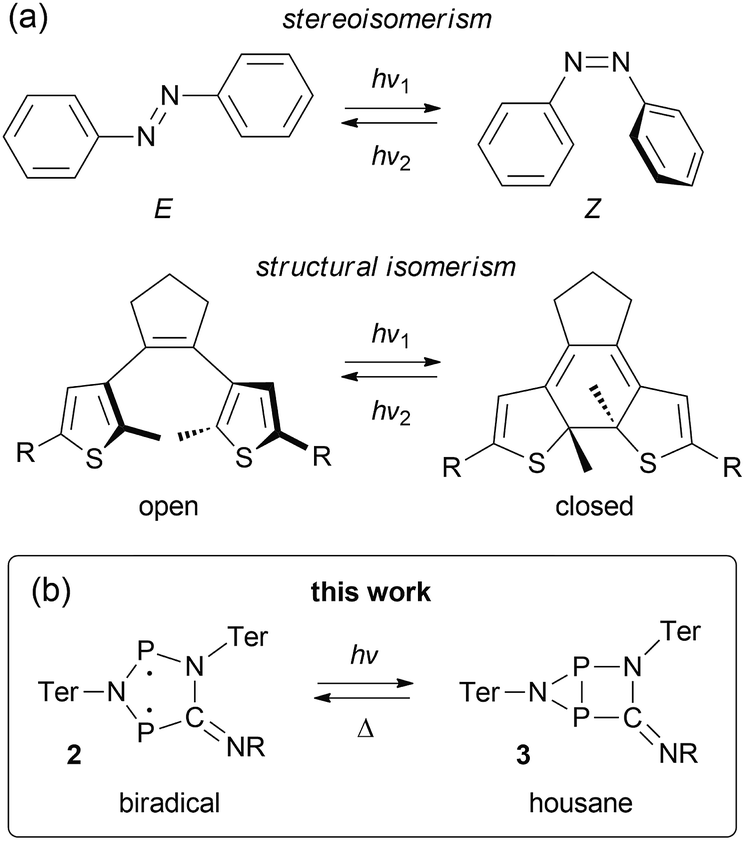 | ||
| Fig. 1 Types of molecular switches. (a) Azobenzenes (top) and stilbenes can be categorized as stereoisomeric switches, while diarylethenes (bottom), spiropyranes, spiroxazines, and fulgimides are typical examples of structural isomeric switches. (b) We investigated the photo-switching behavior of hetero-cyclopentane-1,3-diyls (2), which display considerable singlet biradical character (R = tBu, Dmp (2,6-dimethylphenyl), Ter = 2,6-bis(2,4,6-trimethylphenyl)phenyl). | ||
Understanding bond formation and breaking processes (or the nature of chemical bonds in general) has been a widely investigated topic throughout all areas of chemical science.25–28 Amongst others, singlet biradicals (or biradicaloids) are frequently cited as suitable model systems for bond formation and breaking processes due to their peculiar electronic structure, which is sometimes referred to as comprising a “partial bond” between the formal radical centres.29 In this regard, singlet biradicals are promising candidates for molecular switches, as small structural changes should generally alter the bonding interaction between the radical centres significantly, thereby also affecting the biradical character. The latter being the source of many interesting properties, such as small-molecule activation30–33 or non-linear optical properties,29,34 it should be possible to switch these characteristics as well. Still, there are only few experimental examples in the literature taking advantage of such an approach.35–38
We recently introduced stable hetero-cyclopentane-1,3-diyls based on group 15 elements (2, Fig. 1b).39–42 These five-membered cyclic biradicals can be used to activate small molecules such as alkynes, carbon monoxide or isonitriles. Furthermore, preliminary investigations indicated that they can be isomerized by light to a closed-shell housane-type isomer (3) featuring a P–P single bond. Contrary to carbon based cyclopentane-1,3-diyls,43–46 the isomerization was shown to be reversible. Since we are not aware of molecular switches based on this class of compounds, we sought to study the photo-isomerization in more detail, so as to understand the mechanism of the photo-switching process and control the activation chemistry by switching the biradical character on and off.
Results and discussion
Photo-isomerization and thermal reverse reaction
We chose to begin our investigations on the system 2Dmp (Fig. 1b, R = Dmp = 2,6-dimethylphenyl), since it had proved to be the most stable derivative.40 It was synthesized from the biradical [P(μ-NTer)]2 (1, Ter = 2,6-dimesitylphenyl) by insertion of DmpNC according to published procedures (cf. ESI,†). The UV-Vis spectrum exhibited a broad absorption band centred at 643 nm, i.e. in the red region of the visible spectrum (Fig. 2a). According to time-dependent density functional theory (TD-DFT), complete active space self-consistent field (CASSCF), and model multi-reference configuration interaction (MRCI) calculations, the absorption may be attributed to the formal HOMO → LUMO transition (vide infra; for detailed information on all calculations and proper electronic description of the singlet biradical please refer to the ESI, p. S40†). Since the “LUMO” is transannularly bonding in character between the two P atoms (Fig. S28 and S29;†cf.Fig. 7b), it seemed plausible that population of this orbital due to electronic excitation would lead to P–P bond formation and therefore to isomerization to the housane 3Dmp. Accordingly, irradiation of a blue, dilute solution of 2Dmp with red light (600–650 nm) resulted in quick discoloration (Fig. 3), and the absorption band at 643 nm vanished. When the irradiation was discontinued, the blue solution was fully recovered within 30 minutes (25 °C), as evidenced by UV-Vis measurements (Fig. S18†).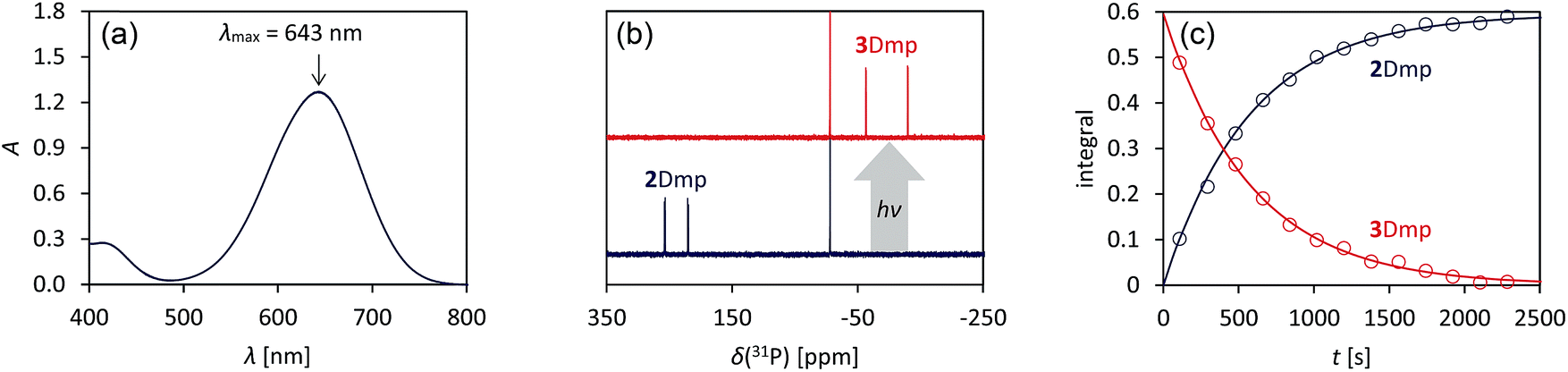 | ||
| Fig. 2 Photo-isomerization of 2Dmp and thermal equilibration of 3Dmp at 25 °C. (a) UV-Vis spectrum of 2Dmp. (b) 31P NMR spectra of a solution of 2Dmp in the dark (bottom) and under irradiation (top). (c) Thermal reverse reaction (3Dmp → 2Dmp) monitored by 31P NMR spectroscopy (please see ESI† for data at different temperatures). | ||
 | ||
| Fig. 3 Left: blue, dilute solution of 2Dmp in benzene. Right: colourless solution of the photo-product 3Dmp in benzene. | ||
Since 31P NMR spectroscopy is exceptionally well suited to study molecules containing P atoms, we decided to irradiate our samples directly in the spectrometer, so we could trace the isomerization reactions as well as determine yields, reaction rates, the stability of the system and possible by-products. We adopted a setup previously published by the Gschwind group,47 using a laser diode instead of an LED as light source. The diode was coupled with an optical fibre, which was inserted into an NMR tube equipped with a coaxial insert, allowing us to measure spectra under light (for technical details, see ESI†). Indeed, irradiation of 2Dmp with red light (638 nm) led to full conversion of the biradical 2Dmp to the housane 3Dmp (Fig. 2b). When the diode was switched off, the thermal equilibration could be monitored (Fig. 4). The thermal reverse reaction was found to be a first-order reaction with a half-life of about 7 min at ambient temperature (k = 1.73(3) × 10−3 s−1), leading to quantitative recovery of 2Dmp (Fig. 2c). The activation barrier was determined according to the Eyring theory by re-determination of the rate constants at several temperatures, giving a Gibbs free energy of activation of 88(4) kJ mol−1 at 25 °C (cf. p. S25†), in good agreement with theoretical predictions (ESI†). No signals of side products were observed in the 31P NMR spectrum after several switching cycles. In a sealed tube, a solution of 2Dmp in benzene was stable for more than a year, and it did not lose its switching capacity.
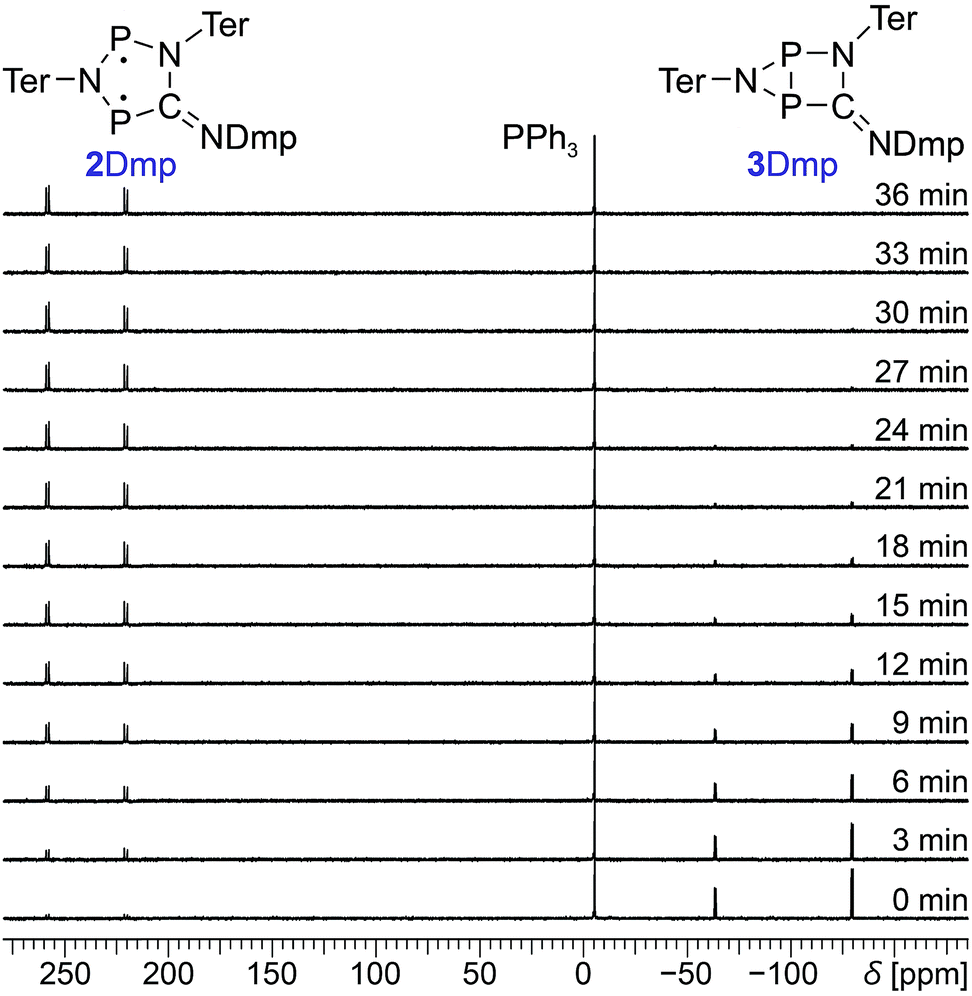 | ||
| Fig. 4 Time-dependent 31P NMR spectra of the thermal reverse reaction (3Dmp → 2Dmp). PPh3 was added as internal standard. | ||
Solid-state properties
Next, we investigated the solid-state properties of the molecular switch 2Dmp. Upon irradiation of a dark blue crystal with a red HeNe laser (633 nm, 0.1 mW), several cracks emanating from a circular, colourless spot could be observed (Fig. 5a–c). Increasing the light intensity (10 mW) led to complete discoloration, but also to fragmentation of the crystal (Fig. 5d and e). We attributed this observation to significant structural changes at the molecular level (vide infra) that caused stress within the crystal lattice and ultimately led to cracking of the crystal. When the light was switched off, a blue, polycrystalline material was recovered (Fig. 5f) that could be identified as 2Dmp by Raman spectroscopy (vide infra). | ||
| Fig. 5 Photos of a single crystal of 2Dmp at several stages of irradiation (25 °C). The cracks in the crystal due to structural changes at the molecular level can be nicely seen. (a) Before irradiation. (b) During irradiation with red laser (633 nm, 0.1 mW). (c) Immediately after irradiation for 1 min (0.1 mW). (d) Immediately after irradiation for 5 min (0.1 mW). (e) Immediately after irradiation for 5 min (10 mW). (f) 10 min after irradiation (reverse reaction is not yet complete). | ||
Notably, ambient light did not affect the integrity of single crystals at room temperature. However, at low temperatures such as −80 °C, even low intensity (white) light eventually led to their disintegration. This can be understood in terms of the thermal reverse reaction: at ambient temperature, the rate of thermal equilibration is much higher than the rate of photo-conversion from ambient light, so the light is completely absorbed at the surface of the crystal. At low temperatures, though, the thermal reverse reaction is much slower than the rate of photo-conversion. Hence, given time, a critical number of molecules will have isomerized at some point, resulting in cracking of the crystal.
With this knowledge, we could now determine the crystal structure of 2Dmp. Notably, the crystal was mounted at ambient temperature, and then cooled to −150 °C for measurement in complete darkness. (All previous attempts to obtain a complete data set failed since the crystals were mounted at low temperatures, leading to their disintegration due to irradiation by the mounting lamp.)40 The molecular structure comprises a nearly planar, five-membered P2N2C ring system (Fig. 6a). All P–N, P–C, and N–C bond lengths within the ring range between typical values48 of a single and a double bond. The transannular P⋯P distance (2.9437(7) Å) is significantly longer than a P–P single bond (Σrcov = 2.22 Å),48 in agreement with the biradical character of the compound (cf. p. S46†).
 | ||
| Fig. 6 (a) Structure of the planar P2N3C scaffold of 2Dmp as determined by single crystal X-ray diffraction. (b) Calculated structure of 3Dmp with a central hetero-bicyclo[2.1.0]pentane motif. | ||
So far, all our efforts to obtain single crystals of the housane 3Dmp have failed. Crystallization under light led to a colourless solid; however, the crystals were too small for single-crystal X-ray analysis. Nonetheless, we performed dispersion corrected49 density functional theory (DFT-D3) calculations to predict the structure (Fig. 6b). Contrary to 2Dmp, the five-membered P2N2C ring system of 3Dmp adopts an envelope conformation with a transannular P–P single bond (2.220 Å). Therefore, the structure changes significantly upon isomerization, explaining the macroscopic changes observed in the material under irradiation.
The isomerization in the solid state could also be observed by Raman microscopy. Apart from visual changes of the material, the Raman spectrum changed significantly after irradiation. Especially, the C–N stretch of the exocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond can be utilized as a probe to identify the isomers (
N double bond can be utilized as a probe to identify the isomers (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) CN = 1640 cm−1 in 3Dmp, Fig. S25†).
CN = 1640 cm−1 in 3Dmp, Fig. S25†).
Mechanism
Comprehensive computations were carried out to understand the mechanism of the photo-isomerization as well as the thermal reverse reaction. Since the correct quantum-mechanical treatment of biradicals requires multi-reference wave functions (which severely limits the size of systems that can be investigated), we opted to study the isomerization process using a model system bearing only hydrogen substituents (2H). Various calculations were performed to ascertain that this is indeed a suitable model (ESI†).The CASSCF and MRCI results indicated that the electronic excitation of 2H leads to a non-stationary point on the S1 potential energy surface (PES). Distortion of the molecule – mainly by folding of the five-membered ring along the P⋯P axis and concurrent shortening of the P⋯P interatomic distance – leads to a conical intersection (CInt) between the S1 and S0 surface, therefore allowing radiationless deactivation of the excited state. The CInt is in the vicinity of a transition state on the S0 PES which connects the biradical 2H and the housane 3H, enabling thermal isomerization of 3H to 2H (Fig. 7a; for more details on the calculations, see p. S42†).
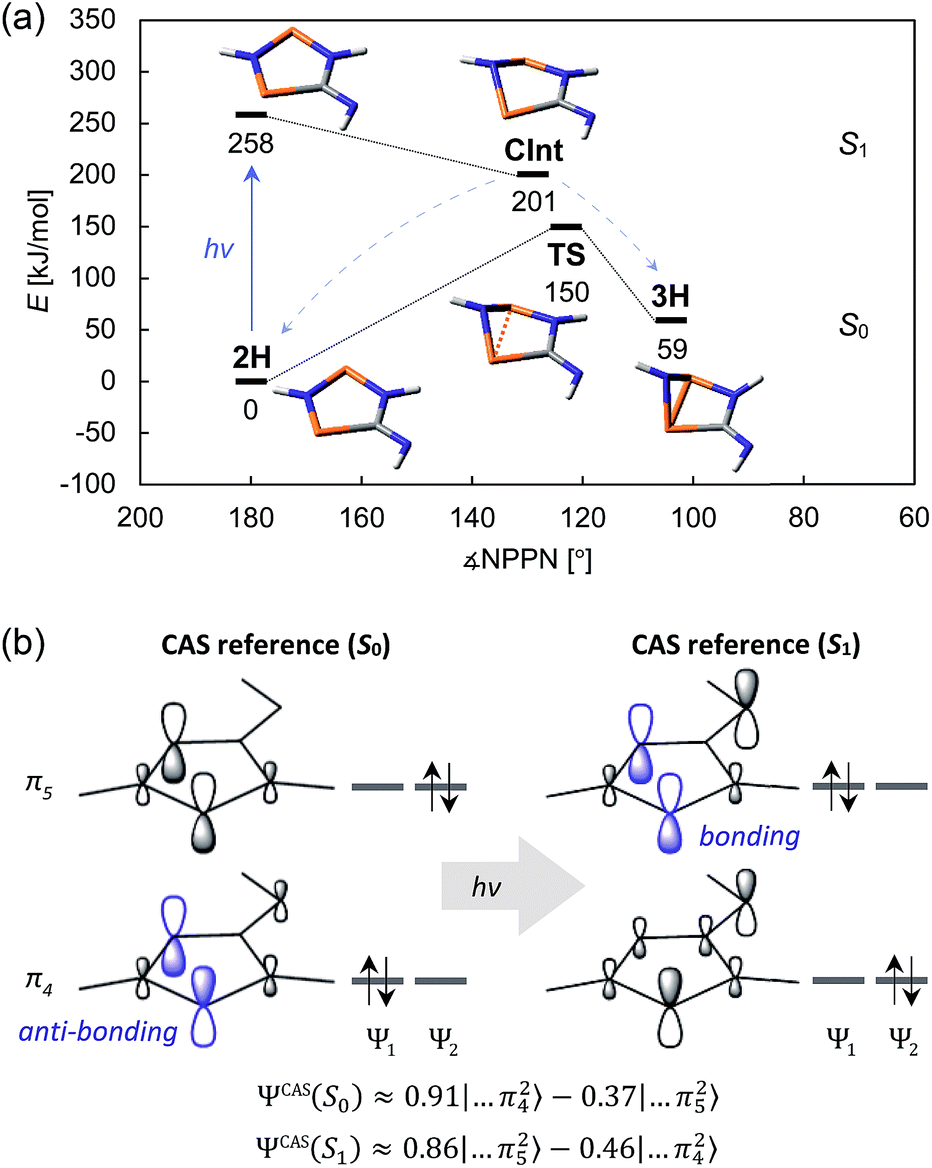 | ||
| Fig. 7 (a) Schematic view of the ground state and first excited state PES of the model system 2H. (b) Simplified depiction of the relevant orbitals of a state-specific CAS (8, 6) calculation. Upon excitation, the electron density is redistributed, resulting in a mostly bonding interaction between the two P atoms. The approximate wave functions are given by ΨCAS ≈ c1Ψ1 + c2Ψ2. | ||
As indicated, a qualitatively correct wave function of a biradical requires multiple determinants. The same is true of excited states, at least if they are modelled as higher energy roots of the CI matrix. However, depending on the reference orbitals used for the CI, the resulting wave functions may be rather difficult to interpret. The more qualitative CASSCF approach (which neglects dynamic electron correlation) is probably best suited to understand the electronic structures of 2H in its ground and first excited singlet state. In particular, a state-specific CASSCF calculation yields a unique set of orbitals for each state. These orbitals are variationally optimized to best represent the electron distribution for a given state, i.e. fewer determinants are needed to describe the wave function, making it easier to interpret. The results for 2H nicely demonstrate that both the S0 and S1 state have considerable biradical character50 (28 and 44%, respectively; cf. 27 and 36% for 2Dmp). The dominant contribution to the wave function of the ground state is transannularly anti-bonding between the two P atoms, whereas the dominant contribution to the S1 state is bonding in nature, in agreement with earlier considerations (Fig. 7b). Note that the CASSCF results also correctly indicate that the electronic excitation involves redistribution of the whole electron density, rather than just one electron being excited into a vacant orbital. A rather detailed discussion of the electronic wave function and electronic excitation including MRCI results can be found in the ESI (p. S46†).
Quantum yield
As demonstrated by the calculations, the excited state can deactivate through a conical intersection. However, this process can lead to the formation of both the starting material (2Dmp) or the photo-product (3Dmp). Thus, we were interested in the quantum yield of the photo-isomerization, i.e. the ratio of successful isomerizations to absorbed photons. Taking advantage of the different absorbances of 2Dmp and 3Dmp, we irradiated a dilute solution with a red HeNe laser (633 nm) and measured the transmitted laser intensity behind the sample. Due to photo-isomerization, the transmitted intensity increased over time, and the time-resolved data could be used to determine the quantum yield (p. S34†). With a value of 24.6(8)%, the photo-isomerization is rather efficient (cf. photosynthesis in plants: 5–12%;51–53 azobenzene: 11% (trans → cis), 42% (cis → trans);54 diarylethenes: 13–59% (cyclization), 0.002–59% (cycloreversion) depending on the substituents;55 retinal: 65%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 56).
56).
Manipulation of an equilibrium reaction
Having understood the photo-switching of the biradical 2Dmp, we were now interested whether it might be possible to manipulate a chemical reaction using this type of molecular switch. In particular, an equilibrium reaction involving activation chemistry of the biradical 2 seemed promising, as this would open the possibility to couple thermal and photochemical processes.As previously shown, the reaction of 1 with tBuNC did not lead to quantitative formation of 2tBu but rather to an equilibrium mixture of 1, 2tBu, and 4tBu2, the addition product of the biradical 2tBu and tBuNC (Scheme 1). The colourless adduct 4tBu2 could be isolated from the mixture by crystallization (ESI†).40 When re-dissolving the colourless crystals, the blue-green equilibrium mixture of 1, 2tBu, and 4tBu2 was recovered, as evidenced by 31P NMR experiments. The UV-Vis spectrum of the solution (Fig. 8a) showed a broad absorption band at 663 nm (2tBu) and two absorption maxima at 463 and 491 nm (1). Accordingly, irradiation of the solution with a red laser diode (638 nm) led to photo-isomerization of the molecular switch 2tBu. Thus, it was completely removed from the thermal equilibrium, resulting in quantitative formation of the housane 3tBu and disappearance of the thermal products 1 and 4tBu2 (Fig. 8b); i.e. the thermal equilibrium involving the activation chemistry of the biradical was entirely suppressed by the photo-isomerization! This might be regarded as “quenching of the biradical character”, as previously proposed by Wang et al.34
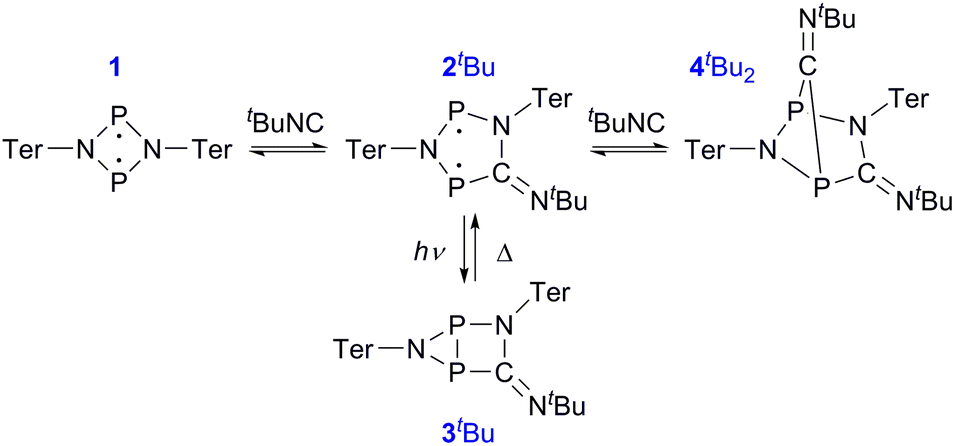 | ||
| Scheme 1 The thermal equilibrium can be completely suppressed by irradiation. | ||
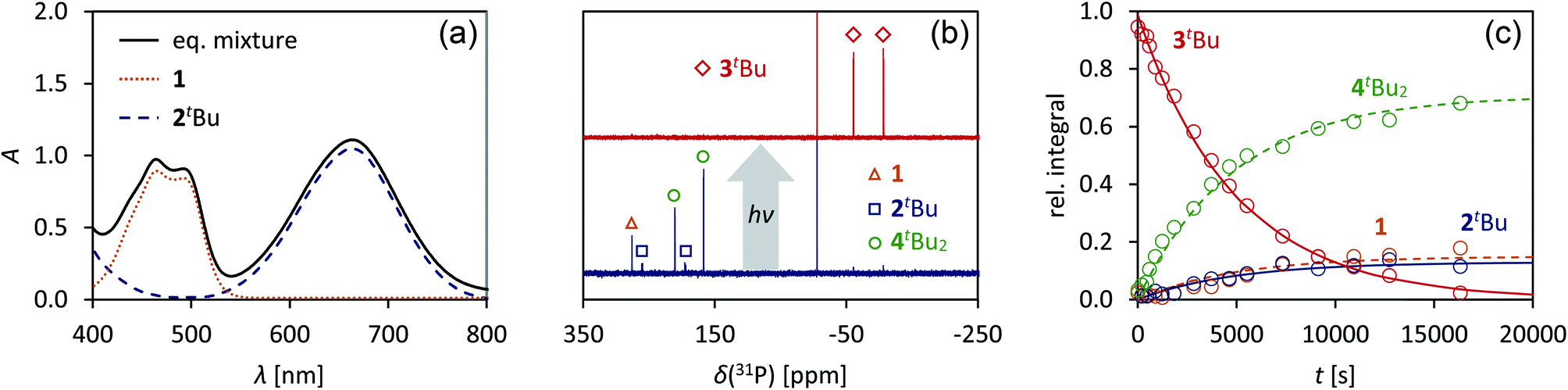 | ||
| Fig. 8 (a) UV-Vis spectrum of an equilibrium mixture of 1, 2tBu, 4tBu2, and tBuNC. (b) Upon irradiation with red light, the complete equilibrium is suppressed and the housane 3tBu is formed exclusively, as evidenced by 31P NMR spectroscopy. (c) The thermal reverse reaction to recover the equilibrium species proceeds as first-order reaction. | ||
When the light source was switched off, the thermodynamic equilibrium mixture was slowly recovered. The housane 3tBu decayed in a first order reaction with a half-life of about 57 min at 25 °C (k = 2.03(4) × 10−4 s−1, Fig. 8c). The process was completely reversible, and even after several switching cycles, no side products could be observed in the 31P NMR spectrum.
Finally, the activation of tBuNC with the more stable biradical 2Dmp was investigated. Indeed, the expected adduct 4DmptBu was formed in an equilibrium with 2Dmp (Scheme 2). To our surprise, irradiation of the blue solution with a red laser diode (638 nm) did not lead to discoloration as in the previous case. Even though formation of the photo-product 3Dmp was observed in the 31P NMR spectrum, quantitative conversion could not be achieved at ambient temperature, indicating that the rate of thermal equilibration was on the same order of magnitude as the rate of photo-conversion and therefore much faster than previously observed (cf. t1/2 ≈ 7 min in the absence of tBuNC). In fact, as soon as the irradiation was discontinued, the thermal equilibrium was restored. Low-temperature investigations indicated that the tBuNC acted as a catalytic agent to accelerate the thermal reverse reaction of the housane 3Dmp (p. S31†). The mechanism of the catalyzed reverse reaction has yet to be investigated; this is outside the scope of this article and will therefore be explored in the future. For now, we just want to point out that it is possible to chemically manipulate the pathway of the thermal reverse reaction and thus influence the switching behaviour.
 | ||
| Scheme 2 Reaction of 2Dmp with tBuNC. | ||
Conclusion
We could demonstrate that biradicals based on hetero-cyclopentane-1,3-diyls (2) are potent and chemically robust systems for application as molecular switches. They could be isomerized by red light to a closed-shell housane-type species (3) with a transannular bond, effectively quenching the biradical character. The biradical (2) was then fully recovered by a thermal reverse reaction in darkness. Substitution of the organic groups at the exocyclic CN scaffold changed the properties of the systems, promising variability of key features such as stability, activation chemistry, and excitation wavelength.
The isomerization could be monitored by UV-Vis, NMR, and Raman spectroscopy. In the solid state, significant structural changes at the molecular level upon irradiation resulted in macroscopic changes such as fragmentation of crystals. Hence, even though the molecular process was fully reversible, the macroscopic crystallinity was lost upon isomerization. In solution, the switching process could be repeated many times without any indications of decomposition. While the rate of photo-isomerization depended on the light intensity, the rate of thermal reverse reaction could be influenced either by changes in temperature or chemically by addition of isonitriles.
In case of the tBu derivative 2tBu, it was possible to completely suppress the thermal equilibrium related to the activation chemistry of the biradical upon irradiation. This serves as a proof-of-principle that chemical reactions can be controlled by molecular switches that take advantage of kinetically stabilized biradicals.
The mechanisms of both photo- and thermal isomerization were studied in detail using multi-reference calculations as well as experimental data. Formal one-electron excitation of 2 led to a transannularly bonding interaction and thus distortion of the molecule on the S1 PES. Deactivation proceeded through a conical intersection and hence radiationlessly. The quantum yield of the photochemical isomerization was determined to be 24.6(8)%. The thermal reverse reaction (3 → 2) proceeded through a single transition state on the ground state PES and was found to be a first-order reaction.
In future studies, we will further investigate the scope of hetero-cyclopentane-1,3-diyls as molecular switches, especially with respect to the control of chemical reactions, such as [2 + 2] additions of alkynes and alkenes.
Experimental
For detailed information on syntheses, equipment, analytical data, computational methods etc. please also consult the ESI.†All manipulations were carried out under oxygen- and moisture-free conditions under an inert atmosphere of argon using standard Schlenk or dry box techniques.
NMR spectroscopy under irradiation
All irradiation experiments were performed on a Bruker AVANCE 250 MHz spectrometer. Light was directed into the sample using a multimode optical fibre with 1000 μm core diameter. To ensure inert conditions, the NMR tubes were filled in a glove box and equipped with a conical insert that could host the optical fibre. Thus, the sample volume could be kept isolated from the outside atmosphere. As light source we used a 700 mW laser diode (638 nm), which was mounted on the far end of the optical fibre. The outer cladding of the fibre was removed for about 5 cm on the sample side and the glass surface was roughened to ensure uniform irradiation of the sample, as previously reported.47UV-Vis spectroscopy
UV-Vis spectra were recorded on a Perkin-Elmer Lambda 19 UV-Vis spectrometer. For irradiation studies, the samples were irradiated outside the spectrometer using white or monochromated light (600–650 nm).Determination of the quantum yield
We used a self-constructed setup mounted on a standard optical table (cf. ESI†). The samples were prepared in a glove box and filled in gas-tight cuvettes with an optical path length of 1 cm. The solutions were irradiated with a HeNe laser (633 nm) and the transmitted laser intensity was measured using a photo diode. The laser intensity was chosen so high that the rate of photo conversion was much larger than the rate of thermal equilibration; hence, the reverse reaction could be neglected. We derived the following time law for the signal sig(t) measured at the photo diode:ϕ is the quantum yield, ε the molar extinction coefficient, I0 the incident laser intensity, NA Avogadro's number, hν the energy of a photon, and a a fitting parameter related to the detection sensitivity. For a derivation of this formula and detailed description of the experimental setup, please refer to the ESI.†
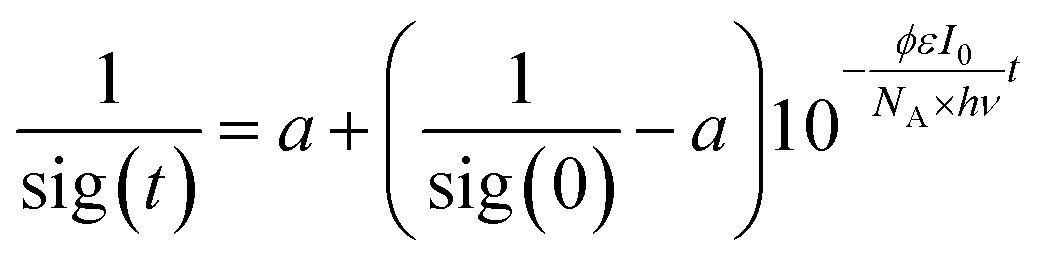
Two different concentrations of 2Dmp in benzene were prepared. The samples were irradiated for 15 seconds, and each of those measurements was repeated multiple times. The data for each of the two concentrations were combined and evaluated by non-linear fitting procedures.
Computational methods
Electronic structure computations were performed using Gaussian0957 (DFT, CASSCF) or ORCA 4.0.158 (CASSCF, MRCI). Structure optimizations were run using pure density functionals in conjunction with the D3BJ49 dispersion correction. The investigation of the PES of the model system was done using CAS(8,6) computations, including all π-type orbitals of 2H (i.e. those belonging to the A′′ irrep, point group CS).
To obtain an idea of the reliability of DFT methods to predict experimental properties of the biradical systems 1 and 2, we performed a survey of different methods and compared their results to experimental as well as high-level ab initio data. Generally, DFT methods are suitable to predict molecular structures and first excitation energies with reasonable accuracy; however, the absolute KS energies are notoriously bad due to single-determinantal treatment of the KS wave function. Notably, the PBE functional59 outperformed all other methods at predicting NMR data. Please refer to the ESI† for in-depth information.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are indebted to Henry Dube for sparking the idea that led to this study. We thank the University of Rostock for access to the cluster computer, and especially Malte Willert for his assistance with the queueing system and software installations. We thank Alexander Villinger for assistance with X-ray crystallography. Special thanks are due to Peter Kumm for his continuous commitment in the technical workshop. Financial support by the DFG (SCHU 1170/11-1) is gratefully acknowledged.Notes and references
- A. D. Johnson, A. R. Poteete, G. Lauer, R. T. Sauer, G. K. Ackers and M. Ptashne, Nature, 1981, 294, 217–223 CrossRef CAS PubMed.
- M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed.
- J. L. Zhang, J. Q. Zhong, J. D. Lin, W. P. Hu, K. Wu, G. Q. Xu, A. T. S. Wee and W. Chen, Chem. Soc. Rev., 2015, 44, 2998–3022 RSC.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- P. C. Knipe, S. Thompson and A. D. Hamilton, Chem. Sci., 2015, 6, 1630–1639 RSC.
- A. F. Hollemann, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, Walter de Gruyter, Berlin, Germany, 102nd edn, 2007 Search PubMed.
- R. W. Sabnis, E. Ross, J. Köthe, R. Naumann, W. Fischer, W.-D. Mayer, G. Wieland, E. J. Newman and C. M. Wilson, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2009 Search PubMed.
- N. A. Rakow and K. S. Suslick, Nature, 2000, 406, 710–713 CrossRef CAS PubMed.
- H. Finkelmann, E. Nishikawa, G. G. Pereira and M. Warner, Phys. Rev. Lett., 2001, 87, 015501 CrossRef CAS PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- T. Ube and T. Ikeda, Angew. Chem., Int. Ed., 2014, 53, 10290–10299 CrossRef CAS PubMed.
- R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054–5075 CrossRef CAS PubMed.
- R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982–1996 RSC.
- E. Orgiu, N. Crivillers, M. Herder, L. Grubert, M. Pätzel, J. Frisch, E. Pavlica, D. T. Duong, G. Bratina, A. Salleo, N. Koch, S. Hecht and P. Samorì, Nat. Chem., 2012, 4, 675–679 CrossRef CAS PubMed.
- M. El Gemayel, K. Börjesson, M. Herder, D. T. Duong, J. A. Hutchison, C. Ruzié, G. Schweicher, A. Salleo, Y. Geerts, S. Hecht, E. Orgiu and P. Samorì, Nat. Commun., 2015, 6, 6330 CrossRef CAS PubMed.
- G. Wald, J. Gen. Physiol., 1948, 31, 489–504 CrossRef CAS PubMed.
- G. Wald, J. Gen. Physiol., 1949, 32, 367–389 CrossRef CAS PubMed.
- G. Wald, Science, 1968, 162, 230–239 CrossRef CAS PubMed.
- B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS PubMed.
- G. S. Hartley, Nature, 1937, 140, 281 CrossRef CAS.
- J. M. Robertson, J. Chem. Soc., 1939, 0, 232–236 RSC.
- Y. Yokoyama, Chem. Rev., 2000, 100, 1717–1740 CrossRef CAS PubMed.
- G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741–1754 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- L. Salem and C. Rowland, Angew. Chem., Int. Ed. Engl., 1972, 11, 92–111 CrossRef CAS.
- Diradicals, ed. W. T.Borden, Wiley-Interscience, New York, 1982 Search PubMed.
- F. Breher, Coord. Chem. Rev., 2007, 251, 1007–1043 CrossRef CAS.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- K. Okuno, Y. Shigeta, R. Kishi and M. Nakano, J. Phys. Chem. Lett., 2013, 4, 2418–2422 CrossRef CAS.
- A. Hinz, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Chem.–Eur. J., 2014, 20, 14659–14673 CrossRef CAS PubMed.
- A. Hinz, R. Kuzora, A.-K. Rölke, A. Schulz, A. Villinger and R. Wustrack, Eur. J. Inorg. Chem., 2016, 2016, 3611–3619 CrossRef CAS.
- J. Bresien, A. Hinz, A. Schulz and A. Villinger, Eur. J. Inorg. Chem., 2018, 2018, 1679–1682 CrossRef CAS.
- J. Bresien, A. Hinz, A. Schulz and A. Villinger, Dalton Trans., 2018, 47, 4433–4436 RSC.
- J.-J. Wang, Z.-J. Zhou, H.-M. He, D. Wu, Y. Li, Z.-R. Li and H.-X. Zhang, J. Phys. Chem. C, 2016, 120, 13656–13666 CrossRef CAS.
- H. Li, A. C. Fahrenbach, A. Coskun, Z. Zhu, G. Barin, Y.-L. Zhao, Y. Y. Botros, J.-P. Sauvage and J. F. Stoddart, Angew. Chem., Int. Ed., 2011, 50, 6782–6788 CrossRef CAS PubMed.
- A. T. Buck, J. T. Paletta, S. A. Khindurangala, C. L. Beck and A. H. Winter, J. Am. Chem. Soc., 2013, 135, 10594–10597 CrossRef CAS PubMed.
- J. Sun, Y. Wu, Y. Wang, Z. Liu, C. Cheng, K. J. Hartlieb, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 13484–13487 CrossRef CAS PubMed.
- P. Ravat, T. Solomek, D. Häussinger, O. Blacque and M. Juricek, J. Am. Chem. Soc., 2018, jacs.8b05465 Search PubMed.
- A. Hinz, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 2776–2779 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, J. Am. Chem. Soc., 2015, 137, 9953–9962 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, Chem. Sci., 2016, 7, 745–751 RSC.
- A. Schulz, Dalton Trans., 2018, 47, 12827–12837 RSC.
- F. Kita, W. Adam, P. Jordan, W. M. Nau and J. Wirz, J. Am. Chem. Soc., 1999, 121, 9265–9275 CrossRef CAS.
- W. Adam, W. T. Borden, C. Burda, H. Foster, T. Heidenfelder, M. Heubes, D. A. Hrovat, F. Kita, S. B. Lewis, D. Scheutzow and J. Wirz, J. Am. Chem. Soc., 1998, 120, 593–594 CrossRef CAS.
- D. Y. Zhang, D. A. Hrovat, M. Abe and W. T. Borden, J. Am. Chem. Soc., 2003, 125, 12823–12828 CrossRef CAS PubMed.
- M. Abe, W. Adam, W. T. Borden, M. Hattori, D. A. Hrovat, M. Nojima, K. Nozaki and J. Wirz, J. Am. Chem. Soc., 2004, 126, 574–582 CrossRef CAS PubMed.
- C. Feldmeier, H. Bartling, E. Riedle and R. M. Gschwind, J. Magn. Reson., 2013, 232, 39–44 CrossRef CAS PubMed.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- E. Miliordos, K. Ruedenberg and S. S. Xantheas, Angew. Chem., Int. Ed., 2013, 52, 5736–5739 CrossRef CAS PubMed.
- J. B. Skillman, J. Exp. Bot., 2007, 59, 1647–1661 CrossRef PubMed.
- S. W. Hogewoning, E. Wientjes, P. Douwstra, G. Trouwborst, W. van Ieperen, R. Croce and J. Harbinson, Plant Cell, 2012, 24, 1921–1935 CrossRef CAS PubMed.
- K. Stranius and K. Börjesson, Sci. Rep., 2017, 7, 41145 CrossRef CAS PubMed.
- G. Zimmerman, L. yung Chow and U. jin Paik, J. Am. Chem. Soc., 1958, 80, 3528–3531 CrossRef CAS.
- K. Morimitsu, S. Kobatake and M. Irie, Mol. Cryst. Liq. Cryst., 2005, 431, 451–454 CrossRef CAS.
- A. Kropf, in Methods Enzymol., 1982, vol. 81, pp. 384–392 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Peterson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Know, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrezewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V Ortiz, J. Cioslowski and D. J. Fox, Gaussian09, Revision E.01, Gaussian Inc, 2013 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental information (incl. experimental setups), structure elucidation, syntheses, spectroscopic details and computational details. CCDC 1869014 and 1869015. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04893b |
| This journal is © The Royal Society of Chemistry 2019 |