
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLocation analysis of 8-oxo-7,8-dihydroguanine in DNA by polymerase-mediated differential coding†
Feng
Tang‡
a,
Shan
Liu‡
a,
Qiao-Ying
Li
a,
Jun
Yuan
b,
Lin
Li
b,
Yinsheng
Wang
b,
Bi-Feng
Yuan
 *a and
Yu-Qi
Feng
a
*a and
Yu-Qi
Feng
a
aKey Laboratory of Analytical Chemistry for Biology and Medicine, Ministry of Education, Department of Chemistry, Wuhan University, Wuhan 430072, P. R. China. E-mail: bfyuan@whu.edu.cn; Fax: +86-27-68755595; Tel: +86-27-68755595
bDepartment of Chemistry and Environmental Toxicology Graduate Program, University of California, Riverside, CA 92521-0403, USA
First published on 11th March 2019
Abstract
Accumulating lines of evidence indicate that reactive oxygen species (ROS) are important signalling molecules for various cellular processes. 8-Oxo-7,8-dihydroguanine (OG) is a prominent oxidative modification formed in DNA by ROS. Recently, it has been proposed that OG may have regulatory and possibly epigenetic-like properties in modulating gene expression by interfering with transcription components or affecting the formation of G-quadruplex structures. Deciphering the molecular mechanisms of OG on regulation of gene expression requires uncovering the location of OG on genome. In the current study, we characterized two commercially available DNA polymerases, Bsu DNA polymerase (Bsu Pol) and Tth DNA polymerase (Tth Pol), which can selectively incorporate adenine (A) and cytosine (C) opposite OG, respectively. By virtue of the differential coding properties of Bsu Pol and Tth Pol that can faithfully or error-prone copy a DNA strand carrying OG, we achieved quantitative and single-base resolution analysis of OG in synthesized DNA that carries OG as well as in the G-rich telomeric DNA from HeLa cells. In addition, the parallel analysis of the primer extension products with Bsu Pol and Tth Pol followed by sequencing provided distinct detection of OG in synthesized DNA. Future application of this approach will greatly increase our knowledge of the chemical biology of OG with respect to its epigenetic-like regulatory roles.
Introduction
Reactive oxygen species (ROS) are typically produced from aerobic metabolism and play important roles in cell signaling and homeostasis.1 ROS are electron deficient and can readily oxidize a variety of macromolecules, such as proteins, lipids, and nucleic acids.2 In DNA, the guanine heterocycle is the most susceptible of the four DNA bases to oxidation because it has the lowest redox potential that makes it the major target of ROS.3,4 A prominent oxidative compound observed is 8-oxo-7,8-dihydroguanine (OG) among these oxidatively modified products (Fig. 1A).3 OG does not block nucleic acid synthesis but rather induces alternate base mispairing, causing G → T transversion mutations that are suspected in disease initiation and progression.5,6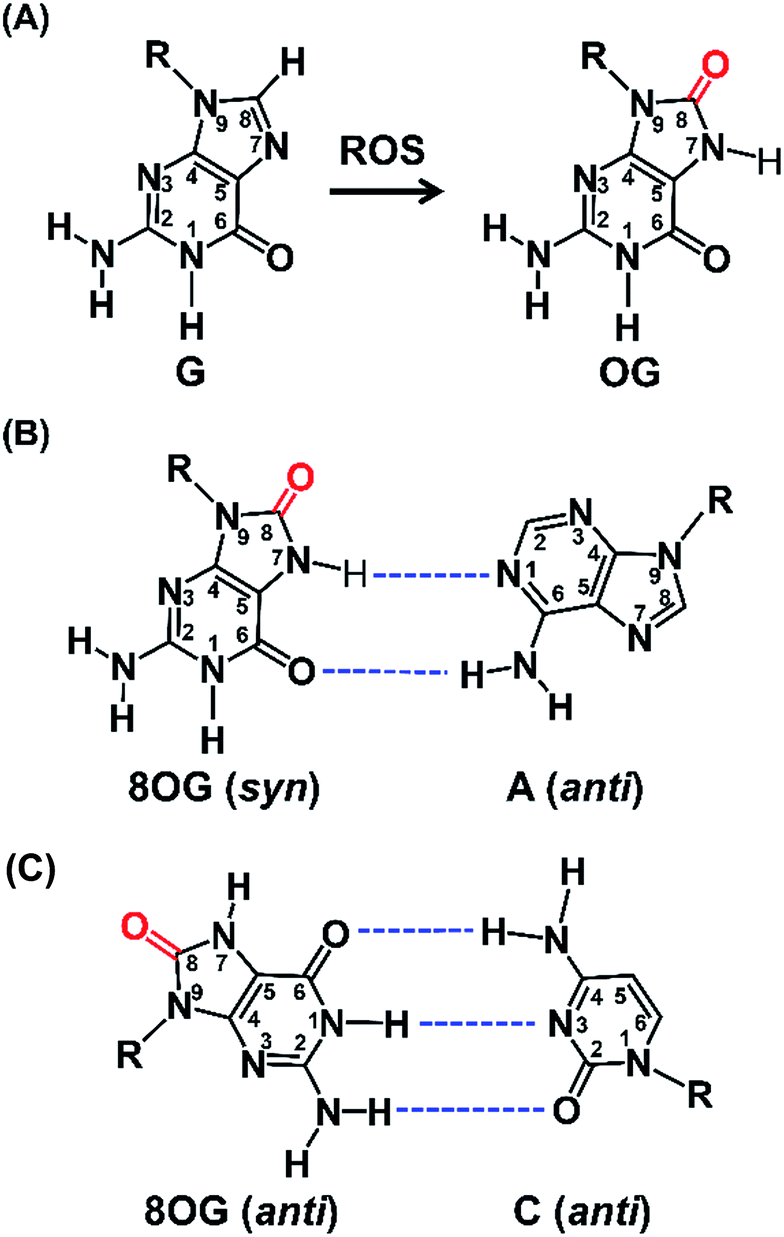 | ||
| Fig. 1 The formation of OG from G by ROS and the base pairing of OG with A and C. (A) Oxidation of G at C8 to form OG by ROS. (B) OG (syn) in a Hoogsteen base pairing with A (anti). (C) OG (anti) in a Watson–Crick base pairing with C (anti). Dashed lines indicate hydrogen bonds. | ||
The long-standing view has been that OG is mutagenic and detrimental to cellular processes.7 Recently, OG has been proposed to be able to modulate gene expression.8 A few examples showed that the increased OG formation in the genome is correlated with increased gene expression via the base excision repair (BER) pathway.9,10 Zarakowska et al.11 reported that the transcriptionally active euchromatin DNA harbored more OG than that in transcriptionally silenced heterochromatin in porcine thymus DNA. Park, et al.12 found that G oxidation to OG could occur site specifically in vivo under oxidative stress. More recently, Fleming et al.13 demonstrated that OG can activate mRNA synthesis by facilitating promoter G-quadruplex formation and cells can harness oxidative modification of G to OG for altering phenotype. These studies indicate that OG may have regulatory and possibly epigenetic-like properties in cells that respond to oxidative stress.
Traditional methods for detection of OG normally involve the comet assay14 or OG was determined by extraction of genomic DNA followed by nuclease digestion and mass spectrometry analysis.15 However, these approaches cannot determine the location of the OG in DNA. Thus, sequencing the genome for locating OGs will provide better understanding of the effects of OG on gene regulation. Direct sequencing for OG has been achieved by single-molecule, real time (SMRT)16 or nanopore-based sequencing methods.17 However, these methods generally are proof-of-concept studies and further improvement is required to realize the mapping of OG in genome. Several studies reported the antibody-based OG sequencing that provided a low-resolution sequence mapping of OG (∼10 kb).18,19 More recently, Ding et al.20 developed an approach (OG-Seq) that utilized amine-terminated biotin to label OG for affinity purification followed by next-generation sequencing to map OG in the mouse genome at ∼0.15 kb resolution. This study showed that gene promoters and untranslated regions (UTRs) harbor more OG-enriched sites. Although these studies offer significant advancement of our knowledge regarding genomic OG, the resolution is not high enough to determine precise genomic elements where OG resides. It is important to provide the information about single-base resolution location of OG in genome-wide scale for understanding the molecular basis of diseases as well as the regulatory roles that are related to OG.
Structural studies demonstrate that OG can adopt two alternative conformations (anti or syn) in the active site of DNA polymerases and therefore OG has dual coding potential.21 The anti conformation of OG allows Watson–Crick base pairing with a cytosine, whereas the syn conformation of OG forms a stable mispairing with an adenine in a normal anti conformation by Hoogsteen base pair (Fig. 1B).21 DNA polymerase β (Pol β) has been shown to be able to incorporate 8-oxo-dGTP opposite adenine in preference to cytosine.22 In contrast, the Y-family enzyme of DNA polymerase ι (Pol ι) prefers error-free bypass of OG and can preferentially insert dCTP opposite OG site.23 Therefore, by virtue of the differential properties of different DNA polymerases that can faithfully or error-prone copy a DNA strand carrying OG, we can develop single-base resolution mapping of OG in DNA.
In the current study, we characterized commercially available DNA polymerases and found that Bsu DNA polymerase (Bsu Pol) predominantly incorporated adenine (A) opposite OG and Tth DNA polymerase (Tth Pol) predominantly incorporated cytosine (C) opposite OG. Using the distinct properties of these two DNA polymerases, we developed an approach for detection of OG in DNA at single-base resolution. This method was then successfully used to quantification of OG in telomeric DNAs from HeLa cells.
Experimental section
Chemicals and reagents
Bsu DNA polymerase (Bsu Pol), Klenow fragment (exo−) (KF Pol), Therminator DNA polymerase (The Pol), Deep Vent (exo−) DNA polymerase (Vent Pol), and Taq DNA polymerase (Taq Pol) were purchased from New England Biolabs (Ipswich, MA, USA). Tth DNA polymerase (Tth Pol) was purchased from Toyobo Life Science Co., Ltd. (Shanghai, China). All of the oligonucleotides were synthesized and purified by Sangon Biotech Co., Ltd. (Shanghai, China). The 134-mer OG-containing single-stranded DNA (L-DNA-OG1, L-DNA-OG2, L-DNA-OG3), the control 134-mer single-stranded DNA (L-DNA-G), and 2′-deoxyribonucleoside triphosphates (dNTPs) were purchased from Takara Biotechnology Co., Ltd. (Dalian, China). The 51-mer DNA (NN-OG-NN), with two randomized bases flanking each side of the OG site was purchased from Integrated DNA Technologies (Iowa, USA). All the DNA sequences are listed in Table S1 in ESI.†Cell culture and H2O2 treatment
HeLa cells were obtained from the China Center for Type Culture Collection (CCTCC) and maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% FBS, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (GIBCO) at 37 °C under 5% CO2 atmosphere. The H2O2 treatment of HeLa cells was performed according to previous method.24 Briefly, the cells were grown to ∼80% confluence in 25 cm2 culture flasks, washed with phosphate-buffered saline (PBS) and then treated with medium including 1000 μM H2O2. After incubation for 30 min in a CO2 incubator, the medium was removed and cells were harvested. Genomic DNA was isolated according to preciously described method with using deferoxamine mesylate as the oxidation inhibitor to protect DNA from excessive oxidation during extraction processing.25 DNA concentration was quantified by NanoDrop 2000c (Thermo Scientific, USA).Single-nucleotide primer extension
Single-nucleotide primer extension assay was performed to screen appropriate DNA polymerase that can most preferentially incorporate dATP opposite OG-containing DNA template. The mixture of FAM-labeled primer (FAM-primer-1,5′-FAM-CGCATAACCCTAACC-3′, 1.0 μM) and template DNA (DNA-G or DNA-OG, 5′-GCGTATTGGGATTGGXATTGACACG-3′, X represents OG or G, 1.0 μM), in buffer containing 10 mM Tris–HCl, 50 mM NaCl, 10 mM MgCl2 and 1 mM DTT, pH 7.9 was annealed at 95 °C for 5 min and then cooled down to room temperature. Then different DNA polymerase was added to the mixture. The reaction was initiated by the addition of dATP (100 μM) in the mixture (10 μL). After incubation at 37 °C for 20 min, the reactions were quenched by adding 20 μL of stop buffer (95% formamide, 25 mM EDTA, pH 8.0), and the solution was immediately heated at 95 °C for 10 min. After cooling down to room temperature, the solution was analyzed by denaturing 20% polyacrylamide gel electrophoresis (PAGE, acrylamide/bisacrylamide = 19/1) in the presence of 7 M urea. All the bands were visualized and quantified using Pharos FX Molecular imager (Bio-Rad, USA).Steady-state kinetics study
The mixture of FAM-primer-1 (1.0 μM) and DNA-G or DNA-OG (1.0 μM), in buffer containing 10 mM Tris–HCl, 50 mM NaCl, 10 mM MgCl2 and 1 mM DTT, pH 7.9 was annealed at 95 °C for 5 min and then cooled down to room temperature. Then DNA polymerase was added to the mixture. The reaction was initiated by the addition of dATP (2–4000 μM) and the mixture (10 μL) was incubated for 1–20 min at 37 °C. The following quenching step and gel electrophoresis analysis are the same as those for single-nucleotide primer extension assay.The relative reaction velocity (v) was calculated from the ratio of the extended product (IE) over the unextended primer (IU) plus the extension product (IE) as follows: v × t = IE/(IU + IE), where t represents the reaction time. The apparent KM and Vmax values were obtained from linear regression analysis of Hanes–Woolf plots using the data points at different dATP concentrations in three independent experiments according to previous described method.26 This assay provides a straightforward method for determining the enzymatic efficiency (Vmax/Km), which is used to describe the selectivity of DNA polymerase for incorporating dATP opposite OG or G. These selectivity values are independent of enzyme activity and thus allow the comparison of different DNA polymerases.
Measurement of the content of OG at telomeric DNA
The control DNA template (T-DNA-1) and three DNA templates (T-DNA-2, T-DNA-3, and T-DNA-4) that carry OG at different sites in DNA of were used to mimic the telomeric DNA (detailed DNA sequences can be found in Table S1 in ESI†). As for the measurement of the content of OG at telomeric DNA, single-nucleotide primer extension was performed with the mixture (10 μL) of HeLa DNA (5 μg) and different fluorophore-labeled primers (Cy3-primer, Cy5-primer, and FAM-primer-2, 5 pmol for each) under the aforementioned conditions. The percentage of the extension of primers was determined based on three independent experiments.
were used to mimic the telomeric DNA (detailed DNA sequences can be found in Table S1 in ESI†). As for the measurement of the content of OG at telomeric DNA, single-nucleotide primer extension was performed with the mixture (10 μL) of HeLa DNA (5 μg) and different fluorophore-labeled primers (Cy3-primer, Cy5-primer, and FAM-primer-2, 5 pmol for each) under the aforementioned conditions. The percentage of the extension of primers was determined based on three independent experiments.
Analysis of OG by Sanger sequencing
The mixture of L-DNA-G (100 nM) or L-DNA-OG1 (100 nM) and L-primer (100 nM) in buffer containing 10 mM Tris–HCl, 50 mM NaCl, 10 mM MgCl2 and 1 mM DTT, pH 7.9 was incubated at 95 °C for 5 min followed by cooling down to 25 °C (detailed DNA sequences can be found in Table S1 in ESI†). dNTPs (final concentration of 100 μM for each), Bsu Pol or Tth pol (0.5 U) were added. The mixture was incubated at 37 °C for 20 min. The extension product was then treated with exonuclease I (15 U) at 37 °C for 15 min to eliminate the L-primer. Then the product was purified by QIAquick PCR Purification Kit (QIAGEN) according to the manufacturer's instruction. The purified extension product was used as template for PCR amplification and then subjected to Sanger sequencing.In addition to the synthesized DNA carrying OG, the sequencing strategy was also applied for the analysis of OG in genomic DNA of HeLa cells. Three human genes, including VEGFA (vascular endothelial growth factor A), TP53 (tumour protein p53), and KRAS (KRAS proto-oncogene, GTPase), were amplified using HeLa genomic DNA as the template. HeLa genomic DNA (500 ng) was denatured and annealed with the corresponding extension primers (VEGFA-L, TP53-L, or KRAS-L, Table S2 in ESI†). The following procedures are the same as those for the analysis of synthesized DNA. In addition, H2O2-treated genomic DNA was also employed as the template for the sequencing analysis. The H2O2 treatment was performed according to a previously described method.27 The detailed sequences of PCR primers can be found in Table S2 in ESI.†
Sequencing of clones
To quantitatively determine the ratio of the G-to-T conversion rates, the primer extension products from DNA templates with different ratios of L-DNA-OG1/L-DNA-G by Bsu Pol were amplified by PCR and the PCR products were then ligated and cloned using pClone007 Blunt Simple Vector System (TSINGKE, Beijing, China) following the manufacturer's instructions. Individual clones were picked, lysed in TE buffer, amplified by PCR using M13 forward and reverse primers, and then sequenced using an ABI3700 (Applied Biosystems, Inc.). Forty positive clones per sample were picked and subjected to sequencing.Analysis of OG by high-throughput sequencing
To quantitatively determine the G-to-T conversion rates, the primer extension products by Bsu Pol from DNA templates of 5 different ratios of L-DNA-OG1/L-DNA-G ranging from 0% to 20% were amplified by PCR and subjected to high-throughput sequencing using the NEB Next® Ultra™ II DNA Library Prep Kit for Illumina® (New England Biolabs, Ipswich, MA, USA). Five pairs of primers with different barcodes were used for indexing the five different groups of the mixture of L-DNA-OG1/L-DNA-G. The pooled DNA was then sequenced on an Illumina HiSeq2500 High-throughput Sequencing system (Illumina). After obtaining the raw sequencing data, the reads that failed to pass the Illumina chastity filter were removed. The low-quality reads which contain >1 nt with a quality score below 20 or any undefined nucleotide ‘N’ were further filtered and removed. Only the reads with perfect match to characteristic strings ‘AGTCGCCTCCCACTTTTGGT’ from the −20th to −1st nucleotides of the OG site for forward sequence reads, or the reads with perfect match to characteristic strings ‘TGTAAATAAACAAGCAGTCA’ from the 1st to 20th nucleotides for reverse sequence reads were selected for calculating the nucleobase (A, T, C or G) frequencies at the original OG site. The mutation rate was calculated using the following formula, %T = (total number of T/total number of reads) × 100%.To investigate potential mutation rate dependency on the sequence context, we also performed the high-throughput sequencing using the synthesized DNA of NN-OG-NN that carries two randomized bases flanking each side of the OG site. The primer extension products from the NN-OG-NN by Bsu Pol were amplified by PCR and subjected to high-throughput sequencing. The library construction, high-throughput sequencing, and processing of raw sequencing data were carried out following the aforementioned protocol. Only the reads with perfect match to characteristic strings ‘GACGACTGGCACTAATG’ from the −19th to −3rd nucleotides of the OG site for forward sequence reads, or the reads with perfect match to characteristic strings ‘TGTCGATCCACGCAGCA’ from the 3rd to 19th nucleotides for reverse sequence reads were used to calculate the frequency of the nucleobases flanking original OG site. The sequence context around the OG site was analyzed and generated by Sequence-logo.
Results and discussion
Bsu Pol incorporates dA and Tth Pol incorporates dC opposite OG
Previous structural studies demonstrated that OG can adopt anti or syn conformation.28 The anti conformation of OG retains the ability to engage in Watson–Crick base pair with cytosine, but oxidation of guanine at C8 position converts a hydrogen bond acceptor (N7) to a hydrogen bond donor, which allows a stable Hoogsteen base pair between syn conformation of OG and adenine.28 This dual coding property of OG being able to pair with both cytosine and adenine offers a possibility to develop appropriate approach for single-base resolution analysis of OG in DNA. DNA polymerase-mediated primer extension may provide a possible method for locating OG during replication of OG-containing DNA due to their sterically sensitive active sites while replicating past OG. We postulated that certain DNA polymerase may selectively incorporate A or C opposite OG, which should be harnessed for detection of OG in DNA.We first carried out a screen of commercially available DNA polymerases by monitoring their ability to extend a FAM-labeled single-stranded DNA (FAM-primer-1) for incorporating dATP opposite either G or OG in DNA template (Fig. 2A). The preliminary result of single-nucleotide primer extension assay demonstrated that Bsu Pol, KF Pol, and The Pol showed good performance on incorporation of dATP opposite OG. But KF Pol and The Pol also can incorporate dATP opposite normal G (Fig. 2B), excluding the potential use of these two DNA polymerases for discriminating G and OG in DNA.
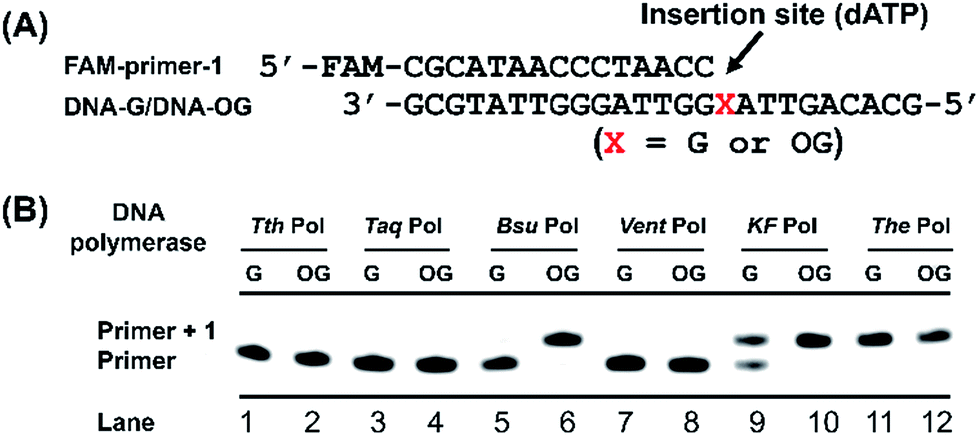 | ||
| Fig. 2 Screen of DNA polymerase for selective incorporation of dATP opposite OG in DNA template. (A) Sequences of DNA templates (DNA-G or DNA-OG) and primer (FAM-primer-1). (B) Single-nucleotide primer extension reaction with dATP. DNA-G was used as DNA template in lanes 1, 3, 5, 7, 9, and 11; DNA-OG was used as DNA template in lanes 2, 4, 6, 8, 10, and 12. The reaction products were analyzed by 20% denaturing PAGE. | ||
We next performed steady-state kinetics study for quantitative comparison of the properties of these DNA polymerases on incorporating dATP opposite G and OG in DNA. As shown in Table 1, Bsu Pol displays the greatest discrimination (34.1-fold) between OG and G in single-nucleotide primer extension reaction with dATP as the substrate. The Vmax value of the DNA-OG template with dATP was higher than that of DNA-G template, and the DNA-OG template had a much more favorable KM value, which resulted in a remarkably better enzyme efficiency (Vmax/KM) for DNA-OG template (27.3 × 10−3 min−1 μM−1) than that for DNA-G template (0.8 × 10−3 min−1 μM−1, Table 1). Given that the adenine base forms a Hoogsteen base pair with OG, it is reasonable to assume that the A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OG base pair is more stable than the A:G base pair. The result of KF Pol also revealed a preference (18.7-fold) for DNA-OG template over the DNA-G template (Table 1). Even though the extension efficiency (Vmax/KM) of KF Pol (66.3 × 10−3 min−1 μM−1) for DNA-OG template was higher than that of Bsu Pol (27.3 × 10−3 min−1 μM−1), it had a higher extension efficiency (Vmax/KM) for DNA-G template (3.5 × 10−3 min−1 μM−1) than that for Bsu Pol (0.8 × 10−3 min−1 μM−1), which leads to a slightly less discrimination (18.7-fold) between OG and G compared with Bsu Pol (34.1-fold). The other DNA polymerases didn't show good discrimination between OG and G in DNA (Table 1).
:OG base pair is more stable than the A:G base pair. The result of KF Pol also revealed a preference (18.7-fold) for DNA-OG template over the DNA-G template (Table 1). Even though the extension efficiency (Vmax/KM) of KF Pol (66.3 × 10−3 min−1 μM−1) for DNA-OG template was higher than that of Bsu Pol (27.3 × 10−3 min−1 μM−1), it had a higher extension efficiency (Vmax/KM) for DNA-G template (3.5 × 10−3 min−1 μM−1) than that for Bsu Pol (0.8 × 10−3 min−1 μM−1), which leads to a slightly less discrimination (18.7-fold) between OG and G compared with Bsu Pol (34.1-fold). The other DNA polymerases didn't show good discrimination between OG and G in DNA (Table 1).
| DNA polymerase | DNA template | V max (% min−1) | K M (μM) | V max/KM (min−1 μM−1) | Discrimination |
|---|---|---|---|---|---|
| a V max, the maximum rate of the enzyme reaction. KM, the Michaelis constant. Discrimination = (Vmax/KM) DNA-OG/(Vmax/KM) DNA-G. | |||||
| Bsu | DNA-G | 14.5 ± 2.3 | 178.6 ± 4.9 | 0.8 × 10−3 | 34.1 |
| DNA-OG | 25.1 ± 2.4 | 9.2 ± 0.6 | 27.3 × 10−3 | ||
| KF | DNA-G | 26.0 ± 2.0 | 73.7 ± 7.2 | 3.5 × 10−3 | 18.7 |
| DNA-OG | 34.5 ± 0.5 | 5.2 ± 1.1 | 66.3 × 10−3 | ||
| The | DNA-G | 56.3 ± 1.7 | 2.9 ± 0.7 | 194.1 × 10−3 | 1.3 |
| DNA-OG | 53.7 ± 1.8 | 2.1 ± 0.8 | 255.7 × 10−3 | ||
| Vent | DNA-G | 9.0 ± 1.1 | 274.8 ± 17.4 | 0.3 × 10−3 | 1.3 |
| DNA-OG | 17.0 ± 5.6 | 451.0 ± 46.7 | 0.4 × 10−3 | ||
| Taq | DNA-G | 12.6 ± 0.4 | 90.5 ± 21.0 | 1.4 × 10−3 | 2.1 |
| DNA-OG | 6.4 ± 0.6 | 21.6 ± 3.5 | 3.0 × 10−3 | ||
| Tth | DNA-G | 5.4 ± 0.5 | 228.3 ± 15.4 | 0.2 × 10−3 | 1.0 |
| DNA-OG | 6.2 ± 0.6 | 275.8 ± 18.2 | 0.2 × 10−3 | ||
It is worth noting that the single-nucleotide primer extension by Tth Pol for both DNA-G and DNA-OG template with dATP scarcely happened (Fig. 2B and Table 1), indicating that Tth Pol has very low tolerance for both A:G and A:OG base pairing. We then further performed the steady-state kinetics study using dCTP by Tth Pol. The result showed that, other than dATP, Tth Pol preferentially incorporated dCTP opposite OG (Table S3 and Fig. S2 in ESI†).
Previous crystal structure of DNA polymerase ι (Pol ι) in complex with OG-containing DNA revealed, in the narrow active site of Pol ι, adenine nucleoside adopts a syn conformation, which destabilizes A:OG base pair and favours C:OG base pair.23 It's possible that Tth Pol may have a similar mechanism to selectively incorporate C opposite OG. Collectively, the properties of Bsu Pol selectively incorporating A opposite OG and Tth Pol selectively incorporating C opposite OG offer the possibility to develop an approach for quantitative analysis of OG through comparing the primer extension product with Bsu Pol and Tth Pol.
Quantitative evaluation of the level of OG in DNA
Next, we evaluated whether the method is capable of quantitative analysis of OG at a specific site in DNA. To this end, we mixed different ratios of DNA-OG/DNA-G with the percentage of DNA-OG ranging from 0% to 100% and measured the yield of primer extension products. The single-nucleotide primer extension assay was performed with 100 μM dATP and 0.5 U Bsu Pol in the 10 μL mixture at 37 °C for 20 min, under which conditions the FAM-primer-1 cannot be extended with DNA-G template. The extension rate of FAM-primer-1 at each percentage of DNA-OG was calculated using the formula of IE/(IU + IE). The result showed that the extension rate of the FAM-primer-1 was linearly proportional to the percentage of OG with the coefficient of determination (R2) being 0.993 (Fig. 3), suggesting that the single-nucleotide primer extension assay can be harnessed for quantitative evaluation of the content of OG at specific sites in DNA.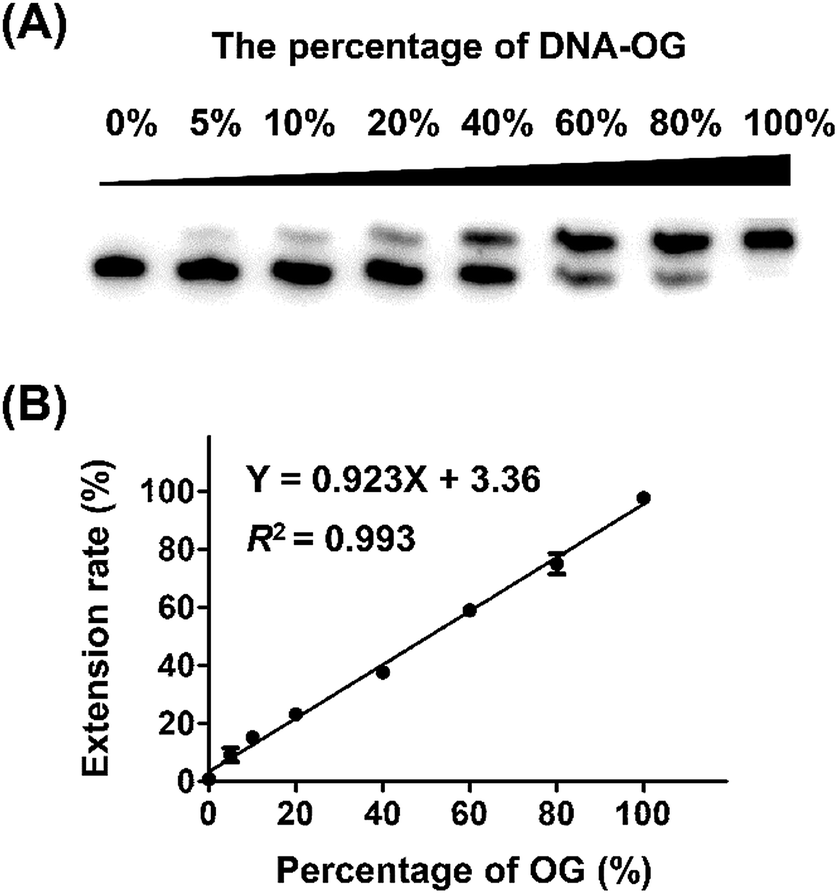 | ||
| Fig. 3 Quantitative evaluation of the level of OG in DNA. (A) Single-nucleotide primer extension reaction using the mixture of DNA-OG and DNA-G with the percentage of DNA-OG ranging from 0% to 100%. (B) The extension rate of FAM-primer-1 is correlated to the relative amounts of OG. Error bars show standard deviations from three independent experiments. | ||
Measurement of the level of OG at telomeric DNA
G-rich sequences in DNA can assemble into G-quadruplex structures.29 Recently, G-quadruplexes have been reported to play critical regulatory roles in a variety of biological processes, including DNA replication, transcription, and translation, which provide new and important mechanisms for controlling gene expression and genome stability.30 Human telomeres carry the repeating sequence of 5′-(TTAGGG)n-3′ that allows formation of G-quadruplex. The G-rich sequences are highly susceptible to oxidation of G to generate OG.31 Since OG is not capable to form Hoogsteen base pairing with G,32 OG can therefore cause significant perturbation of the structure of G-quadruplex, which may impose effects on the regulatory functions of G-quadruplex. In this respect, elucidation of the content of OG at telomeric DNA will provide a better understanding of the regulatory roles of OG in response to stresses through perturbing the structure of G-quadruplexes.First, we used four synthetic DNA with (TTAG1G2G3)4 repeat sequence, including DNA-1 (G1, G2, and G3 represent guanine), DNA-2 (G1 and G2 represent guanine; G3 represents OG), DNA-3 (G1 and G3 represent guanine; G2 represents OG), and DNA-4 (G2 and G3 represent guanine; G1 represents OG) as well as three primers labeled with different fluorophores (Table S1 in ESI†) to evaluate the quantification (Fig. 4A). FAM-primer-2, Cy5-primer, and Cy3-primer target G1, G2, and G3 in the repeat sequences, respectively. With the use of Cy3-primer, Bsu Pol can incorporate dATP opposite DNA-2 that contains OG at G3 position (Fig. 4B, Lane 2), but not other DNA templates (DNA-1, DNA-3 and DNA-4). Similarly, Bsu Pol can incorporate dATP opposite DNA-3 that contains OG at G2 position with Cy5-primer (Fig. 4B, Lane 7) and incorporate dATP opposite DNA-4 that contains OG at G1 position with FAM-primer-2 (Fig. 4B, Lane 12). However, no extended band was observed in the DNA templates containing G (Fig. 4B, Lane 1, Lane 5 and Lane 9). The results suggest that the single-nucleotide primer extension assay by Bsu Pol has good selectivity for quantitative analysis of OG in G-rich sequence of DNA.
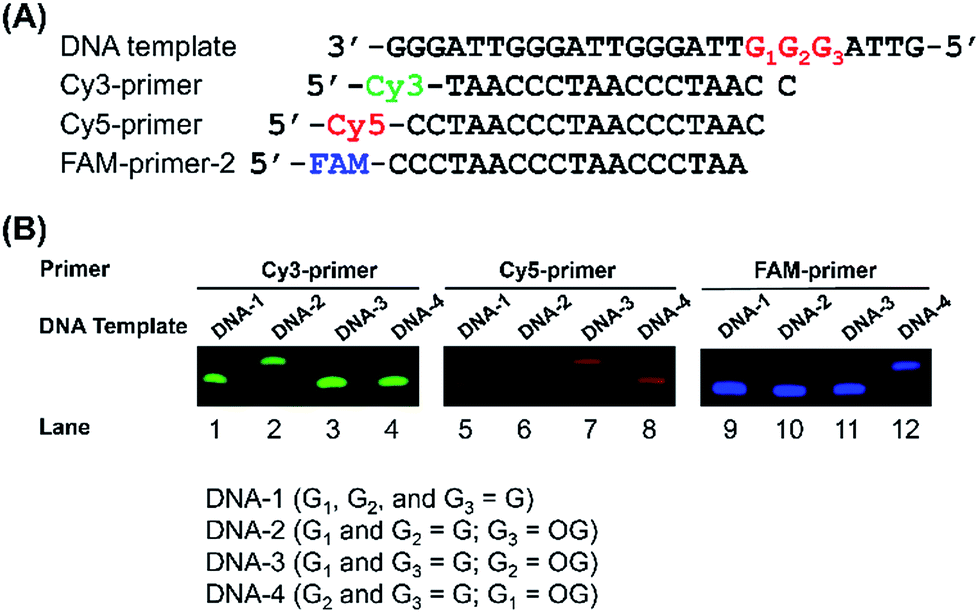 | ||
| Fig. 4 Sequence-specific incorporation of dATP opposite OG. (A) The sequences of the DNA templates and different fluorophores-labeled primers. (B) The single-nucleotide primer extension reaction using different fluorophores-labeled primers that target different guanosine sites in DNA templates. | ||
We next performed quantitative analysis of OG in telomeric DNA from HeLa cells. Similarly, we used the three primers (Cy3-primer, Cy5-primer, and FAM-primer-2) that target the different G in the repeating sequence of 5′-(TTAG1G2G3)n-3′ in telemetric DNA (Fig. 5). The single-nucleotide primer extension assay was performed with 100 μM dATP and 0.5 U Bsu Pol in the 10 μL mixture at 37 °C for 20 min, where the fluorescent primers cannot be extended while opposite G in DNA (Fig. 4B). The result showed that stronger signals of the primer extension products were observed by all the three primers in H2O2-treated HeLa DNA compared to control DNA (Fig. 5), suggesting that H2O2 treatment can induce the generation of OG residues in all of the guanine sites of telomeric DNA. In addition, we also observed weak signals of the primer extension products in the control DNA, indicating that OG might exist endogenously in telomeric DNA.
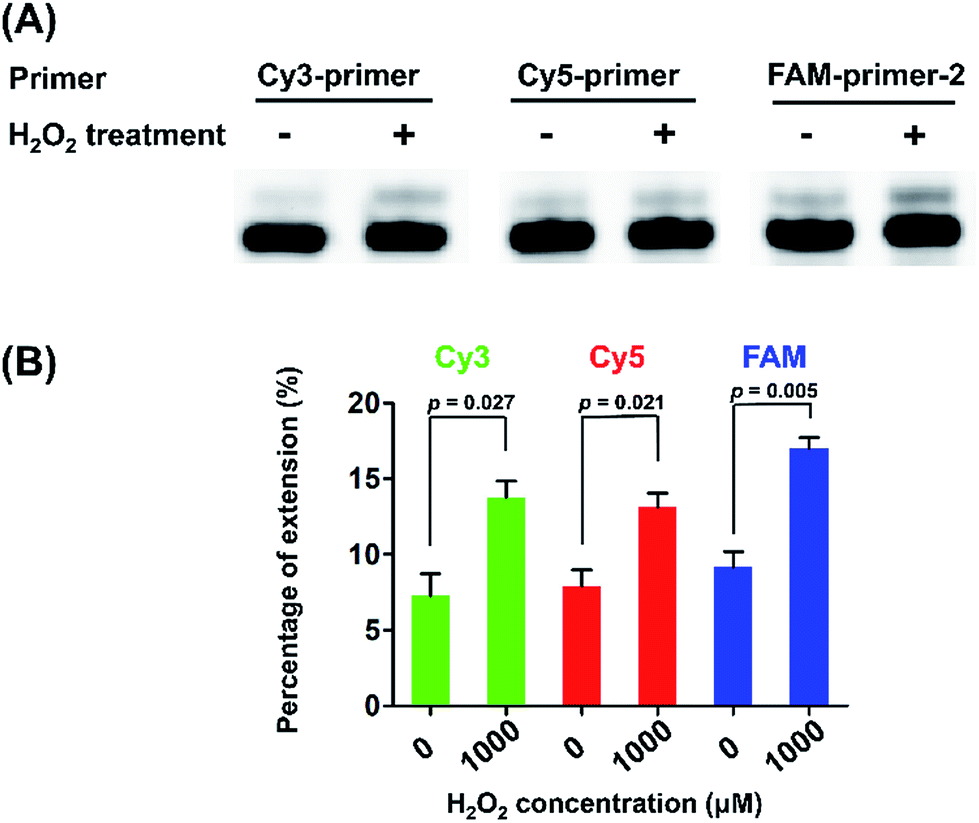 | ||
| Fig. 5 Measurement of the level of OG in telomeric DNA. (A) The single-nucleotide primer extension reaction using 5 μg HeLa DNA and different fluorophore-labeled primers (5 pmol for each) that target different guanosine sites in telomeric DNA. (B) The quantification result for the measurement of the level of OG at telomeric DNA. Error bars show standard deviations from three independent experiments. The unpaired t-test was performed to evaluate the differences of extension rate. All p values were two-sided. | ||
It is possible that endogenous OG sites may be converted to thymine through replication. The products from the primer extension assay may therefore be attributed to the existence of OG or thymine in HeLa DNA since Bsu Pol can incorporate adenine to OG or thymine (Fig. S3 in ESI†). In this respect, we further used Tth Pol instead of Bsu Pol to perform the same single-nucleotide primer extension reaction because Tth Pol cannot incorporate adenine opposite OG (as can be seen from Fig. 2) and only incorporate adenine opposite thymine (Fig. S4 in ESI†). We first mixed the DNA-G and DNA-T templates (detailed sequence information can be found in Table S1 in ESI†) at different ratios and performed the primer extension assay by Tth Pol with dATP as the substrate. The result showed that the extension product can be observed with as low as 0.2% DNA-T (DNA-T/(DNA-T + DNA-G)), suggesting that even low amount of thymine can be detected by using Tth Pol (Fig. S5 in ESI†). The result of the single-nucleotide primer extension reaction using Tth Pol and HeLa genomic DNA showed that no extension product was observed by different fluorophores-labeled primers (Fig. S6 in ESI†), indicating that no detectable OG-converted thymine was present in vivo. Collectively, these results demonstrated that the single-nucleotide primer extension assay by Bsu Pol can be employed for quantitative analysis of OG in telomeric DNA.
Analysis of OG in DNA by sequencing
Inspired by the good performance of Bsu Pol to discriminate OG and G in DNA, we next evaluated whether Bsu Pol could be used to map the site of OG in DNA at single-base resolution through inducing G-to-T mutation. In this respect, a 134-mer single-stranded DNA that contains a single OG (L-DNA-OG1) as well as the 134-mer control DNA (L-DNA-G) were synthesized and used for the primer extension by Bsu Pol. The extension product was then amplified by PCR and subjected to sequencing (Fig. 6A). The sequencing result showed that a distinct thymine signal was observed at the original OG site (Fig. 6B). The primer extension was performed with all the presence of four dNTPs, indicating that OG in DNA has a preference to pair with adenine instead of cytosine, which was consistent with the steady-state kinetics study (the Vmax/Km values for incorporation of dATP and dCTP opposite OG are 27.3 × 10−3 and 2.6 × 10−3 min−1 μM−1, respectively, Table S3 in ESI†).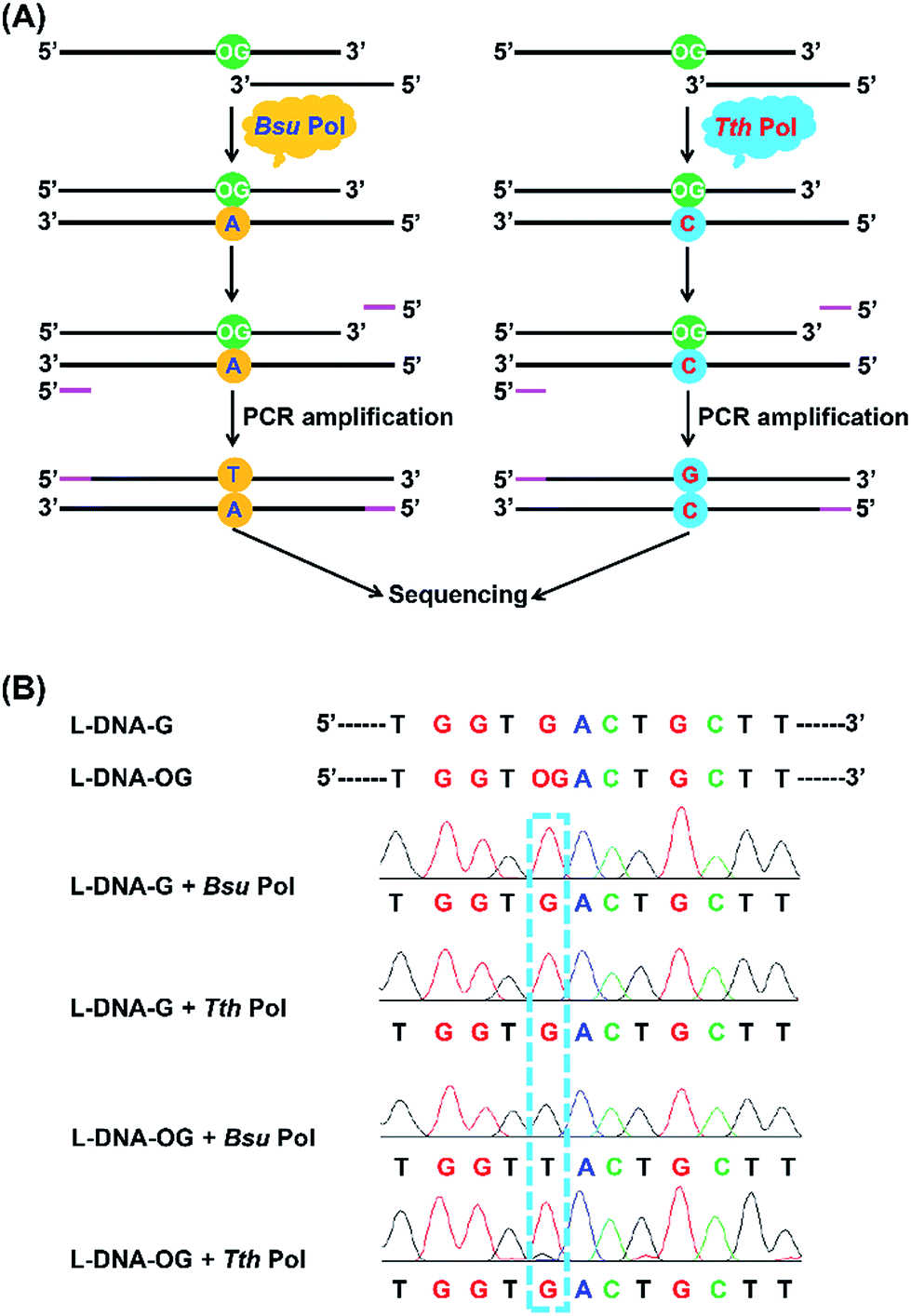 | ||
| Fig. 6 Analysis of OG in DNA by sequencing. (A) Schematic illustration of the strategy for single-base resolution analysis of OG in DNA by sequencing. (B) The results of sequencing analysis for the primer extension products by Bsu Pol and Tth Pol with L-DNA-G and L-DNA-OG1 templates. | ||
The steady-state kinetics study also showed that Tth Pol preferentially incorporates dCTP other than dATP opposite OG (the Vmax/Km are 1.7 × 10−3 and 0.2 × 10−3 min−1 μM−1, respectively, for incorporation of dCTP and dATP opposite OG, Table S3 in ESI†). Here we also performed the primer extension using Tth Pol followed by PCR amplification and sequencing (Fig. 6A). The result showed that a distinct guanine but not thymine signal was observed at the original OG site (Fig. 6B).
We also synthesized DNA carrying multiple sites of OG (L-DNA-OG2 and L-DNA-OG3, Table S1 in ESI†) and performed the same sequencing analysis. The results showed that thymine signals were observed at all the original OG sites by Bsu Pol extension (Fig. S7 in ESI†); however, only guanine signals were observed at the original OG sites and no thymine signal was observed by Tth Pol extension (Fig. S8 in ESI†). This result demonstrated that multiple OG sites in DNA can be distinctly detected by sequencing. Therefore, by comparing the primer extension product using Bsu Pol and Tth Pol, mapping of OG in DNA can be achieved.
Guanine base at a given site of DNA may not be fully oxidized to OG, thus it is important to obtain the frequency of conversion of guanine to OG at specific site in genomic DNA. Hence, we performed the quantitative evaluation of the level of OG in a given site in DNA using the primer extension followed by sequencing. Various ratios of L-DNA-OG1/L-DNA-G were mixed as the DNA template for primer extension by Bsu Pol with the presence of four dNTPs. The Sanger sequencing result showed that only G signal was observed from the 100% L-DNA-G template and only T signal was observed from the 100% L-DNA-OG1 template at the original OG site; however, both G and T signal were observed from the 35% and 70% OG-containing DNA template at the original OG site (Fig. 7A). To evaluate quantitatively the G-to-T conversion rates at the original OG site, we further performed the clone sequencing. The result showed that the ratios of T/(T + G) in the original OG site increased linearly with the increased ratios of L-DNA-OG1/L-DNA-G (Fig. 7B), indicating that the method allowed for site-specific determination of modification stoichiometry of OG in DNA.
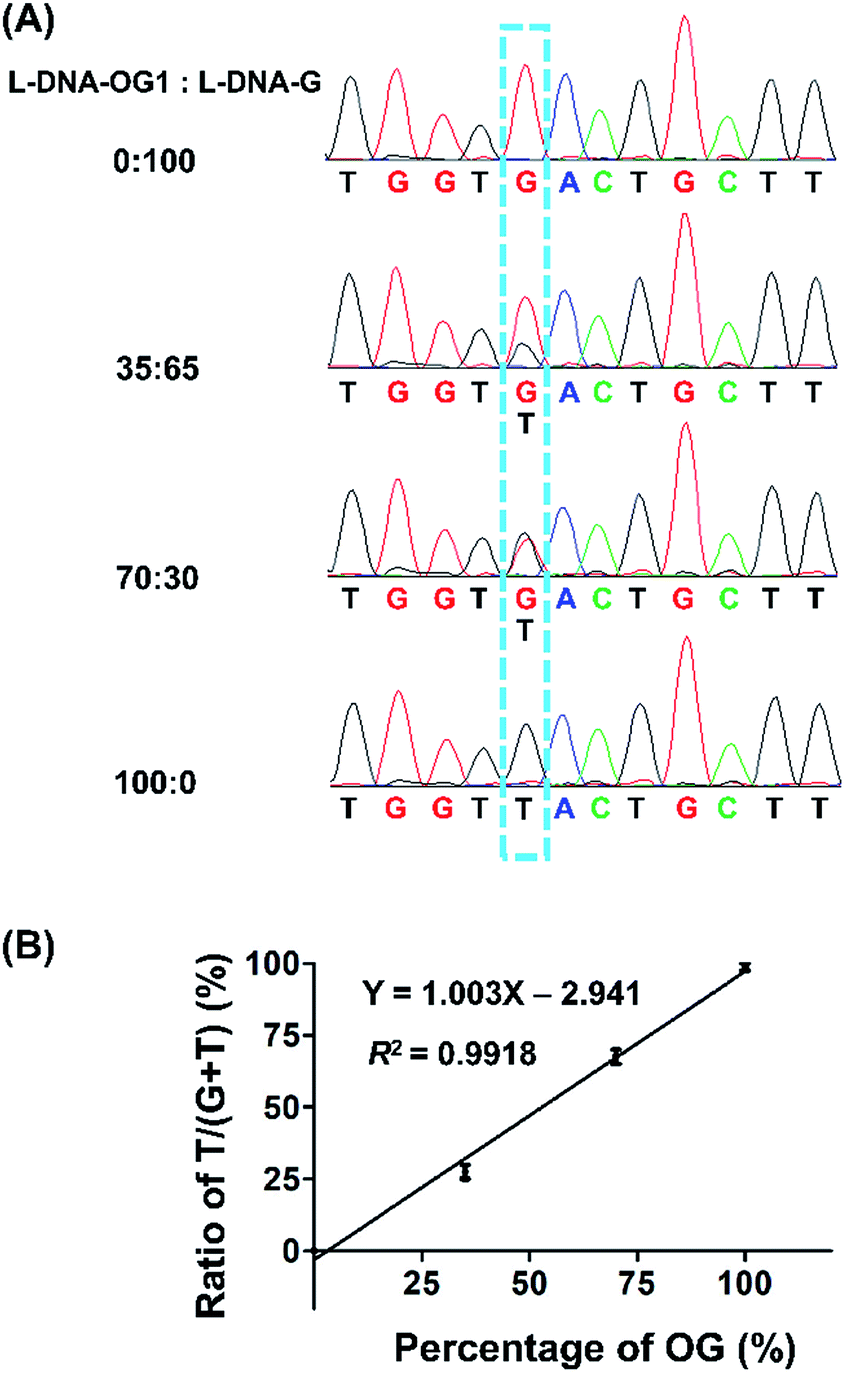 | ||
| Fig. 7 Quantitative evaluation of the level of OG at a given site of DNA using the primer extension followed by sequencing. (A) Various ratios of L-DNA-OG1/L-DNA-G were mixed and used as the DNA template for primer extension with Bsu Pol in the presence of four dNTPs followed by Sanger sequencing. (B) Quantitative evaluation of the G-to-T conversion rates at the original OG site by Sanger sequencing. Forty positive clones per sample were picked and subjected to sequencing. Three independent measurements were performed. | ||
We further employed the high-throughput sequencing to assess quantitatively the level of OG in DNA using various ratios of L-DNA-OG1/L-DNA-G as the DNA template. The result showed that the percentage of the “T” signal at OG site (total number of T/total number of reads) increased linearly with the increased level of OG (Fig. S9 in ESI†), suggesting the capability of high-throughput sequencing on the quantitative measurement of OG in DNA. In addition, we explored whether the OG-induced mutation rate could be modulated by flanking sequence context by employing high-throughput sequencing analysis of the primer extension products of synthesized DNA containing NN-OG-NN, where N is an equimolar mixture of A, T, C and G. Shown in Fig. S10 in ESI† is a schematic illustration of the procedures for the library construction. The sequencing result showed similar frequencies of the four natural nucleosides flanking the G-to-T mutation site (Fig. 8), indicating that the sequence context of flanking nucleosides does not play an apparent role on the Bsu-mediated G-to-T mutation at the OG site. Collectively, the above results suggested that, in conjugation with the Bsu-mediated DNA replication, the high-throughput sequencing is capable of mapping OG sites in DNA.
 | ||
| Fig. 8 High-throughput sequencing revealed the lack of influence of flanking nucleosides on OG-induced G-to-T mutation. Shown are similar frequencies of incorporation of A, T, C, and G nucleotides opposite the N in template DNA with OG being flanked with two random nucleotides at both the 5′ and 3′ side (i.e. NN-OG-NN). | ||
We next applied the Sanger sequencing method to detect OG at three genes (VEGFA, TP53, and KRAS) from DNA of HeLa cells. Because the endogenous level of OG in genomic DNA is relatively low and may not be easily detected by Sanger sequencing, we also performed the analysis of OG in H2O2-treated DNA of HeLa cells. Shown in Fig. S11 in ESI† is the schematic illustration of the analytical procedure. The results showed an obviously increased signal of thymine occurred in certain site of the H2O2-treated DNA of HeLa cells (Fig. 9), suggesting that this sequencing method can be applied for the analysis of OG at specific sites of interest in DNA. In addition, we noticed that the H2O2 treatment induced the formation of OG at the 5′-G in GG motif and at the middle G of GGG motif. This observation is in line with previous studies showing that the 5′-G in GG motif and the 5′- or middle G of GGG motif are more susceptible to oxidation.33,34
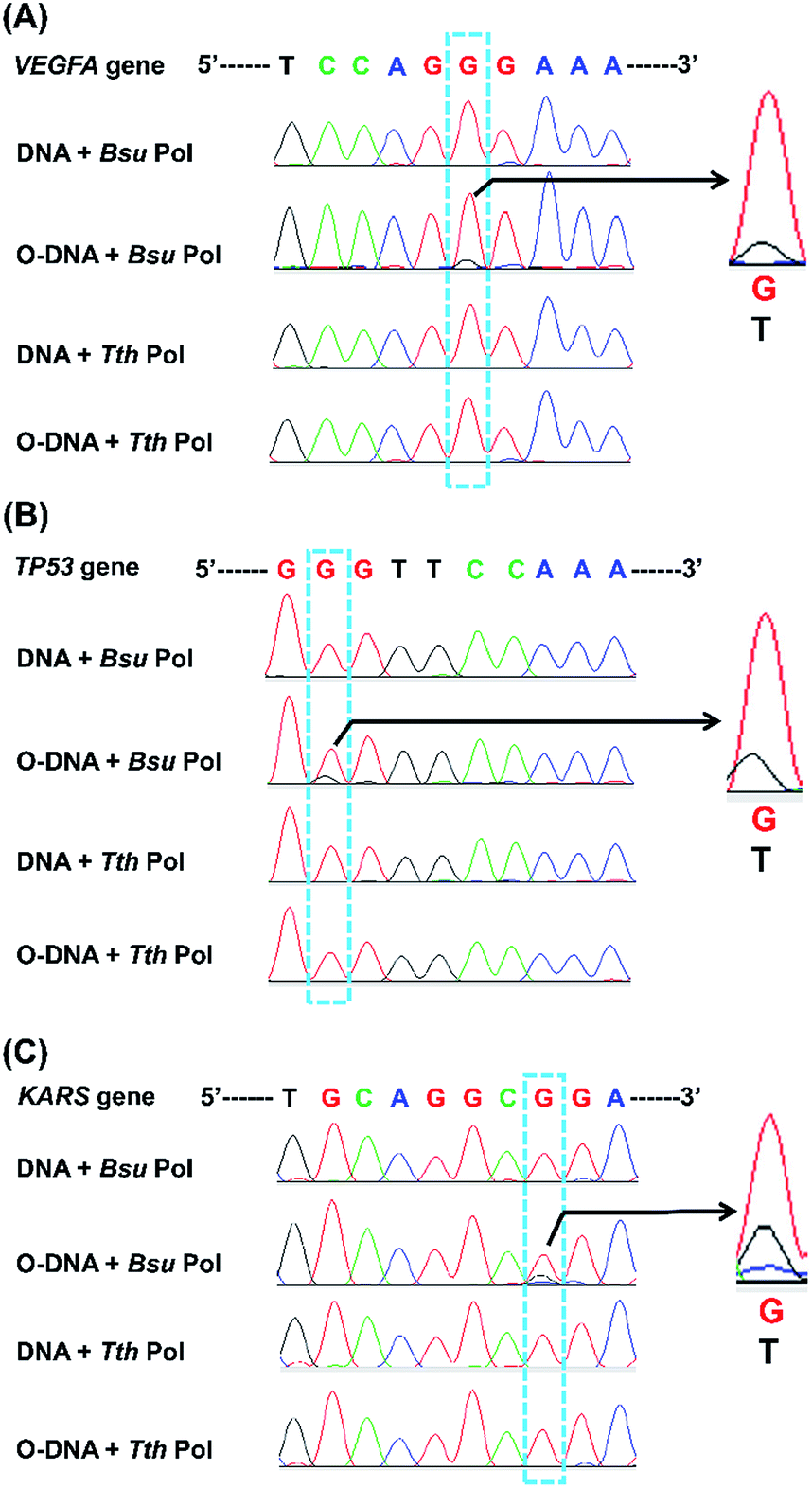 | ||
| Fig. 9 Analysis of OG in genomic DNA of HeLa cells by Sanger sequencing. Three genes, including VEGFA (A), TP53 (B), and KRAS (C), were amplified using HeLa genomic DNA as the template. HeLa genomic DNA or H2O2-treated HeLa genomic DNA was annealed with the corresponding extension primers and then extended by Bsu or Tth Pol. The extension products were amplified with PCR and subjected to Sanger sequencing. O-DNA, H2O2-treated HeLa genomic DNA. Shown in right are the enlarged peaks with G-to-T conversion. | ||
Sanger sequencing is appropriate for exploring the sequence information of small specific regions and is not suitable to afford sequence information of the whole genome. On the other hand, parallel analysis of the primer extension products with Bsu Pol and Tth Pol followed by high-throughput sequencing should be useful for detection of OG in genomic DNA. High-throughput sequencing can provide vast quantities of DNA sequencing data. However, sequencing errors can be introduced during PCR amplification including that mediated by OG.35 Lou et al.36 reported a circle-sequencing library preparation method that improved the error rate associated with high-throughput sequencing. They found that the addition of formamidopyrimidine-DNA glycosylase (Fpg) during rolling circle amplification dramatically eliminated the majority of errors caused by OG, which could be due to the removal of OG. In addition, Costello et al.37 and Chen et al.38 used the fact that OG leads to a global imbalance between variants detected in read 1 (R1) and read 2 (R2) in paired-end sequencing. The degree of this imbalance is correlated with the amount of OG present in a sample, which can be potentially used for genome-wide mapping of OG in DNA.
However, these studies mainly focus on lowering the error rate associated with high-throughput sequencing and the genome-wide mapping of OG in DNA has not yet been performed. Genome-wide mapping of OG in DNA with these methods entail repair-enzyme system to repair OG followed by comparing base calls between repaired and un-repaired samples, which requires sophisticated algorithm to analyse the sequencing data and then identify the precise location of OG in genomic DNA. In addition, the repair enzyme of Fpg does not act exclusively on OG. Apart from OG base, other modified bases can also be recognized and removed by Fpg, such as fapy-adenine (4,6-diamino-5-formamidopyrimidine), 5-hydroxycytosine and 5-hydroxyuracil,39,40 which will complicate the readouts of sequencing. It was also reported that this enzymatic repair is not totally accurate and might be a source of error.41 Moreover, different DNA polymerases may have different properties in incorporating nucleotides opposite OG. Not all DNA polymerases incorporate adenine nucleotide opposite OG during DNA replication and induce G-to-T transversion. Therefore, the properties of DNA polymerases used in the replication of OG-containing DNA fragments in library preparation need to be carefully evaluated if these analytical strategies will be used.
Extensive efforts in recent years have been made to modify standard high-throughput sequencing protocols to increase the fidelity. The sensitivity to detect rare variants has been reported in the range of 10−8 to 10−7 per base pair,42,43 with error rates being estimated to be as low as <10−11.42 Endogenous OG in DNA is approximately several OG per million nucleosides,25,44,45 which is in the similar range as those of 5-fdC (5-formyl-2′-deoxycytidine), 5-acdC (5-carboxy-2′-deoxycytidine), and m6dA (N6-methyl-2′-deoxyadenosine) in genomic DNA.46–51 Although the frequencies of these modifications are low in genomic DNA, genome-wide mapping of these modified nucleosides were successfully achieved by high-throughput sequencing.49,52 Hence, genome-wide mapping of OG in DNA should be achievable by high-throughput sequencing. Along this line, antibody-based enrichment of OG-containing DNA fragments can be carried out in the same way as that for mapping m6dA in DNA in the library preparation.49 The enrichment can further improve the abundance of OG in the DNA sample and then alleviate the burden of high-throughput sequencing.
Since we found that Tth polymerase can faithfully insert a cytosine opposite OG, the repair-enzyme treatment system is not needed in the library preparation with using Tth polymerase. The parallel analysis of the primer extension products with Bsu and Tth polymerase followed by high-throughput sequencing can provide distinct detection of OG in DNA. The analytical strategy is straight-forward and the analysis of sequencing data should be relatively easy to perform without the need of complex algorithm. Future application of this approach at the genome-wide scale will greatly increase our knowledge of the chemical biology of OG with respect to its epigenetic-like regulatory roles. Moreover, analysis of the dynamic changes of contents and sites of OG in genome under a variety of conditions (i.e., different stressors) will reveal the regulatory functions of OG in response to stresses.
Conclusions
In the current study, we characterized two commercially available DNA polymerases, Bsu Pol and Tth Pol, which can selectively incorporate adenine and cytosine opposite OG, respectively. Using the properties of these two DNA polymerases, we achieved quantitative analysis of OG in synthesized DNA that carries OG as well as in telomeric DNA from HeLa cells. In addition, parallel analysis of the extension products with Bsu Pol and Tth Pol followed by sequencing can provide distinct detection of OG in DNA. Taken together, the developed method can promote the functional study of OG in future.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The work is supported by the National Key R&D Program of China (No. 2017YFC0906800), the National Natural Science Foundation of China (No. 21672166, 21728802, 21635006, 21721005), and the National Institutes of Health (R01 CA210072).References
- S. J. Forrester, D. S. Kikuchi, M. S. Hernandes, Q. Xu and K. K. Griendling, Circ. Res., 2018, 122, 877–902 CrossRef CAS PubMed.
- P. T. Schumacker, Cancer Cell, 2015, 27, 156–157 CrossRef CAS PubMed.
- A. Alenko, A. M. Fleming and C. J. Burrows, Biochemistry, 2017, 56, 5053–5064 CrossRef CAS PubMed.
- J. C. Genereux and J. K. Barton, Chem. Rev., 2010, 110, 1642–1662 CrossRef CAS PubMed.
- S. A. Roberts and D. A. Gordenin, Nat. Rev. Cancer, 2014, 14, 786–800 CrossRef CAS PubMed.
- B. F. Yuan, Y. Jiang and Y. Wang, Chem. Res. Toxicol., 2010, 23, 11–19 Search PubMed.
- A. Tubbs and A. Nussenzweig, Cell, 2017, 168, 644–656 CrossRef CAS PubMed.
- A. M. Fleming and C. J. Burrows, DNA Repair, 2017, 56, 75–83 CrossRef CAS PubMed.
- L. Pan, B. Zhu, W. Hao, X. Zeng, S. A. Vlahopoulos, T. K. Hazra, M. L. Hegde, Z. Radak, A. Bacsi, A. R. Brasier, X. Ba and I. Boldogh, J. Biol. Chem., 2016, 291, 25553–25566 CrossRef CAS PubMed.
- G. Antoniali, L. Lirussi, C. D'Ambrosio, F. Dal Piaz, C. Vascotto, E. Casarano, D. Marasco, A. Scaloni, F. Fogolari and G. Tell, Mol. Biol. Cell, 2014, 25, 532–547 CrossRef PubMed.
- E. Zarakowska, D. Gackowski, M. Foksinski and R. Olinski, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2014, 764–765, 58–63 CrossRef CAS PubMed.
- J. Park, J. W. Park, H. Oh, F. S. Maria, J. Kang and X. Tian, PLoS One, 2016, 11, e0155792 CrossRef PubMed.
- A. M. Fleming, Y. Ding and C. J. Burrows, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2604–2609 CrossRef CAS PubMed.
- A. Azqueta, S. Shaposhnikov and A. R. Collins, Mutat. Res., 2009, 674, 101–108 CAS.
- Y. Yu, P. Wang, Y. Cui and Y. Wang, Anal. Chem., 2018, 90, 556–576 CrossRef CAS PubMed.
- T. A. Clark, K. E. Spittle, S. W. Turner and J. Korlach, Genome Integr., 2011, 2, 10 CrossRef CAS PubMed.
- A. E. Schibel, N. An, Q. Jin, A. M. Fleming, C. J. Burrows and H. S. White, J. Am. Chem. Soc., 2010, 132, 17992–17995 CrossRef CAS PubMed.
- M. Yoshihara, L. Jiang, S. Akatsuka, M. Suyama and S. Toyokuni, DNA Res., 2014, 21, 603–612 CrossRef CAS PubMed.
- M. Ohno, T. Miura, M. Furuichi, Y. Tominaga, D. Tsuchimoto, K. Sakumi and Y. Nakabeppu, Genome Res., 2006, 16, 567–575 CrossRef CAS PubMed.
- Y. Ding, A. M. Fleming and C. J. Burrows, J. Am. Chem. Soc., 2017, 139, 2569–2572 CrossRef CAS PubMed.
- W. J. Wu, W. Yang and M. D. Tsai, Nat. Rev. Chem., 2017, 1, 1–16 CrossRef CAS.
- B. D. Freudenthal, W. A. Beard, L. Perera, D. D. Shock, T. Kim, T. Schlick and S. H. Wilson, Nature, 2015, 517, 635–639 CrossRef CAS PubMed.
- K. N. Kirouac and H. Ling, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3210–3215 CrossRef CAS PubMed.
- Y. Taniguchi, Y. Kikukawa and S. Sasaki, Angew. Chem., Int. Ed., 2015, 54, 5147–5151 CrossRef CAS PubMed.
- C. M. Gedik, A. Collins and Escodd, FASEB J., 2005, 19, 82–84 CrossRef CAS PubMed.
- A. Horhota, K. Zou, J. K. Ichida, B. Yu, L. W. McLaughlin, J. W. Szostak and J. C. Chaput, J. Am. Chem. Soc., 2005, 127, 7427–7434 CrossRef CAS PubMed.
- H. Cao and Y. Wang, Nucleic Acids Res., 2007, 35, 4833–4844 CrossRef CAS PubMed.
- G. W. Hsu, M. Ober, T. Carell and L. S. Beese, Nature, 2004, 431, 217–221 CrossRef CAS PubMed.
- D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627–8637 CrossRef CAS PubMed.
- C. K. Kwok and C. J. Merrick, Trends Biotechnol., 2017, 35, 997–1013 CrossRef CAS PubMed.
- J. Zhou, A. M. Fleming, A. M. Averill, C. J. Burrows and S. S. Wallace, Nucleic Acids Res., 2015, 43, 4039–4054 CrossRef CAS PubMed.
- N. An, A. M. Fleming and C. J. Burrows, ACS Chem. Biol., 2016, 11, 500–507 CrossRef CAS PubMed.
- Y. Margolin, V. Shafirovich, N. E. Geacintov, M. S. Demott and P. C. Dedon, J. Biol. Chem., 2008, 283, 35569–35578 CrossRef CAS PubMed.
- I. Saito, T. Nakamura, K. Nakatani, Y. Yoshioka, K. Yamaguchi and H. Sugiyama, J. Am. Chem. Soc., 1998, 120, 12686–12687 CrossRef CAS.
- D. B. Sloan, A. K. Broz, J. Sharbrough and Z. Wu, Trends Biotechnol., 2018, 36, 729–740 CrossRef CAS PubMed.
- D. I. Lou, J. A. Hussmann, R. M. McBee, A. Acevedo, R. Andino, W. H. Press and S. L. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19872–19877 CrossRef CAS PubMed.
- M. Costello, T. J. Pugh, T. J. Fennell, C. Stewart, L. Lichtenstein, J. C. Meldrim, J. L. Fostel, D. C. Friedrich, D. Perrin, D. Dionne, S. Kim, S. B. Gabriel, E. S. Lander, S. Fisher and G. Getz, Nucleic Acids Res., 2013, 41, e67 CrossRef CAS PubMed.
- L. Chen, P. Liu, T. C. Evans Jr and L. M. Ettwiller, Science, 2017, 355, 752–756 CrossRef CAS PubMed.
- J. Tchou, V. Bodepudi, S. Shibutani, I. Antoshechkin, J. Miller, A. P. Grollman and F. Johnson, J. Biol. Chem., 1994, 269, 15318–15324 CAS.
- Z. Hatahet, Y. W. Kow, A. A. Purmal, R. P. Cunningham and S. S. Wallace, J. Biol. Chem., 1994, 269, 18814–18820 CAS.
- B. Arbeithuber, K. D. Makova and I. Tiemann-Boege, DNA Res., 2016, 23, 547–559 CrossRef CAS PubMed.
- M. L. Hoang, I. Kinde, C. Tomasetti, K. W. McMahon, T. A. Rosenquist, A. P. Grollman, K. W. Kinzler, B. Vogelstein and N. Papadopoulos, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9846–9851 CrossRef CAS PubMed.
- S. R. Kennedy, M. W. Schmitt, E. J. Fox, B. F. Kohrn, J. J. Salk, E. H. Ahn, M. J. Prindle, K. J. Kuong, J. C. Shen, R. A. Risques and L. A. Loeb, Nat. Protoc., 2014, 9, 2586–2606 CrossRef CAS PubMed.
- J. L. Ravanat, T. Douki, P. Duez, E. Gremaud, K. Herbert, T. Hofer, L. Lasserre, C. Saint-Pierre, A. Favier and J. Cadet, Carcinogenesis, 2002, 23, 1911–1918 CrossRef CAS PubMed.
- C. Bolin, T. Stedeford and F. Cardozo-Pelaez, J. Neurosci. Methods, 2004, 136, 69–76 CrossRef CAS PubMed.
- Y. Tang, J. Xiong, H. P. Jiang, S. J. Zheng, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2014, 86, 7764–7772 CrossRef CAS PubMed.
- Y. Tang, S. J. Zheng, C. B. Qi, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2015, 87, 3445–3452 CrossRef CAS PubMed.
- M. Bachman, S. Uribe-Lewis, X. Yang, H. E. Burgess, M. Iurlaro, W. Reik, A. Murrell and S. Balasubramanian, Nat. Chem. Biol., 2015, 11, 555–557 CrossRef CAS PubMed.
- T. P. Wu, T. Wang, M. G. Seetin, Y. Lai, S. Zhu, K. Lin, Y. Liu, S. D. Byrum, S. G. Mackintosh, M. Zhong, A. Tackett, G. Wang, L. S. Hon, G. Fang, J. A. Swenberg and A. Z. Xiao, Nature, 2016, 532, 329–333 CrossRef CAS PubMed.
- W. Huang, M. D. Lan, C. B. Qi, S. J. Zheng, S. Z. Wei, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2016, 7, 5495–5502 RSC.
- J. Xiong, T. T. Ye, C. J. Ma, Q. Y. Cheng, B. F. Yuan and Y. Q. Feng, Nucleic Acids Res., 2019, 47, 1268–1277 CrossRef PubMed.
- B. Xia, D. Han, X. Lu, Z. Sun, A. Zhou, Q. Yin, H. Zeng, M. Liu, X. Jiang, W. Xie, C. He and C. Yi, Nat. Methods, 2015, 12, 1047–1050 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04946g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |