
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonomeric Cp3tAl(I): synthesis, reactivity, and the concept of valence isomerism†
Alexander
Hofmann
ab,
Tobias
Tröster
ab,
Thomas
Kupfer
ab and
Holger
Braunschweig
 *ab
*ab
aInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 14th January 2019
Abstract
With the isolation of Cp3tAl (1), the first monomeric Cp-based Al(I) species could be realized in a pure form via a three-step reaction sequence (salt elimination/adduct formation/adduct cleavage) starting from readily available AlBr3. Due to its monomeric structure, reactions involving 1 were found to proceed more selectively, faster, and under milder conditions than for tetrameric (Cp*Al)4. Thus, 1 readily formed simple Lewis acid–base adducts with tBuAlCl2 (6) and AlBr3 (7), reactions that before have always been interfered with by the presence of aluminum halide bonds. In addition, the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of 1 with AlBr3 enabled the realization of the very rare trialuminum adduct species 8. 1 also reacted rapidly with N2O and PhN3 at room temperature to afford Al3O3 and Al2N2 heterocycles 9 and 10, respectively. With the structural characterization of products 4 and 5, the reaction of monovalent 1 with Cp3tAlBr2 (2) provided the first experimental evidence for the concept of valence isomerism between dialanes and their Al(I)/Al(III) Lewis adducts.
:1 reaction of 1 with AlBr3 enabled the realization of the very rare trialuminum adduct species 8. 1 also reacted rapidly with N2O and PhN3 at room temperature to afford Al3O3 and Al2N2 heterocycles 9 and 10, respectively. With the structural characterization of products 4 and 5, the reaction of monovalent 1 with Cp3tAlBr2 (2) provided the first experimental evidence for the concept of valence isomerism between dialanes and their Al(I)/Al(III) Lewis adducts.
Introduction
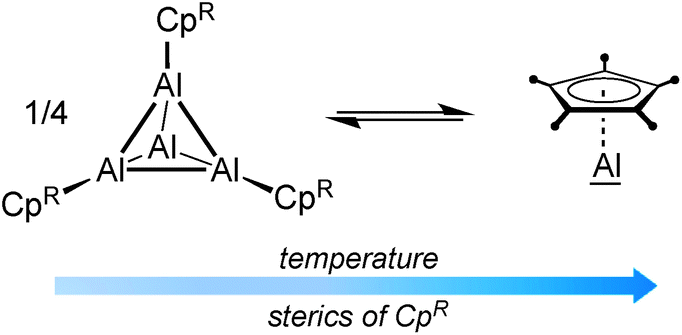
The organometallic chemistry of aluminum is strongly dominated by the oxidation state +III, and subvalent aluminum species are quite rare.1–4 Even though currently on the transition from chemical curiosities to a rather well-established class of compounds, the synthesis of molecules with aluminum in the low oxidation states +I and +II is still experimentally challenging and consistently hampered by their high reactivity and pronounced tendency to disproportionation.5,6 Thus, only a handful of examples of dialanes with Al(II) centers have been reported so far.6–12 Monovalent aluminum was first realized by Schnöckel and coworkers with their seminal discoveries of metastable AlX (X = halide)13,14 and (Cp*Al)4.15,16 These studies have inspired many main group element chemists, and several other Al(I) species have been made available,17,18 including other Cp-based derivatives,19–22 β-diketiminate-stabilized systems,23 the recently reported NHC-stabilized dialumenes,24 and aluminyl anions.25 Among these, Schnöckel's (Cp*Al)4 and derivatives derived thereof still represent the best-studied subvalent aluminum systems. It was shown that (CpRAl)4 tend to be tetrameric in the solid state. In solution, however, an equilibrium with their monomeric form (CpRAl) is established, which is highly dependent on the sterics of CpR and temperature.19–22For (Cp*Al)4, this equilibrium lies almost completely on the side of the tetrameric aggregate at ambient temperatures, which together with the low solubility of the aggregate in common organic solvents efficiently masks the high reactivity of the Al(I) centers. Accordingly, (Cp*Al)4 appears rather unreactive, and often higher temperatures and/or longer reaction times are required. For this reason, much efforts have been made to develop monomeric CpRAl species by introducing bulky CpR ligands, which are expected to prevent any aggregation. As shown in Table 1, increasing the steric demand of CpR to C5(CH2Ph)5, C5H2(SiMe3)3, and C5iPr4H proved actually successful, and no tetramer (CpRAl)4 was detected in solution via27Al NMR spectroscopy. However, the synthetic approaches proved either tedious (long reaction times) and/or required special equipment (generation of metastable AlX precursors), and did not allow for the isolation of CpRAl.20
|
|
||||
|---|---|---|---|---|
| CpR | Species in solution | Isolated | Ref. | |
| (CpRAl)4 | CpRAl | |||
| a Decomposes above −60 °C. b CpRAl detectable at 60 °C. c Decomposes above −30 °C. | ||||
| C5H5a | ✓ | — | — | 20 |
| C5Me4H | ✓ | — | ✓ | 21 |
| Cp*b | ✓ | — | ✓ | 15 |
| C5Me4Prb | ✓ | — | — | 22 |
| C5Me4iPr | ✓ | ✓ | — | 22 |
| C5(CH2Ph)5 | — | ✓ | — | 19 |
| C5H4tBuc | ✓ | — | — | 20 |
| C5iPr4H | — | ✓ | — | 20 |
| C5H3(SiMe3)2 | ✓ | ✓ | — | 20 |
| C5H2(SiMe3)3 | — | ✓ | — | 20 |
| C5H2tBu3 | — | ✓ | ✓ | This work |
Recently, we reported an alternative approach towards the generation of such CpRAl(I) species by Lewis base-induced disproportionation of a Cp*-substituted dialane into Cp*Al(I) and Cp*Al(III)Br2·L (L = Lewis base).26 We envisaged that it might be feasible to transfer this strategy to systems with bulkier Cp groups, eventually allowing for a straightforward isolation of pure monomeric CpRAl. Thus, we initially set out to study the suitability of 1,3,5-tri-tert-butylcyclopentadienyl (Cp3t) as a stabilizing ligand for subvalent CpRAl(I).
Results and discussion
The synthetic protocol used for the generation of Cp3tAl (1) is outlined in Scheme 1. Accordingly, we first developed a convenient synthesis for the precursor Cp3tAlBr2 (2). While simple salt elimination reactions of MCp3t (M = Li, Na, and K) with AlBr3 were found to be inefficient showing only poor conversion and were, in part, accompanied by decomposition processes, 2 was isolated in good yields by applying (Cp3t)2Mg in pentane solution (Scheme 1).27 Due to the high solubility of the magnesium reagent in hydrocarbon solvents, the reaction proceeded quickly and quantitatively within 30 minutes. Characterization of 2 in solution provided no difficulties and NMR spectroscopic parameters agreed very well with the anticipated structure. Thus, the chemical shift of the 27Al NMR resonance of 2 (δ −42) strongly resembles that of Cp*AlBr2 (δ −46).28 Also, the 1H NMR spectrum features a signal for the aromatic protons at δ 6.64, and two signals for the tBu groups at δ 1.41 and 1.24. Colorless crystals of 2 were obtained by cooling a saturated hexane solution. 2 is highly sensitive towards water and oxygen, which is evident by an immediate color change from colorless to slightly brownish.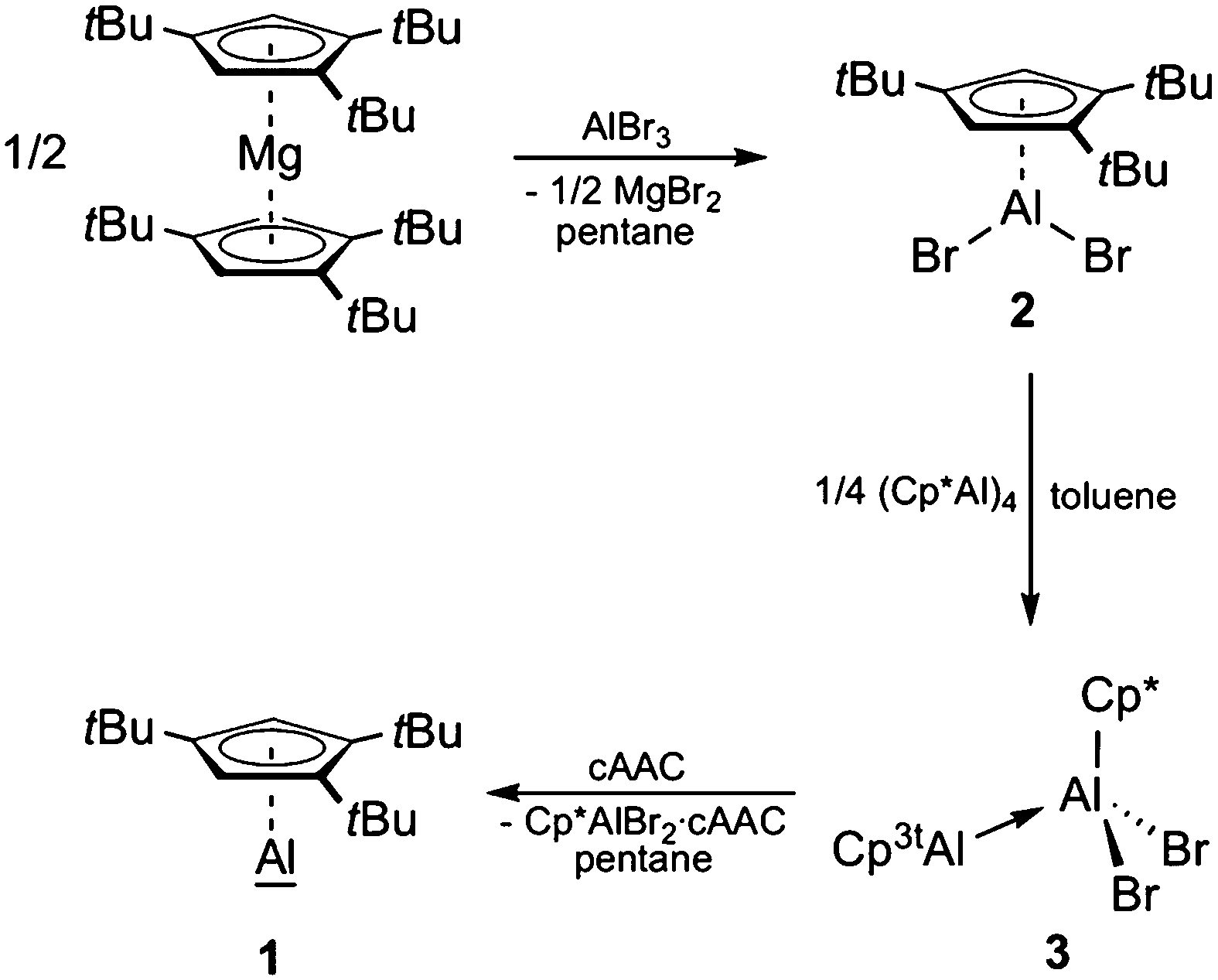 | ||
| Scheme 1 Experimental approach towards the synthesis of 1. | ||
The solid state structure of 2 nicely reflects the increased steric requirements of the Cp3t ligand with respect to Cp* as evidenced by its monomeric structure (Fig. 1).27 By contrast, Cp*AlBr2 is a halide-bridged dimer in the solid state. However, both the Al–C bond lengths (2.175(4)–2.239(4) Å), as well as the distance between the aluminum center and the Cp centroid (Al–cent 1.835 Å) compare very well with the values found in Cp*AlBr2 (av. Al–C 2.223 Å; Al–cent 1.865 Å).28
 | ||
| Fig. 1 Molecular structures of 2 (left) and 3 (right) in the solid state. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å): 2 Al1–C1 2.196(4), Al1–C2 2.214(4), Al1–C3 2.175(4), Al1–C4 2.187(4), and Al1–C5 2.239(4); 3 Al1–Al2 2.533(1), Al1–C1 2.005(3), Al2–C11 2.196(3), Al2–C12 2.180(3), Al2–C13 2.136(3), Al2–C14 2.192(3), and Al2–C15 2.220(3). | ||
With Cp3tAlBr2 (2) in hand, we next tried to make Cp3tAl (1) accessible via direct reductive dehalogenation, a route that has been successfully applied to the synthesis of (Cp*Al)4, for instance.16 However, reduction of 2 with alkali metal-based reductants such as KC8, NaK2.8, Na, Na[C10H8], and Li under various conditions consistently failed, and no traceable materials could be isolated. Thus, we next attempted the reductive elimination pathway recently introduced by Fischer and Frenking for an alternative synthesis of (Cp*Al)4.29 This approach requires (Cp3t)2AlH as the reagent, though, which upon heating might be susceptible to Cp3tH elimination to generate the desired monovalent Cp3tAl (1). Again, all our attempts to selectively prepare (Cp3t)2AlH remained unsuccessful. Heating a mixture of (Cp3t)2Mg and HAlCl2 always resulted in the formation of elemental Al among several other undefined species. According to 27Al NMR spectroscopy, only trace amounts of a compound with a chemical shift of δ −161, indicative of the presence of 1, were present in the reaction mixtures. Thus, we were not able to separate and isolate 1.
After these rather disappointing results we reviewed our most recent findings on the related Cp* system, i.e. the Lewis base-induced cleavage of dialane Cp*(Br)Al–Al(Br)Cp* into (Cp*Al)4 and Cp*Al(III)Br2·L.26 Unfortunately, no Cp3t-substituted dialane was accessible by stoichiometric reduction of 2. However, keeping in mind that (i) DFT calculations have established an equilibrium between dialane Cp*(Br)Al–Al(Br)Cp* and the corresponding Lewis acid–base adduct Cp*Al·Al(Br)2Cp*,26 which is in line with the valence isomerism between these two classes of compounds, as already suggested by Cowley,30 and (ii) Al–Al bond formation has also been accomplished by the reaction of Al(I) species with H2AlNacNac (NacNac = [ArNC(Me)CHC(Me)NAr]−; Ar = 2,6-iPr2C6H3)31 and AlX3·cAAC (X = Cl and I; cAAC = 1-[2,6-iPr2C6H3]-3,3,5,5-tetramethyl-2-pyrrolidinylidene),32 we wondered if an asymmetric dialane is available by the comproportionation of (Cp*Al)4 and Cp3tAlBr2, or if an initial Lewis acid–base adduct is already susceptible to release the targeted Cp3tAl (1) upon treatment with Lewis bases. To this end, a solution of 2 in toluene was treated with (Cp*Al)4 under sonication, whereupon the color of the suspension turned from yellow to colorless (Scheme 1).27 The progress of the reaction was easily monitored by the disappearance of the 27Al NMR signals of 2 (δ −46) and (Cp*Al)4 (δ −78), while no product resonances were apparent. 1H NMR spectroscopic studies on the reaction mixture evidenced the selective formation of a single product with Cp* and Cp3t ligands in a 1:1 ratio, thus pointing to the formation of either an asymmetric dialane or the asymmetric Al(I)–Al(III) Lewis acid–base adduct 3. In its 1H NMR spectrum, the reaction product features one signal at δ 1.90 for the Cp* protons, as well as signals at δ 6.25, δ 1.38, and 1.24 for the Cp3t ligand. In particular, the highfield shift of the aromatic Cp3t protons in comparison to 2 is noteworthy and favors a Cp3t ligand as part of an Al(I) fragment, thus suggesting the presence of 3 (Scheme 1).
X-ray diffraction studies on suitable single crystals of 3 eventually verified the adduct formation.27 As evident from Fig. 1, (Cp*Al)4 acted as a reductant in its reaction with 2, and the Cp* fragment now features a Lewis-acidic Al(III) center containing the two bromides formerly belonging to 2. Accordingly, it is the Cp3t fragment that plays the Lewis-basic Al(I) part in adduct 3, while the Cp3t ligand adopts a η5 coordination mode with bonding parameters (Al–C 2.136(3)–2.220(3) Å, Al–cent 1.815 Å) similar to those in 2 (Al–C 2.175(4)–2.239(4) Å, Al–cent 1.835 Å). By contrast, the Cp* ligand adopts a η1 coordination mode with the Al(III) center with an Al–C distance (2.005(3) Å) in the same region as observed before. The Al–Al separation distance in 3 (2.533(1) Å) is somewhat shorter than that in the Al(I)–Al(III) adducts Cp*Al·AltBu3 (2.689(2) Å)33 or Cp*Al·Al(C6F5)3 (2.591(2) Å),30 which indicates a quite strong dative Al–Al interaction in 3. The Al–Al distance in 3 is rather reminiscent of the Al–Al bond length found in dialane Cp*(Br)Al–Al(Br)Cp* (2.530(2) Å),12 which might be taken as another piece of evidence for the close relationship between dialanes and the corresponding Al(I)–Al(III) adducts as valence isomers.
Adduct 3 appears to be an ideal precursor for the release of Cp3tAl (1) by the addition of strong Lewis bases, for which reason we reacted solutions of 3 in C6D6 with PMe3, IPr (1,3-diisopropyl-imidazole-2-ylidene), and cAAC. In all cases, 27Al NMR spectroscopy showed the formation of a new signal at δ −161, indicative of a monomeric Al(I) species. In addition, the expected resonances of Cp*AlBr2·PMe3 (δ 48) and Cp*AlBr2·IPr (δ 106) could be found in the respective 27Al NMR spectra of the reaction mixtures, while the signal of Cp*AlBr2·cAAC was not detected, most likely due to its broadness. The formation of all three adducts was, however, clearly verified by 1H NMR spectroscopy, which showed the expected signal patterns for one equivalent of Cp*AlBr2·L, as well as signals at δ 5.94, 1.36, and 1.20 for monovalent 1. Compound 1 was eventually isolated on a larger scale by the reaction of 3 with cAAC in pentane, making use of the poor solubility of Cp*AlBr2·cAAC under these conditions (Scheme 1).27 After filtration and removal of the solvent under reduced pressure, a yellow oil remained, which contained essentially pure 1. Even though all attempts to obtain suitable single crystals of 1 for X-ray diffraction failed, its 27Al NMR resonance (cf. monomeric Cp*Al(I) shows a signal at δ −150 at 60 °C),20 the absence of characteristic signals for a tetrameric aggregate (usually between δ −60 and δ −110),20 and the results of 1H DOSY NMR spectroscopic studies (single species, diffusion coefficient D = 8.703 × e−10 m2 s−1; see Fig. S32 in the ESI†) leave no doubt on its constitution and its description as a monomeric, monovalent aluminum species.
The isolation of pure 1 also allowed us to evaluate the concept of valence isomerism between dialanes and their respective Al(I)–Al(III) adducts, and to probe the possibility of generating a Cp3t-based dialane by simply reacting 1 with one equivalent of Cp3tAlBr2 (2; Scheme 2).27 The 27Al NMR spectrum of the reaction mixture features a single broad resonance at δ −64 with a chemical shift intermediate between those of 1 (δ −161) and 2 (δ −46). Also, only one set of signals is evident in the 1H NMR spectrum, while the chemical shift of the aromatic Cp3t protons (δ 6.39) is found again intermediate between the values of 1 (δ 5.94) and 2 (δ 6.64). Hence, while the observation of a single set of NMR spectroscopic parameters would suggest the formation of a symmetric dialane (5), the observed 27Al NMR chemical shift (δ −64) significantly differs from that of dialane Cp*(Br)Al–Al(Br)Cp* (δ −11).12 On the other hand, for adduct 4, two sets of signals are to be expected with substantially different 27Al NMR chemical shifts as observed in this case (cf. Cp*Al·Al(C6F5)3δ −116, 107).30 For a related gallium system comprising Cp*Ga and GaX2Cp* (X = Cl and I), Jutzi and co-workers similarly reported only one set of NMR signals for the Cp* moieties upon mixing of the components, even at −80 °C,34 which indicates a corresponding valence isomerism in the case of gallium. Thus, our NMR spectroscopic studies rather indicate the presence of a rapid equilibrium between adduct 4 and dialane 5, which we tried to verify by VT NMR spectroscopic studies in toluene solution (−90 °C to +100 °C). Unfortunately, we were not able to derive a clear picture of this process, most likely because the exchange process is too fast on the NMR time scale across the entire temperature range. Nevertheless, a few hints can be extracted from the measurements. While low temperature 27Al NMR spectroscopy was not very helpful in solving this puzzle (the signal only broadened, eventually disappearing in the baseline; see Fig. S28–S31 in the ESI†), the 27Al NMR resonance at δ −64 is shifted to a higher field (δ −90) at increased temperatures. This chemical shift is reminiscent for Al(I) centers in Lewis acid–base adducts (cf. Cp*Al·Al(C6F5)3δ −116).30 In addition, a low intensity signal appears at δ 90, which is found in the same region as the tetracoordinate aluminum atom of Cp*Al·Al(C6F5)3 (δ 106) or adducts 6 (δ 146) and 7 (δ 95; see below). These findings suggest that at higher temperatures, the equilibrium mixture of 4/5 is pushed to the side of adduct 4. By contrast, the 1H NMR signals of the aromatic C5H2tBu3 protons are gradually shifted to a lower field upon cooling the sample (δrt 6.39; δ−60°C 6.59) and finally became chemically non-equivalent at −80 °C, as evidenced by two low-field signals (δ−80°C 6.89, 6.64). It should be noted that this decoalescence is not related to the 4/5 isomerization process, but only to the rotation of the Cp3t rings, which gets frozen at low temperatures. Thus, we still see only one Cp3t ligand environment, which in combination with the low-field chemical shift of these protons (characteristic for three-coordinate aluminum species such as 2) indicates an equilibrium mixture shifted to the side of the symmetric dialane 5. In order to evaluate the thermodynamic stabilities of both species 4 and 5, we performed DFT calculations at the M06L/Def2-SVP level of theory (Scheme 2 (bottom)). Accordingly, dialane 5calc is energetically more stable by 4.0 kcal mol−1 than the asymmetric adduct structure 4calc, and both species are separated by a low-lying transition state TS with an activation barrier of only 5.6 kcal mol−1 for the transformation of 4 into 5. These results fit very well to our experimental findings that only mixtures of adduct 4/dialane 5 can be generated in solution, and that the interconversion is possible over the entire temperature range. Eventually, both isomers could be characterized in the solid state by X-ray diffraction (Fig. 2). Consistent with the spectroscopic results and the calculated stabilities, we were able to selectively crystallize dialane 5 at −30 °C and adduct 4 at room temperature from the same pentane reaction mixture.27 In agreement with the equilibrium shown in Scheme 2, both isolated species showed the same averaged NMR spectra as the initial reaction mixture. Importantly, we were able to isolate again 4 and 5 from both of these solutions under the same conditions as described above, thus providing a definite experimental proof for this isomerization process. To the best of our knowledge, this represents the first instance, in which the suggested valence isomerism between dialanes and their respective Al(I)–Al(III) adducts could be demonstrated directly within a single molecular system. It should be noted, however, that Nikonov and co-workers had previously observed the reversible disproportionation of a dialane to the corresponding pair of discrete Al(I) and Al(III) species, which, in contrast to 4, did not form an Al(I)–Al(III) Lewis pair.31
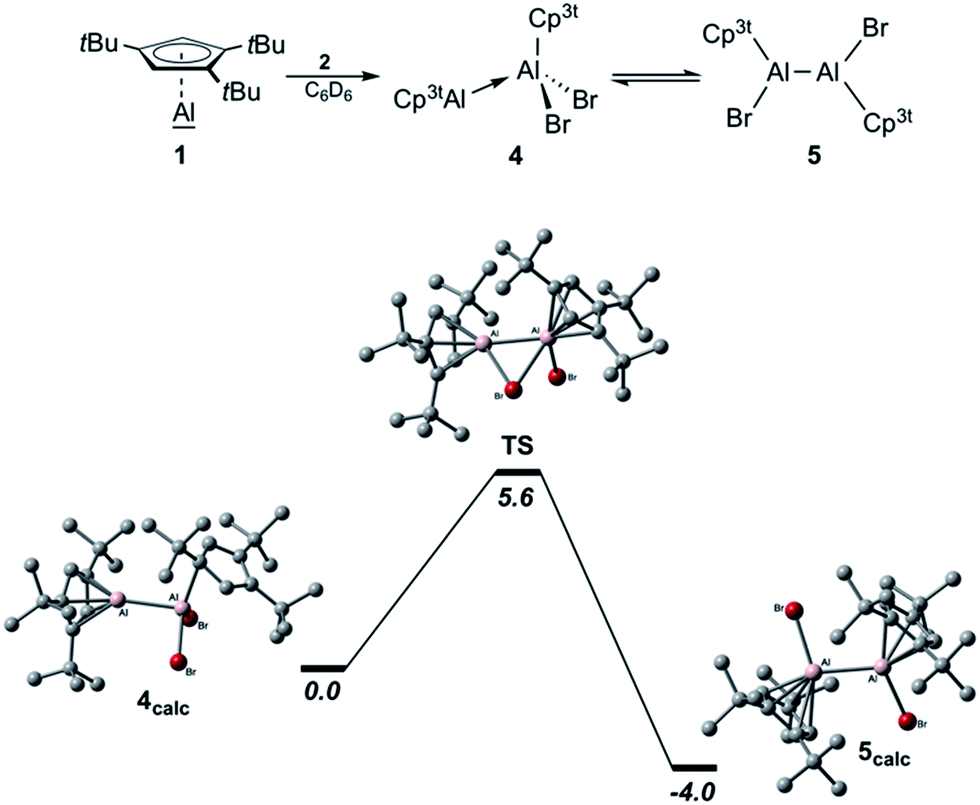 | ||
| Scheme 2 Valence isomerism between adduct 4 and dialane 5 (top). The energy profile of the isomerisation from 4 to 5via transition state TS (bottom; M06L/Def2-SVP; free energies given in kcal mol−1). | ||
 | ||
| Fig. 2 Molecular structures of 4 (left) and 5 (right) in the solid state. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) of 4 Al1–Al2 2.599(3), Al1–C1 2.209(7), Al1–C2 2.183(7), Al1–C3 2.216(7), Al1–C4 2.192(7), Al1–C5 2.178(7), and Al2–C11 2.063(7); 5 Al1–Al2 2.586(3), Al1–C1 2.231(8), Al1–C2 2.343(8), Al1–C3 2.392(8), Al1–C4 2.247(8), and Al1–C5 2.172(8). | ||
The structural parameters of adduct 4 are comparable to those of 3, but illustrate the increased steric demand of the Cp3t ligand. Accordingly, the Al–Al separation distance of 4 (2.599(3) Å) is slightly larger than that of the less bulky adduct 3 (2.533(1) Å), but in the same region as in Cp*Al·Al(C6F5)3 (2.591(2) Å).30 In analogy to 3, the Cp3t ligand of the monovalent aluminum center of 4 (Al1; Al–C 2.178(7)–2.216(7) Å, Al–cent 1.824 Å) adopts a η5 coordination mode with essentially the same bonding parameters as in 3 (Al–C 2.136(3)–2.220(3) Å, Al–cent 1.815 Å). Similarly, the Cp3t ligand of the Al(III) fragment of 4 (Al2) is again bound in a η1 fashion, while the Al2–C11 distance (2.063(7) Å) is slightly elongated with respect to 3 (2.005(3) Å). The bulkier nature of the Cp3t ligand is also evident in the molecular structure of dialane 5. Thus, the torsion angle Br1–Al1–Al2–Br2 (−152.71(8)°) and the Al–cent distances of 5 (1.925 Å, 1.930 Å) are significantly larger than in the related dialane Cp*(Br)Al–Al(Br)Cp* (Br1–Al1–Al2–Br2 102.04(5)°; Al–cent 1.902 Å, 1.904 Å).12 Other relevant structural features of 5 strongly resemble those found for Cp*(Br)Al–Al(Br)Cp*, i.e. both species show η5-coordination modes for the Cp rings and similar Al–Al distances (5: Al–C 2.164(8)–2.392(8) Å, Al–Al 2.586(3) Å; Cp*(Br)Al–Al(Br)Cp*: Al–C 2.169(4)–2.365(4) Å, Al–Al 2.530(2) Å).12
The reaction of isolated 1 with simple haloalanes aimed at elucidating similarities and differences of the reactivity patterns of 1, (Cp*Al)4, and other monovalent Al(I) species. It has been shown that halide substituents are usually non-innocent in the reaction of Al(I) species with haloalanes, leading to ionic or comproportionation products. Thus, the reaction of (Cp*Al)4 with AlCl3 enabled the isolation of the decamethylaluminocenium cation [Cp*2Al]+,35 while β-diketiminate-stabilized LAl(I) asymmetric dialanes were formed via comproportionation.32 Only when reacted with halide-free alanes such as Al(C6F5)3 (ref. 30) or AltBu3,33 (Cp*Al)4 acts as a Lewis base. In our work, monovalent 1 did not show any evidence for reduction chemistry upon treatment with tBuAlCl2 and AlBr3, but rather afforded adducts 6 and 7 quantitatively within seconds (Scheme 3).27 As expected, 1H NMR spectroscopic parameters of 6 and 7 strongly resemble each other and those of adducts 3 and 4, both featuring the typical set of three signals for the monovalent Cp3tAl part (6: δ 6.13, 1.30, and 1.22; 7: δ 6.10, 1.13, and 0.99). The 1H NMR resonance for the tBu group of 6 (δ 1.09) integrates very well with 9H, thus confirming the presence of a 1:1 ratio of the Al(I) and Al(III) fragments. The 27Al NMR spectra of 6 (δ 146) and 7 (δ 95) showed only a single resonance each for the tetracoordinate Al(III) fragments, while the signals of the Al(I) centers were not detected presumably because of their large full widths at half maximum. The molecular structures of 6 and 7 are unobtrusive and reminiscent of known Al(I)/Al(III) Lewis acid–base adducts such as 3 and 4, or the aforementioned literature examples.27 The Al–Al separation distances (6: 2.621(2) Å; 7: 2.554(1) Å), as well as the Al–C (6: 2.176(4)–2.203(4) Å; 7: 2.142(2)–2.188(2) Å) and Al–cent distances (6: 1.820 Å; 7: 1.796 Å) are found in the expected regions, while the Cp3t ligand in 7 appears to be more tightly bound to the Al(I) center as in all other adducts (Fig. 3).
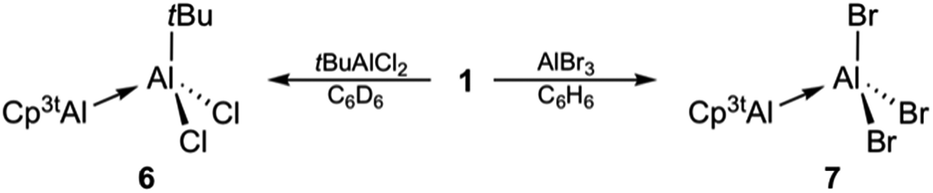 | ||
| Scheme 3 Lewis base reactivity of monovalent 1. | ||
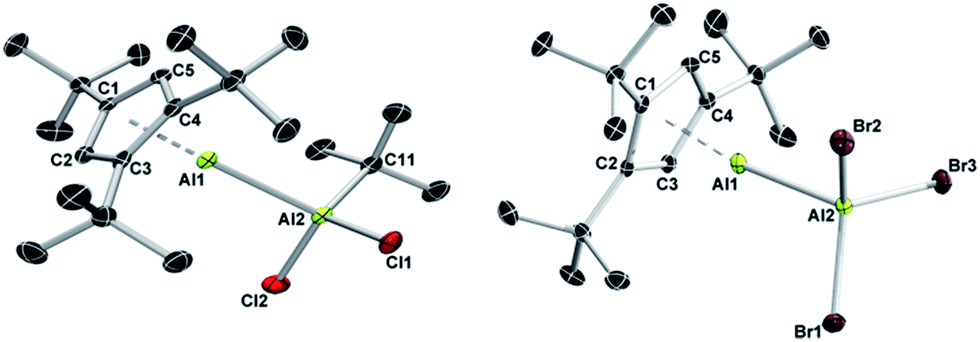 | ||
| Fig. 3 Molecular structures of 6 (left) and 7 (right) in the solid state. Hydrogen atoms are omitted for clarity. The asymmetric unit of 6 contains two independent molecules with similar metrical parameters; only one molecule is shown. Selected bond lengths (Å): 6 Al1–Al2 2.621(2), Al1–C1 2.202(3), Al1–C2 2.174(4), Al1–C3 2.203(4), Al1–C4 2.194(4), Al1–C5 2.176(4), and Al2–C11 1.973(4); 7 Al1–Al2 2.554(1), Al1–C1 2.188(2), Al1–C2 2.177(2), Al1–C3 2.142(2), Al1–C4 2.178(2), and Al1–C5 2.172(2). | ||
A closer inspection of the synthesis of 7 revealed another highly interesting reactivity of 1. When a solution of 1 was added dropwise to a solution of AlBr3, 7 was formed quantitatively as a single product. By reverse addition, however, a minor amount of a second aluminum-containing species was observed by 27Al NMR spectroscopy (δ −71), which we could eventually isolate as colorless crystals in low yield. This compound was subsequently identified by X-ray diffraction as the trialuminum species 8 (Fig. 4) with a very rare structural motif. In the solid state, 8 features three distinct aluminum centers with aluminum in two different oxidation states, being best described as the Lewis pair of the asymmetric dialane Br2Al–Al(Br)Cp3t and 1. Thus, the Lewis-basic Al(I) atom Al3 of 1 forms a dative interaction with the sterically less crowded and more Lewis-acidic Al(II) center Al2 of the dialane fragment (Br2Al–Al(Br)Cp3t), while the second Al(II) center Al3 remains ‘three-coordinate’ and covalently bound to Al2 (Br2Al–Al(Br)Cp3t). The observed Al–Al bond lengths are consistent with this picture, and Al1–Al2 (2.538(1) Å) and Al2–Al3 distances (2.601(1) Å) show typical values for covalent and dative Al–Al bonds, respectively. To the best of our knowledge, only one related, even though ionic, non-cluster-type trialuminum species has been mentioned in the literature, [Cp*2Al3I2]+[Cp*Al2I4]−.36 The selective, large scale synthesis of 8 was accomplished by either reacting adduct 7 with one equivalent of 1 or by dropwise addition of 0.5 equivalents of AlBr3 to a solution of 1 (Scheme 4). The 27Al NMR spectrum of 8 features only a single resonance for a three-coordinate aluminum atom (δ −71), which clearly substantiates the formulation of 8 as a dialane adduct. The absence of resonances for the other two aluminum atoms can be rationalized in combination with 1H NMR data, which both suggest the fluxional behavior of the bromide atoms. Thus, only one set of signals for the Cp3t protons is evident in the 1H NMR spectrum. These findings strongly indicate scrambling of the bromide substituents between the two Cp3tAl units. While the exact mechanism for the formation of 8 is not known, the fluxional behaviour in solution is consistent with a mechanism proceeding either (i) by reversible oxidative addition of one Al–Br bond of 7 to the Al(I) center of 1, (ii) by reversible coordination of the Lewis-basic Al(I) center of 1 to the hypothetical valence isomer of 7 (i.e. Cp3t(Br)Al–AlBr2), or (iii) by initial formation of the bis(adduct) Cp3tAl·AlBr3·AlCp3t and subsequent bromide migration. So far, we do not have any spectroscopic evidence for a favoured reaction pathway.
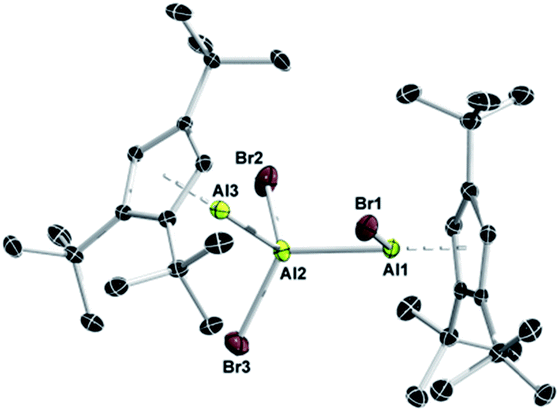 | ||
| Fig. 4 Molecular structures of 8 in the solid state. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å): 8 Al1–Al2 2.538(1), Al2–Al3 2.601(1), Al1–Br1 2.473(1), Al2–Br2 2.319(1), and Al2–Br3 2.319(1). | ||
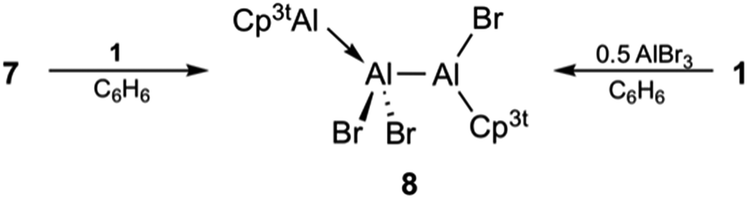 | ||
| Scheme 4 Synthesis of 8. | ||
Monovalent aluminum compounds have also been used to study Al![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E multiple bonding (E = N, P, As, O, Se, and Te). Thus, (Cp*Al)4 was shown to form tetrameric heterocubane type clusters (Cp*AlE)4 when reacted with elemental O2, N2O,37 Se, and Te,16,38 respectively. Similarly, treatment of (tBu3SiAl)4 with molecular oxygen afforded (tBu3SiAlO)4 among other products.39 Employing β-diketiminate-stabilized LAl(I) in the reaction with O2 led to a dimeric product, (LAlO)2.40 These results imply a strong impact of the electronic and steric parameters of the Al(I) species on the product structure, while a truly monomeric AlO compound is still absent in the literature.41 Accordingly, we became curious about what kind of product 1 might generate in view of its larger steric bulk as compared to (Cp*Al)4. To this end, a degassed solution of 1 in C6D6 was subjected to an atmosphere of N2O, which instantaneously resulted in the formation of the oxygenated species 9 (Scheme 5).27 NMR spectroscopic studies suggested the presence of a symmetric compound in solution, as evidenced by a single set of signals for the Cp3t ligand in the 1H NMR spectrum of 9 (δ 6.49, 1.61, and 1.39). However, NMR data were not sufficient to clarify the identity of 9, particularly because no 27Al NMR resonance was found.
E multiple bonding (E = N, P, As, O, Se, and Te). Thus, (Cp*Al)4 was shown to form tetrameric heterocubane type clusters (Cp*AlE)4 when reacted with elemental O2, N2O,37 Se, and Te,16,38 respectively. Similarly, treatment of (tBu3SiAl)4 with molecular oxygen afforded (tBu3SiAlO)4 among other products.39 Employing β-diketiminate-stabilized LAl(I) in the reaction with O2 led to a dimeric product, (LAlO)2.40 These results imply a strong impact of the electronic and steric parameters of the Al(I) species on the product structure, while a truly monomeric AlO compound is still absent in the literature.41 Accordingly, we became curious about what kind of product 1 might generate in view of its larger steric bulk as compared to (Cp*Al)4. To this end, a degassed solution of 1 in C6D6 was subjected to an atmosphere of N2O, which instantaneously resulted in the formation of the oxygenated species 9 (Scheme 5).27 NMR spectroscopic studies suggested the presence of a symmetric compound in solution, as evidenced by a single set of signals for the Cp3t ligand in the 1H NMR spectrum of 9 (δ 6.49, 1.61, and 1.39). However, NMR data were not sufficient to clarify the identity of 9, particularly because no 27Al NMR resonance was found.
 | ||
| Scheme 5 Synthesis of Al3O3 (9) and Al2N2 (10) heterocycles from 1. | ||
It was an X-ray diffraction study on suitable single crystals of 9 that eventually helped to assign the exact composition.27 As can be seen from Fig. 5, oxygenation of 1 with N2O goes along with the formal oligomerization of hypothetical monomeric Cp3tAlO to afford the six-membered Al3O3 heterocycle 9. Thus, a new member of the [LAlO]n family with n = 3 is accessible making use of the steric demand of the Cp3t ligand. The central Al3O3 ring of 9 is almost planar with all Al–O distances within a narrow range (1.697(2)–1.705(2) Å). The Al–O distances of 9 are thus significantly shorter than those in dimeric (LAlO)240 and tetrameric (R*AlE)4 (R = SitBu3 1.836 Å;39 R = Cp*calc 1.845 Å),37 which might indicate larger ionic contributions to the Al–O bonds in 9. As a consequence, the Al–C distances (2.216(3)–2.295(3) Å) of all three exocyclic η5-bound Cp3t ligands are somewhat elongated in comparison to all other Cp3t based compounds described in this study.
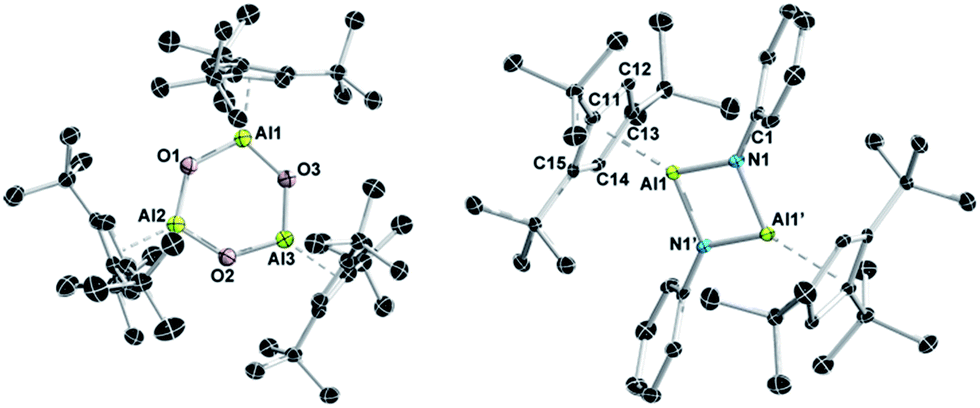 | ||
| Fig. 5 Molecular structures of 9 (left) and 10 (right) in the solid state. Hydrogen atoms are omitted for clarity. Symmetry operation for the conversion of Al1 to Al1′ in 10: –x + 1, –y + 1, –z + 1. Selected bond lengths (Å): 9 Al1–O1 1.701(2), Al1–O3 1.703(2), Al2–O1 1.702(2), Al2–O2 1.697(2), Al3–O2 1.705(2), Al3–O3 1.701(4), Al–C 2.216(3)–2.296(3); 10 Al1–N1 1.809(2), Al1–N1′ 1.827(2), Al1–C11 2.295(2), Al1–C12 2.244(2), Al1–C13 2.216(2), Al1–C14 2.198(2), Al1–C15 2.249(2), N1–C1 1.396(3). | ||
The related RAlNR′ chemistry appears even more unpredictable due to the presence of a substituent at the nitrogen center. Hence, the reaction of (Cp*Al)4 with different azides R′N3 was used to realize a variety of Al–N-based compounds, the formation of which can be rationalized by the stability of the initial iminoalane Cp*AlNR′ and the nature and size of R′. While for R′ = SiiPr3, SiPh3, and SitBu3 symmetric iminoalane dimers with a regular Al2N2 core have been obtained,42 the reaction with Me3SiN3 was more complicated affording an irregular iminoalane dimer.43 With the bulky azide MesN3, the monomeric iminoalane did not participate in oligomerization reactions, but was rather stabilized by a C–H bond activation/proton migration sequence.43 Other (RAlNR′)n structures ranging from monomeric iminoalanes (n = 1)44 to species with n = 16 were also described in the literature.45–47 However, either β-diketiminate-stabilized LAl(I) was used as a reagent in the reaction with azides, or other approaches were applied in their synthesis.
In our work, the reaction of 1 with PhN3 proceeded smoothly at room temperature to afford dimeric iminoalane 10 in good yields (Scheme 5).27 Again, the degree of Cp3tAlNPh aggregation is not apparent from NMR spectroscopy in solution, which pointed to either the presence of a monomeric iminoalane, or a symmetric species with a single set of 1H NMR signals for the Cp3t ligand (δ 6.78, 1.57, and 1.18) and the phenyl group (δ 7.35, 7.11, and 6.97) in a relative ratio of 1:1. No 27Al NMR resonance was observed for 10. X-ray diffraction eventually established a dimeric iminoalane structure of 10 in the solid state with the Cp3t ligands in typical η5 coordination modes (Al–C 2.198(3)–2.295(2) Å, Al–cent 1.883 Å; Fig. 5). The basic structural parameters of 10 are very similar to those of the iminoalane (Mes*AlNPh)2 described by Power,48 while noticeable differences are found in Roeskys' actually more related iminalane (Cp*AlNSitBu3)2.42 In analogy to (Mes*AlNPh)2 (Al–N 1.824(2) Å, ΣN 360°), the Al2N2 core of 10 is perfectly planar, as expected for a centrosymmetric heterocycle, with Al–N bond lengths of 1.809(2) and 1.827(2) Å, and trigonal planar nitrogen atoms (ΣN 359.9°). By contrast, (Cp*AlNSitBu3)2 features somewhat elongated Al–N bonds (1.835(2)–1.842(2) Å) and nitrogen atoms that significantly deviate from planarity (ΣN 353.4°, 353.7°), and most importantly η1-coordinated Cp* ligands. These structural distinctions most likely trace back to the steric demand of the extremely bulky NSitBu3 fragments.
Conclusions
In this contribution, we demonstrated that Cp3tAl (1) can be obtained by a three-step protocol involving the Lewis base-induced release of 1 from an Al(I)/Al(III) adduct as the key step. Thus, it was possible for the first time to isolate an analytically pure monomeric Cp-based Al(I) compound, a task that has consistently been hampered by synthetic difficulties. Detailed reactivity studies clearly furnished evidence that reactions involving 1 proceed more selectively (adducts 6 and 7), faster, and under milder conditions (heterocylces 9 and 10) than for (Cp*Al)4, which is in line with our expectations that the monomeric nature of 1 might provide some experimental benefits. Thus, monovalent 1 is expected to show a rich chemistry, particularly with respect to the realization of novel structural motifs (dialane adduct 8), and the activation of small molecules, two areas that are part of our current research efforts. One finding that deserves special attention is the experimental verification (reaction of 1 with 2) of the concept of valence isomerism between dialanes and the respective Al(I)/Al(III) Lewis acid–base adducts (structural characterization of 4 and 5), which so far has only been proposed on the basis of computational studies.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support from the Julius-Maximilians-Universität Würzburg is gratefully acknowledged.References
-
T. Blümke, Y.-H. Chen, S. Dagorne, M. Dieǵuez, V. A. D'yakonov, V. U. M. Dzhemilev, C. Fliedel, K. Groll, P. Knochel, A. Kolb, J. LewinśkiK. Maruoka, Y. Naganawa, O. Pàmies, S. Schulz, P. von Zezschwitz, R. J. Wehmschulte and A. E. H. Wheatley, Modern Organoaluminum Reagents: Preparation, structure, Reactivity and Use, ed. S. Woodward and S. Dalgarno, Springer-Verlag, Berlin/Heidelberg, 2013 Search PubMed
.
- M. Witt and H. W. Roesky, Curr. Sci., 2000, 78, 410 CAS
- H. W. Roesky, Inorg. Chem., 2004, 43, 7284 CrossRef CAS PubMed
- P. Bag, C. Weetman and S. Inoue, Angew. Chem., Int. Ed., 2018, 57, 14394 CrossRef CAS PubMed
- A. Schnepf and H. Schnöckel, Angew. Chem., Int. Ed., 2002, 41, 3532 CrossRef CAS
- W. Uhl, Adv. Organomet. Chem., 2004, 51, 53 CrossRef CAS
- W. Uhl, Angew. Chem., Int. Ed., 1993, 32, 1386 CrossRef
- N. Wiberg, K. Amelunxen, T. Blank, H. Nöth and J. Knizek, Organometallics, 1998, 17, 5431 CrossRef CAS
- R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 10784 CrossRef CAS PubMed
- C. Cui, X. Li, C. Wang, J. Zhang, J. Cheng and X. Zhu, Angew. Chem., Int. Ed., 2006, 45, 2245 CrossRef CAS PubMed
- T. Agou, K. Nagata, T. Sasamori and N. Tokitoh, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 588 CrossRef CAS
- A. Hofmann, A. Lamprecht, O. F. González-Belman, R. D. Dewhurst, J. O. C. Jiménez-Halla, S. Kachel and H. Braunschweig, Chem. Commun., 2018, 54, 1639 RSC
- H. Schnöckel, Z. Naturforsch. B Chem. Sci., 1976, 31, 1291 Search PubMed
- M. Tacke and H. Schnöckel, Inorg. Chem., 1989, 28, 2895 CrossRef CAS
- C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, Angew. Chem., Int. Ed., 1991, 30, 564 CrossRef
- S. Schulz, H. W. Roesky, H. J. Koch, G. M. Sheldrick, D. Stalke and A. Kuhn, Angew. Chem., Int. Ed., 1993, 32, 1729 CrossRef
- M. N. Sudheendra Rao, H. W. Roesky and G. Anantharaman, J. Organomet. Chem., 2002, 646, 4 CrossRef CAS
- H. W. Roesky and S. S. Kumar, Chem. Commun., 2005, 4027 RSC
- C. Dohmeier, E. Baum, A. Ecker, R. Köppe and H. Schnöckel, Organometallics, 1996, 15, 4702 CrossRef CAS
- H. Sitzmann, M. F. Lappert, C. Dohmeier, C. Üffing and H. Schnöckel, J. Organomet. Chem., 1998, 561, 203 CrossRef CAS
- M. Huber and H. Schnöckel, Inorg. Chim. Acta, 2008, 361, 457 CrossRef CAS
- W. W. Tomlinson, D. H. Mayo, R. M. Wilson and J. P. Hooper, J. Phys. Chem. A, 2017, 121, 4678 CrossRef CAS PubMed
- C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoescu, Angew. Chem., Int. Ed., 2000, 39, 4274 CrossRef CAS PubMed
- P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, J. Am. Chem. Soc., 2017, 139, 14384 CrossRef CAS PubMed
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92 CrossRef CAS PubMed
- A. Hofmann, A. Lamprecht, J. O. C. Jiménez-Halla, T. Tröster, R. D. Dewhurst, C. Lenczyk and H. Braunschweig, Chem.–Eur. J., 2018, 24, 11795 CrossRef CAS PubMed
- Experimental details, and details on the X-ray diffraction studies are included in the ESI.†.
- M. Schormann, K. S. Klimek, H. Hatop, S. P. Varkey, H. W. Roesky, C. Lehmann, C. Röpken, R. Herbst-Irmer and M. Moltemeyer, J. Solid State Chem., 2001, 162, 225 CrossRef CAS
- C. Ganesamoorthy, S. Loerke, C. Gemel, P. Jerabek, M. Winter, G. Frenking and R. A. Fischer, Chem. Commun., 2013, 49, 2858 RSC
- J. D. Gorden, C. L. B. Macdonald and A. H. Cowley, Chem. Commun., 2001, 75–76 RSC
- T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195 CrossRef CAS PubMed
- B. Li, S. Kundu, H. Zhu, H. Keil, R. Herbst-Irmer, D. Stalke, G. Frenking, D. M. Andrada and H. W. Roesky, Chem. Commun., 2017, 53, 2543 RSC
- S. Schulz, A. Kuczkowski, D. Schuchmann, U. Flörke and M. Nieger, Organometallics, 2006, 25, 5487 CrossRef CAS
- P. Jutzi, B. Neumann, G. Reumann, L. O. Schebaum and H.-G. Stammler, Organometallics, 2001, 20, 2854 CrossRef CAS
- C. Dohmeier, H. Schnöckel, C. Robl, U. Schneider and R. Ahlrichs, Angew. Chem., Int. Ed., 1993, 32, 1655 CrossRef
- C. Üffig, E. Baum, R. Köppe and H. Schnöckel, Angew. Chem., Int. Ed., 1998, 37, 2397 CrossRef
- A. C. Stelzer, P. Hrobárik, T. Braun, M. Kaupp and B. Braun-Cula, Inorg. Chem., 2016, 55, 4915 CrossRef CAS
- A monomeric L2·RAl Te has been reported recently by Inoue and coworkers. However, no Al(I) species was directly involved in the multi-step synthesis: D. franz, T. Szilvási, E. Irran and S. Inoue, Nat. Commun., 2015, 6, 10037 CrossRef CAS PubMed
- N. Wiberg, T. Blank, K. Amelunxen, H. Nöth, H. Schnöckel, E. Baum, A. Purath and D. Fenske, Eur. J. Inorg. Chem., 2002, 341 CrossRef CAS
- H. Zhu, J. Chai, V. Jancik, H. W. Roesky, W. A. Merrill and P. P. Power, J. Am. Chem. Soc., 2005, 127, 10170 CrossRef CAS PubMed
- A monomeric LAl O compound of the type LAl–O–B(C6F5)3 has been reported by Roesky and coworkers. However, no Al(I) species was involved in the synthesis. Here, a custom-designed β-diketiminate ligand with pendant amino groups was used, which provided additional stabilization to the reactive aluminium center by Lewis acid–base interaction: D. Neculai, H. W. Roesky, A. M. Neculai, J. Magull, B. Walfort and D. Stalke, Angew. Chem., Int. Ed., 2002, 41, 4294 CrossRef CAS PubMed
- S. Schulz, A. Voigt, H. W. Roesky, L. Häming and R. Herbst-Irmer, Organometallics, 1996, 15, 5252 CrossRef CAS
- S. Schulz, L. Häming, R. Herbst-Irmer, H. W. Roesky and G. M. Sheldrick, Angew. Chem., Int. Ed., 1994, 33, 969 CrossRef
- J. Li, X. Li, W. Huang, H. Hu, J. Zhang and C. Cui, Chem.–Eur. J., 2012, 18, 15263 CrossRef CAS PubMed
- K. M. Waggoner, H. Hope and P. P. Power, Angew. Chem., Int. Ed., 1988, 27, 1699 CrossRef
- H. Zhu, Z. Yang, J. Magull, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, Organometallics, 2005, 24, 6420 CrossRef CAS
-
M. Cesari and S. Cucinella, The Chemistry of Inorganic Homo and Heterocycles, ed. I. Haiduc and D. B. Sowerby, Academic Press, London, 1987, vol. 1 Search PubMed
- R. J. Wehmschulte and P. P. Power, J. Am. Chem. Soc., 1996, 118, 791 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of new compounds, NMR spectra, crystallographic details. CCDC 1878104–1878111 and 1879323. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05175e |
| This journal is © The Royal Society of Chemistry 2019 |