
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterolytic bond activation at gold: evidence for gold(III) H–B, H–Si complexes, H–H and H–C cleavage†
Luca
Rocchigiani
 *a,
Peter H. M.
Budzelaar
*b and
Manfred
Bochmann
*a
*a,
Peter H. M.
Budzelaar
*b and
Manfred
Bochmann
*a
aSchool of Chemistry, University of East Anglia, Norwich Research Park, Norwich NR4 7TJ, UK. E-mail: l.rocchigiani@uea.ac.uk; m.bochmann@uea.ac.uk
bDepartment of Chemistry, University of Naples Federico II, Via Cintia, 80126 Naples, Italy. E-mail: p.budzelaar@unina.it
First published on 16th January 2019
Abstract
The coordinatively unsaturated gold(III) chelate complex [(C^N–CH)Au(C6F5)]+ (1+) reacts with main group hydrides H–BPin and H–SiEt3 in dichloromethane solution at −70 °C to form the corresponding σ-complexes, which were spectroscopically characterized (C^N–CH = 2-(C6H3But)-6-(C6H4But)pyridine anion; Pin = OCMe2CMe2O). In the presence of an external base such as diethyl ether, heterolytic cleavage of the silane H–Si bond leads to the gold hydrides [{(C^N–CH)AuC6F5}2(μ-H)]+ (2+) and (C^N–CH)AuH(C6F5) (5), together with spectroscopically detected [Et3Si–OEt2]+. The activation of dihydrogen also involves heterolytic H–H bond cleavage but requires a higher temperature (−20 °C). H2 activation proceeds in two mechanistically distinct steps: the first leading to 2 plus [H(OEt2)2]+, the second to protonation of one of the C^N pyridine ligands and reductive elimination of C6F5H. By comparison, formation of gold hydrides by cleavage of suitably activated C–H bonds is very much more facile; e.g. the reaction of 1·OEt2 with Hantzsch ester is essentially instantaneous and quantitative at −30 °C. This is the first experimental observation of species involved in the initial steps of gold catalyzed hydroboration, hydrosilylation and hydrogenation and the first demonstration of the ability of organic C–H bonds to act as hydride donors towards gold.
Introduction
Gold catalysts, both heterogeneous and homogeneous, have experienced an explosive growth in interest in recent years.1–3 In the absence of direct evidence, their mode of action tends to be explained in analogy to the well-known chemistry of other noble metals, notably palladium and platinum, and indeed gold and platinum systems are often compared side-by-side.4,5 There are significant differences, however, not least in the distinct reluctance of gold to undergo oxidative addition reactions,6,7 and an experiment-based outline of the reactivity of gold, and in particular of the much less well explored chemistry of gold(III), is only now beginning to emerge.8–10 Heterogeneous gold catalysts show high activity in a multitude of reactions, for example in hydrogenations,11–16 including the hydrogenation of nitro compounds17–19 and hydrogen transfer,20 in acetylene hydrochlorination,21 in the water–gas shift reaction,22 in hydrosilylations and in catalytic dehydrogenative Si–O coupling reactions.23–28 Similarly, homogeneously gold-catalyzed hydrogenations and hydrosilylations29–31 as well as alkyne hydroborations32 have been reported.Details of the reactions of H2, silanes and boranes with gold compounds, and particularly of their modes of activation and reaction pathways, are however rather scarce. The common feature of all these reactions is that they involve, or are postulated to involve, Au–H species. While in a number of heterogeneous systems surface gold hydride species could indeed be detected spectroscopically,33–35 the mechanisms of gold-catalysed reactions have been explored by computational modelling. Naturally, most studies are based on mechanistic analogies to better known noble metal catalysts; for example, homolytic H–H and H–Si bond scission and oxidative addition of dihydrogen or of silanes to surface gold atoms are assumed, to give Au–H and Au–SiMe3 species which initiate the catalytic cycles.24–28,35–37 There are however also cases where the possible involvement of polar solvents in the catalytic process has been recognized, leading to models of heterolytic, solvent-facilitated H–H bond cleavage.38–41
We present here the first experimental evidence for detectable gold(III) adducts with borane H–B and silane H–Si bonds, elucidate the important role of the solvent and of basic ligand sites in the formation of Au–H species via heterolytic H–Si and H–H bond scission, and demonstrate the ability of gold(III) to generate gold hydrides by H–C(sp3) bond cleavage.
Results and discussion
As we showed recently,42 the reaction of the pincer complex (C^N^C)AuX with the strong Brønsted acid [H(OEt2)2]+[H2N{B(C6F5)3}2]− (HAB2)43 creates the ether-stabilized C^N chelate complex [(C^N–CH)AuX(OEt2)]+AB2− (1·OEt2) (C^N^C = 2,6-(C6H3But)2pyridine dianion). In the present work the pentafluorophenyl derivative (X = C6F5) was chosen, firstly because this ligand provides additional stability, and secondly because the fluorine atoms can act as additional reporter nuclei and aid spectroscopic characterization. The ether ligand is labile and can be removed to afford ether-free 1 with a “dangling” –C6H4But substituent, of a “Pacman”-type structure and which is capable of supporting ligand binding to the coordination pocket, such that adducts of weak ligands with 1 are accessible which would not be feasible in C^N complexes without this dangling aryl moiety. We have also shown44 that treatment of 1·OEt2 with HSi(OMe)3 leads quantitatively to hydride transfer and the formation of the hydrido-bridged binuclear gold(III) complex 2 (Scheme 1).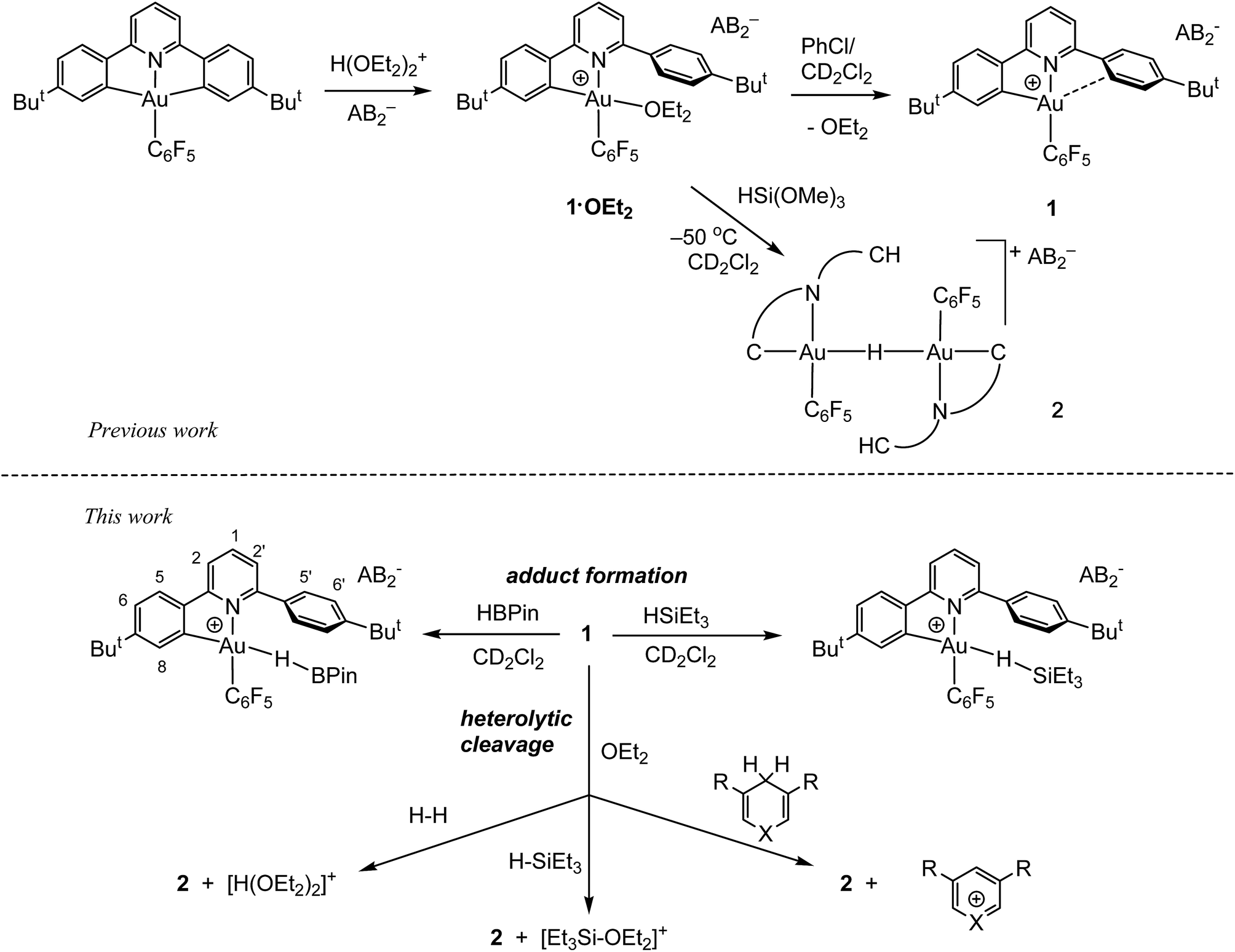 | ||
| Scheme 1 Reaction outlines including the atom labelling scheme used in NMR assignments. | ||
By contrast, we now find that gold hydride formation is preceded by formation of observable adducts between Au(III) and H–B and H–Si bonds.
Borane σ-complexes
In contrast to the reaction of 1·OEt2 with HSi(OMe)3, no reaction took place when this gold complex was mixed with 2 molar equiv. of HBPin at −70 °C, suggesting that the borane is unable to displace the coordinated ether ligand. However, when the solution was warmed to −20 °C, the starting material reacted slowly over a period of 30 min, affording the bridging hydride 2 quantitatively.On the other hand, adding 1 equiv. of HBPin to the ether-free complex 1 in dichloromethane at −70 °C immediately afforded a mixture of the hydride 2 (15%), together with a new species 3 (85%). The relative positions of the borane and the C^N ligand framework are most clearly indicated by nuclear Overhauser effect (NOE) experiments, which show selective interactions between the “dangling” –C6H4But substituent of the C^N ligand and the methyl groups of the boron-pinacolato moiety (Fig. 1). These data suggest that 3 is a gold(III) σ-borane adduct, to our knowledge the first such observation. By contrast, as a reviewer pointed out, M–HB interactions in copper(I) and silver(I) complexes of hydroborates and anionic borane clusters are well known,45–49 while intramolecular Au(I)–HB interactions were observed in heterometallic Au–Rh carborane clusters.50 Further characterization of 3 is hampered by its poor thermal stability, even at very low temperatures. On warming the sample to −20 °C 3 is quickly and quantitatively converted into the hydride 2. This suggests that heterolytic B–H splitting occurs readily without the involvement of solvent (in contrast to silanes, see below), and it seems reasonable to assume that the B–H cleavage process is assisted by the O-donor of a second molecule of HBPin. However, the 11B NMR spectrum is uninformative and no clear indication about the nature of the boron side-product could be obtained. The formation of a gold–HB σ-complex, as well as its lability, are in agreement with computational results (vide infra).
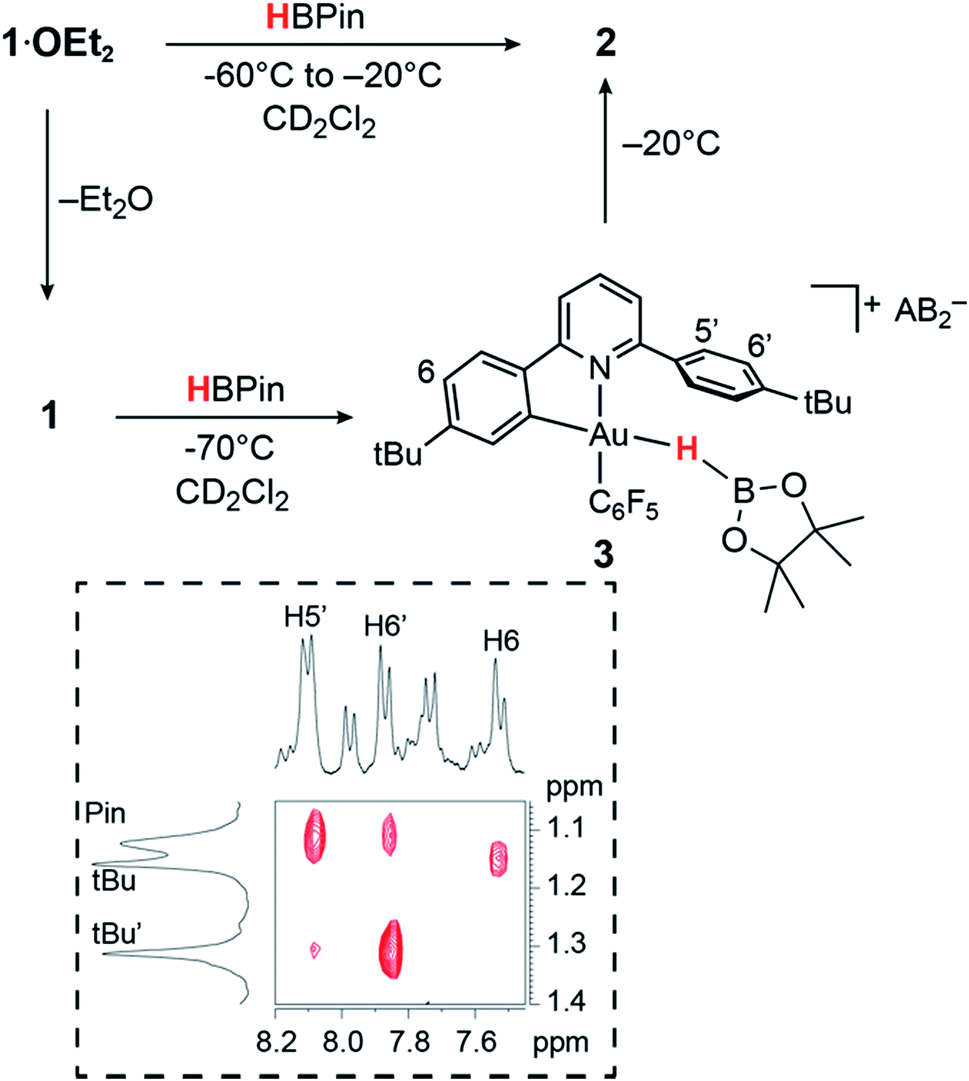 | ||
| Fig. 1 Reactivity of 1 and 1·OEt2 with HBPin. The inset shows a section of the 1H NOESY NMR spectrum of 3 (−70 °C, CD2Cl2). | ||
Gold–silane σ-complexes
As stated above, the reaction of 1·OEt2 with HSi(OMe)3 provided a convenient route to the μ-H gold hydride 2. However, a very different course of reaction was observed when the ether-free cation 1 was treated with HSiEt3 at −70 °C in CD2Cl2. Under these conditions, there is no HSiEt3 cleavage and no hydride transfer. Instead, the Au(III) σ-silane complex 4 is formed in essentially quantitative yield (Fig. 2). The identity of 4 was confirmed by multinuclear and multidimensional NMR spectroscopy. The H–Si moiety shifted by 2.19 ppm with respect to free silane, to δH = 1.27 ppm, while the ethyl substituents showed only moderate shifts of Δδ = 0.25 and 0.28 ppm for the CH2 and CH3 groups, respectively. The coordination of HSiEt3 in 4 is readily reversible; resonances are broad even at −70 °C, and the 1H NOESY NMR spectrum shows extensive chemical exchange between free and coordinated silane. On the basis of these data it is therefore not possible to discriminate between side-on and end-on coordination modes, although DFT calculations indicate that end-on Au–H–Si bonding is preferred (vide infra).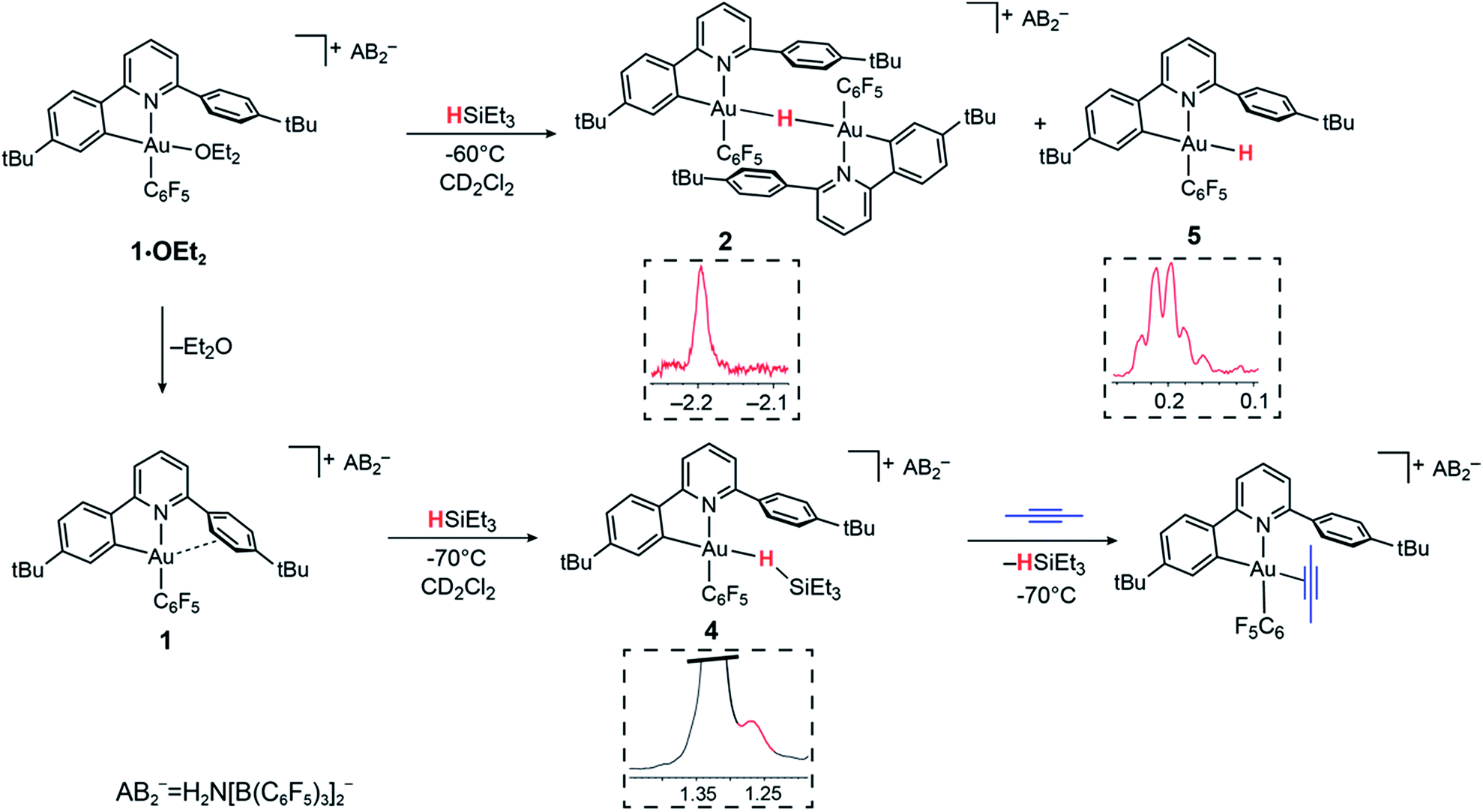 | ||
| Fig. 2 Reactivity of 1·OEt2 and 1 with HSiEt3. Insets show sections of the 1H NMR spectra highlighting the hydride signals. | ||
The coordination of HSiEt3 to the Au(III) centre in 4 is weak enough to be displaced by other weakly coordinated ligands such as 2-butyne. Under these conditions, no hydrosilylation of the alkyne was observed. Instead, the alkyne complex evolved by carbocationic alkyne cyclodimerization, as we observed previously in reactions of Au(III) centres with sterically undemanding alkynes,53 while HSiEt3 remains unreacted.
In the presence of diethyl ether hydride transfer to gold involving heterolytic splitting of the H–SiEt3 bond takes place immediately, as shown by the quantitative formation of the Et2O-stabilized triethylsilylium cation [Et3SiOEt2]+ which is stable at −60 °C and was characterized spectroscopically. Relative to free Et2O the ethyl groups of Et2O in [Et3SiOEt2]+ are high-frequency shifted, to δH = 4.55 ppm (CH2, Δδ = 1.14) and δH = 1.57 ppm (Δδ = 0.45), respectively.
Contrary to what we observed for HSi(OMe3), when 1·OEt2 was treated with 8 equiv. of HSiEt3 at −30 °C, a new major product 5 was formed, accompanied by only small traces of 2 (<5%). Compound 5 is characterized by an Au–H signal at δH = 0.20 ppm; this chemical shift is in good agreement with the value expected for a neutral Au(III) hydride, with H in trans-position to an anionic aryl-C.44 The 1H NMR signal of the hydride moiety in 5 appears as a pseudo-quartet, due to the simultaneous coupling of the hydride with one o-fluorine atom of the pentafluorophenyl ligand and the α proton H8 of the cyclometalated C^N chelate (for the numbering system used for NMR assignments see Scheme 1). Probing the hydrogen–fluorine heterocoupling by means of 1H{19F} and 19F{1H} experiments revealed a coupling constant 4JHF = 5.7 Hz, which might arise from a through-space correlation mechanism.54 This pattern is not observed in 2, where the steric constraints imposed by μ-H dimer formation hold the pentafluorophenyl rings tightly in positions perpendicular to the square-planar coordination plane. On the basis of these data 5 is identified as a neutral mononuclear gold(III) hydride complex.
The 19F NMR spectrum of the mixture shows only one signal for both the ortho fluorine atoms of 5, suggesting that the monomeric environment makes aryl tilting and C6F5 rotation possible, thus enabling close H–F proximity. Homocoupling between the hydride signal and H8 has previously been observed in biphenyl-based Au(III) dihydrides Li[(C^C)AuH2];44 however, the coupling in 5 is larger (4JHH = 5.0 versus 2.3 Hz). This suggests that minor alteration of the ligand structure has a dramatic impact on ligand flexibility and hence the spectroscopic features of this class of compounds.
Species 2 and 5 do not show chemical exchange in the 1H NOESY NMR spectrum recorded at −60 °C, suggesting that they form as kinetic products upon the addition of the silane. Warming the samples at temperatures above −30 °C results in the decomposition of 5 by reductive elimination, to give C6F5H. On the other hand, 2 remains unaltered up to −10 °C, as observed previously. Evidently, terminal neutral gold(III) hydrides are less stable and more reactive than cationic H-bridged analogues. On warming the sample at room temperature C6F5H was formed quantitatively, indicating that ArF–H reductive elimination is the only decomposition pathway accessible to both 2 and 5.
The observation of Au(III) σ-silane complexes as detectable intermediates is unprecedented in gold chemistry. There are apparently no previous examples of silane–gold complexes,51 and Bourissou and co-workers pointed out recently that whereas copper(I) forms intramolecular σ-Si–H adducts, gold(I) does not.52 Mechanistically, their formation under base-free conditions suggests that the heterolytic Si–H bond cleavage requires the presence of a Lewis base, which is capable of reacting with and stabilizing silylium cations generated after the hydride abstraction (see DFT section for details). This process contrasts with the reactions of heterogeneous gold nanoparticle hydrosilylation catalysts, for which the evidence points to the formation of H˙ radicals and Au–silyl surface species.26
Hydrogen activation
The ability of gold(III) cations to promote the heterolytic cleavage of Si–H and B–H bonds via a base-assisted non-redox mechanism is reminiscent of the behaviour of frustrated Lewis pairs.55 We therefore envisioned that same reaction principle might enable the heterolytic splitting of molecular hydrogen. When a solution of 1·OEt2 in CD2Cl2 was saturated with H2 (1 bar) at −50 °C, no reaction was observed over a period of 1 h. However, upon warming the sample to −20 °C, a slow reaction took place and gave a mixture containing 2 (41%), C6F5H (43%) and a minor component of a second hydride complex 6 with a hydride chemical shift at δH = −4.47 ppm (16%). At the same time the evolution of H(OEt2)2+ (δH = 16.6 ppm) was observed. These results show that heterolytic hydrogen splitting had occurred, and that some of the gold(III) hydride formed had undergone reductive elimination of C6F5H.The 1H NMR spectrum of the mixture shows the presence of two AA′XX′ systems for 6 at δH = 7.52/7.35 and δH = 7.18/6.66 ppm, integrating as 8 and 4 protons, respectively, which give dipole interactions with the Au–H signal in the 1H NOESY NMR spectrum (see ESI†). The chemical shift of the Au–H moiety in 6 matches that of Au(III) bridging hydrides, where the hydrogen is simultaneously trans to an Au–C(aryl) and an Au–N(pyridine) bond.44 Consistent with this, the 19F NMR spectrum shows only one signal for the two ortho fluorine atoms, suggesting that the Au–C6F5 ring in 6 can rotate freely. These observations suggest that 6 is an H-bridged mixed-valence Au(III)/Au(I) complex, formed from 2 by a second heterolytic H–H cleavage step followed by reductive elimination (Scheme 2).
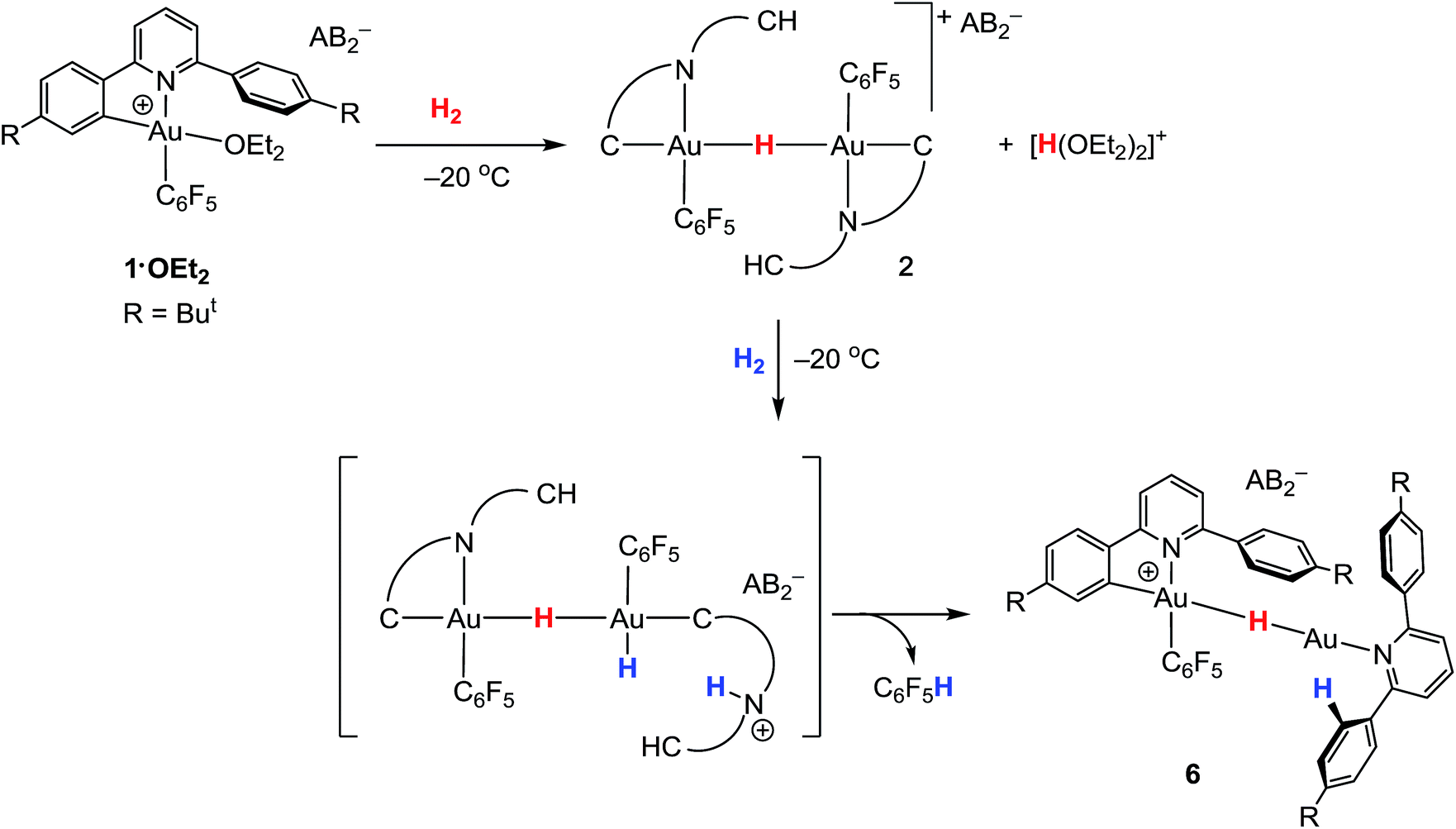 | ||
| Scheme 2 Formation of gold hydrides 2 and 6 by two successive heterolytic H–H cleavage steps. | ||
A control experiment in which pre-formed 2 was mixed with [H(OEt2)2][AB2] at −50 °C revealed no formation of 6, suggesting that the latter is not the product of a side reaction of 2 with protons. We suggest that a dissociated pyridine moiety of one C^N chelate ligand in 2 acts as base and enables a second, slower heterolytic H–H cleavage step. This leads to an unstable transient Au(III)/Au(III) dihydride, which undergoes fast reductive elimination of C6F5H coupled with internal protodeauration of the remaining Au–aryl bond of that phenylpyridine ligand, to give 6. Finally, upon warming the sample to room temperature a clean and quantitative conversion to C6F5H and protonated bis(4-t-butylphenyl)pyridine was observed, without any trace of C–C reductive elimination products (Fig. 3).56
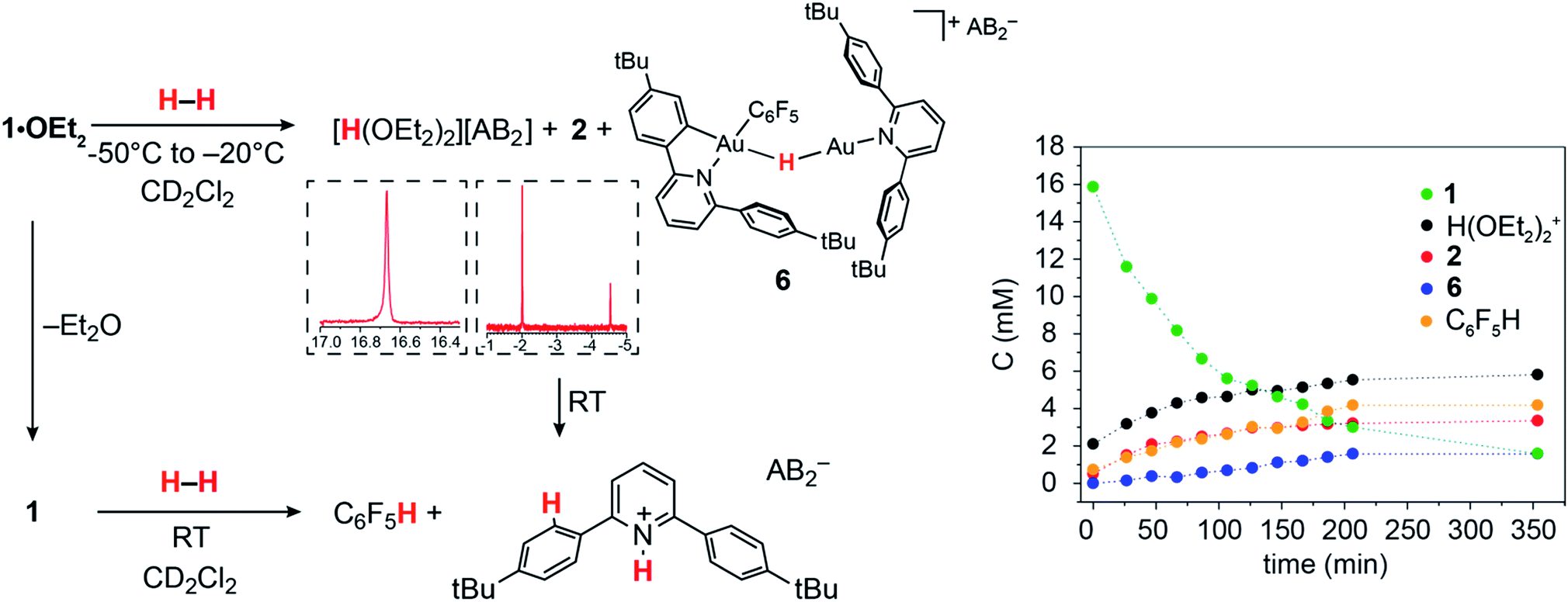 | ||
| Fig. 3 Left: Reactions of 1·OEt2 and of 1 with H2. Insets show sections of the 1H NMR spectrum highlighting protic and hydride signals. Right: Kinetic profile for the reaction with 1·OEt2 at −20 °C. | ||
Ether-free 1 is unreactive towards H2 at −10 °C over a period of several hours. No gold dihydrogen complex was observed, suggesting that H2 binding to gold(III) is not strong enough to displace the π-interaction with the dangling aryl ring. However, 1 and H2 do react slowly over a period of 1 week at room temperature to generate quantitatively C6F5H and protonated bis(4-t-butylphenyl)pyridine, as was the case in the reaction with 1·OEt2. It seems reasonable to assume that H2 is activated by the cooperation between gold and base (following pyridine dissociation) to give pyridinium salts and Au(III)–H, which then decomposes upon further reaction with H2 as suggested in Scheme 2.
We note in this context that Corma and co-workers prepared heterogenised Schiff-base complexes of gold(III) which proved to be highly active in olefin hydrogenation.38 The possible active site was probed by DFT calculations which assumed formation of Au(III) hydrides and persistence of the Schiff-base structure. In light of the mechanistic results described above, it seems probable however that ligands with N and O-donor sites are far from innocent and may facilitate both H2 activation and reduction of the gold catalyst.
Heterolytic C–H bond cleavage
Organic hydride donors can exceed dihydrogen in their ability to generate gold hydrides. For example, 1·OEt2 reacts with 1 equiv. of Hantzsch ester at −30 °C essentially instantaneously and much faster than with H2, to give 2 in quantitative yield (Scheme 3). Since the by-product is the corresponding pyridinium salt, the heterolytic C–H bond cleavage does not require the action of an external base. Indeed, ether-free 1 reacts with Hantzsch ester readily to give 2 quantitatively even at −70 °C.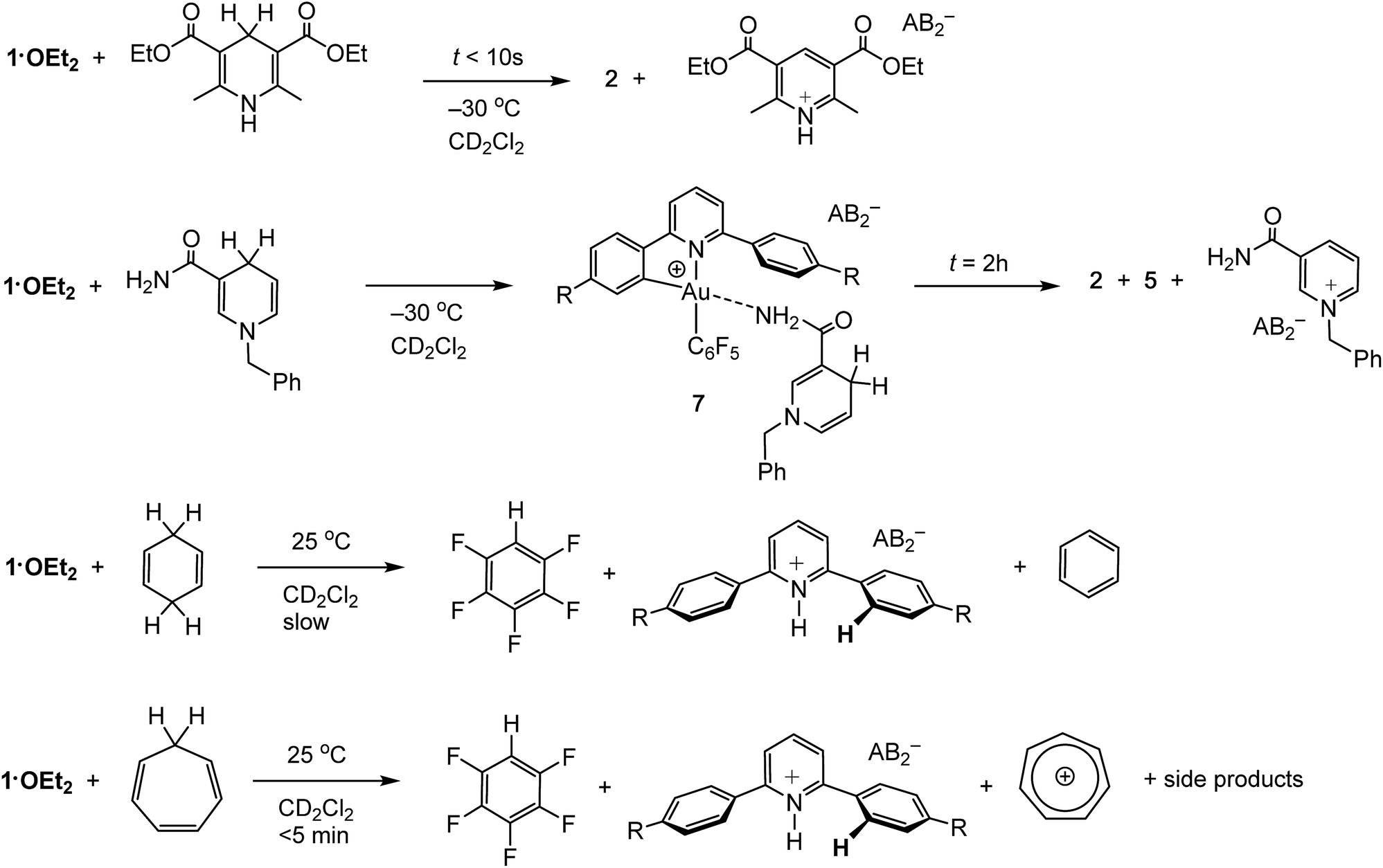 | ||
| Scheme 3 Reactions of 1·OEt2 with activated organic hydride donors. | ||
N-Benzyl nicotinamide reacts rather more slowly under the same conditions, to give a mixture containing 2 and 5 in a 30/70 molar ratio. Interestingly, the 1H NMR spectrum recorded soon after mixing at −30 °C reveals the formation of the Au–nicotinamide adduct 7. The coordination of the amide group to the metal was revealed by the chemical shift changes of the NH2 signal (Δδ = 0.4 ppm) and those of the benzyl group and C–H moiety α to the nitrogen. The formation of 7 affects the kinetics of C–H activation and product distribution: as most of 1·OEt2 is captured by the nicotinamide, the concentration of gold cations with sufficient electrophilicity for hydride abstraction is reduced, leading to a slower C–H activation rate and formation of the mononuclear terminal hydride 5 as the major product.
Less activated C–H compounds react only at higher temperature and significantly more slowly. For example, 1,4-cyclohexadiene (8.5 equiv.) undergoes hydride transfer to 1·OEt2 over the period of 2 weeks. Since the gold hydrides formed in these reactions are not thermally stable under these conditions, their intermediacy is deduced from the appearance of the reductive elimination product, C6F5H. Under similar conditions the analogous reaction with cycloheptatriene proceeds significantly faster and is complete in <5 min, to give C6F5H and the tropylium cation C7H7+ in about 40% yield, accompanied by a number of side products including evidence for reductive C6F5–phenylpyridine C–C coupling (Scheme 3). NMR monitoring at −15 °C confirmed the formation of the gold hydride 2 as reaction intermediate.
The facile formation of gold hydrides from substrates such as benzyl-nicotinamide is remarkable and may have wider implications. Compounds like the Hantzsch ester and nicotinamide are well-known analogues of NADH, which is involved in electron transport in the mitochondrial respiratory chain.57 Many gold(III) chelate complexes exhibit pronounced cytotoxicity, although their modes of action remain to be elucidated.58,59 The hydride abstraction ability of Au(III) demonstrated here raises the possibility that interference in the NAD/NADH intracellular redox pathways may have to be added to the pathways responsible for the anti-tumour activity of gold(III) compounds.
DFT studies
The reactions of several substrates HE (E = BPin, SiMe3 or H) with [(C^N–CH)Au(C6F5)]+ (here abbreviated to LAu+) were studied by Density Functional Theory (DFT) calculations. Geometry optimization and thermal corrections were carried out at B3LYP/def2-SVP/PCM(CH2Cl2) level, final electronic energies from M06/cc-pVTZ/PCM(CH2Cl2), with free energies calculated at the compromise temperature of 250 K. The model ligand L has the tBu substituents of the C^N–CH ligand replaced by hydrogens, and substrates were replaced by minimal models. For further details see the ESI.†The basic reaction studied is given in eqn (1):
| LAu+ + HE → [LAu⋯H⋯E]+ → LAuH + E+ | (1) |
Table 1 lists the most relevant free energies for different choices of E; complete listings can be found in Table S1.†
| HE | LAu(HE)+ | [LAu⋯H⋯E⋯B]+ | LAuH + BE+ | [LAu]2H+ + BE+ |
|---|---|---|---|---|
| a LAu+ + HE as reference (0 kcal mol−1); B = OMe2; reference energy of LAu(B)+ is −8.86 kcal mol−1 if the reaction is done in presence of ether. b Transition state. c Intermediate (local minimum). d No assistance by ether. e Not separated products but complex LAuH⋯BE+. f Forming H[OMe2]2+ instead of HOMe2+. g Barrierless to reactants. | ||||
| HSiMe3 | −5.28 | −0.87b | −7.15 | −25.83 |
| HBPin | −0.72 | −1.71c | −4.72 | −23.40 |
| H2 | +6.21 | +9.72b | +10.51e | −16.06f |
| PyCarH | — | +3.67b,d | −7.57d | −26.25 |
| CHDH | — | +16.22b,d | +15.83d | −2.85 |
| Ph3CH | — | — | +19.24d | +0.56 |
| PhCH3 | — | +45.80d | +27.12 | |
Before going into specific reactions, a few general points should be noted: (i) several reactions start with the ether complex LAu(OR2)+, see Scheme 4. The initial dissociation of coordinated ether costs 8.9 kcal mol−1. (ii) The initially produced LAuH would have the hydride trans to the aryl-C atom of the C^N–CH ligand. The isomer with H trans to N(pyridine) is actually preferred by 6.5 kcal mol−1, although it is not clear how such an isomerization would happen. However, any LAuH formed can be quickly trapped by unreacted LAu+ to form the dimer [(LAu)2(μ-H)]+, for which a structure with H trans to both aryl-C atoms is preferred. (iii) In many of the cases studied the basic reaction (1) is endergonic. However, the trapping of the initial product LAuH by LAu+ adds −18.7 kcal mol−1 to the reaction free energy, pulling the equilibrium to the right (Scheme 4).
 | ||
| Scheme 4 H–E activation equilibria. | ||
(iv) The formation of free E+ is energetically unfavourable. However, the presence of a Lewis base B (ether in this case) may help trap and stabilize the H abstraction product E+ as an adduct BE+. Stabilization by B may start at various stages of reaction (1): by initial coordination of B to HE, or by having B as a nucleophile displacing LAuH from E, or even after hydride displacement has already completed. This results in a set of similar but subtly different “mechanisms” as detailed below.
It is convenient to start with a silane (HSiMe3) substrate. The relevant energy profile is shown in Fig. 4. Reaction of HSiMe3 with LAu+ produces a weakly bound silane complex (−5.3 kcal mol−1 relative to “naked” LAu+), but this low binding energy is misleading: both Si–H and Au–H bonds are clearly elongated relative to the parent molecules HSiMe3 and LAuH, indicating extensive electronic reorganization to the separated hydrides. Relevant bond lengths and Wiberg bond indexes (WBIs) are shown in Fig. 5; the WBI suggests that at this stage H transfer has progressed to about 30%. Simple dissociation of SiMe3+ is not feasible, and even trapping of LAuH by LAu+ is not enough to make this reaction exergonic. However, if the same reaction is carried out in the presence of ether, an SN2-like attack of OMe2 on the Si atom of the coordinated silane leading to loss of [Me2OSiMe3]+ is remarkably easy, requiring only 4.4 kcal mol−1. The rate-limiting step for this process is actually the initial ether dissociation, which costs 8.9 kcal mol−1, corresponding to a reaction that is extremely fast even at −60 °C.
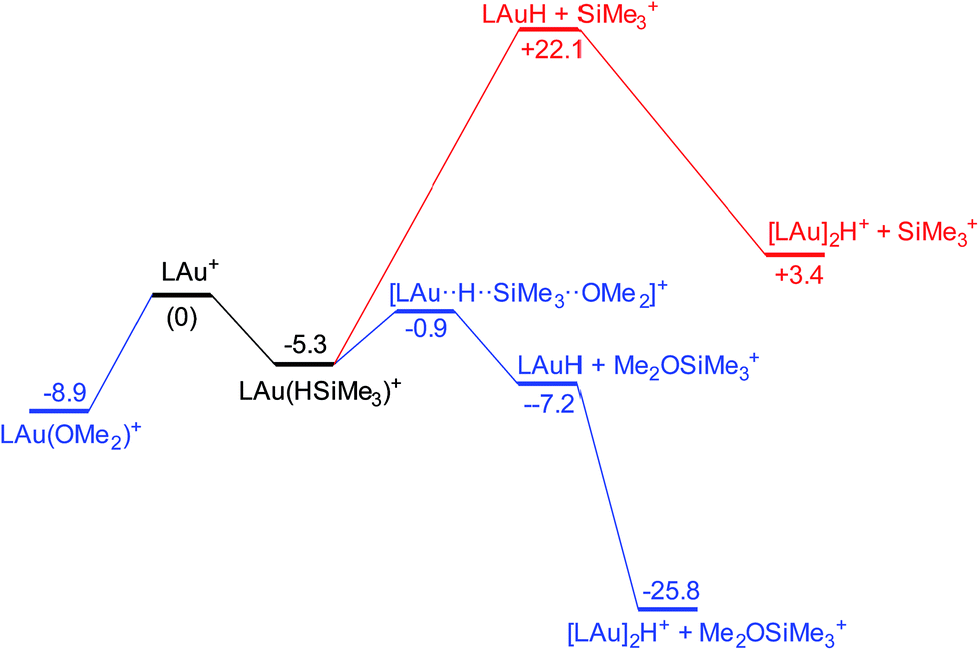 | ||
| Fig. 4 Free energy profile of the reaction on LAu+ with HSiMe3 in the presence (black and blue parts) and absence (black and red parts) of OMe2. | ||
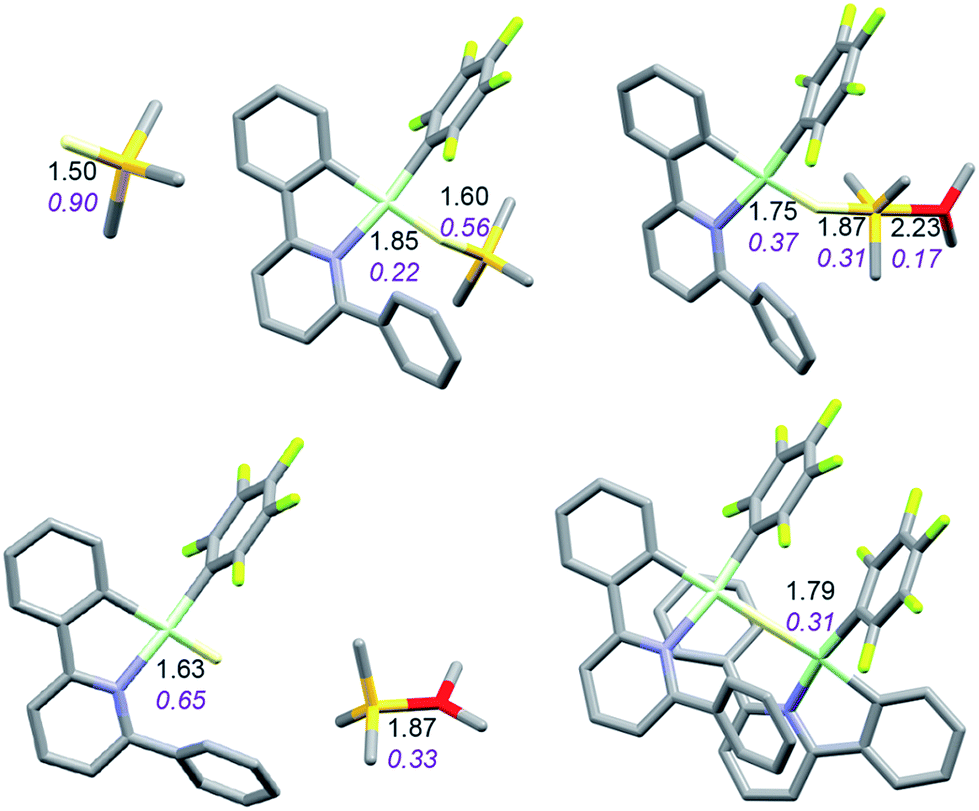 | ||
| Fig. 5 Structure of the silane complex [LAu(HSiMe3)]+ and activation of HSiMe3 by LAu+/OMe2. Bond lengths in Å, Wiberg bond indexes shown in purple and italics. | ||
Coordination of borane HBPin to LAu+ (see profile in Fig. 6) is even weaker than coordination of silane (less than 1 kcal mol−1). The resulting species is best regarded as a borane σ-complex, with a short B–H bond and a large Au–H distance, as is clear from the B–H/Au–H bond distances and the corresponding WBIs (Fig. 7). Again, direct dissociation of BPin+ is not feasible. Approach of OMe2 does not result in displacement of the hydride from B, the ether simply coordinates weakly (∼1 kcal mol−1) to the B atom. This results in considerable reorganization and has the effect of making the borane a better hydride donor, as is clear from the elongation of the B–H bond and considerable shortening of the Au–H bond: the ether complex can now be seen as a gold hydride donating density to the B atom of (Me2O)BPin+. Finally, dissociation of (Me2O)BPin+ produces the free gold hydride, to be captured by LAu+. The subtle difference with the silane case is that here the LAu(HBPin)(OMe2)+ represents a local minimum (intermediate) instead of an SN2-like transition state. Similar to the silane case, this is predicted to be a very fast reaction.
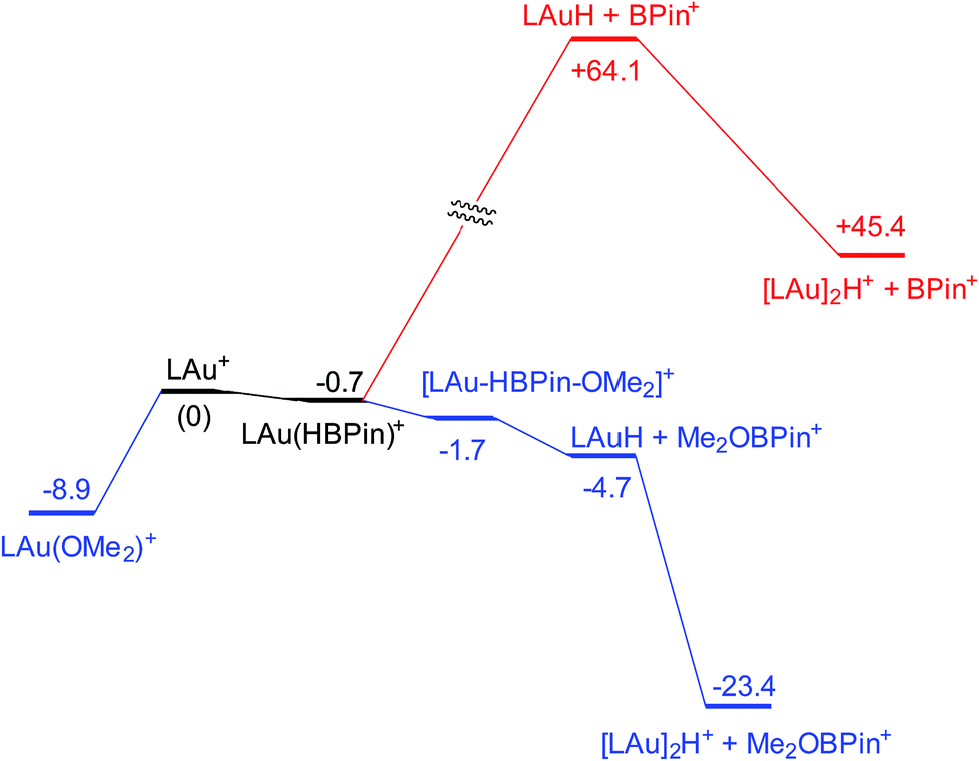 | ||
| Fig. 6 Free energy profile (kcal mol−1) for the reaction of HBPin with LAu+ in the presence (black and blue parts) and absence (black and red parts) of ether OMe2. | ||
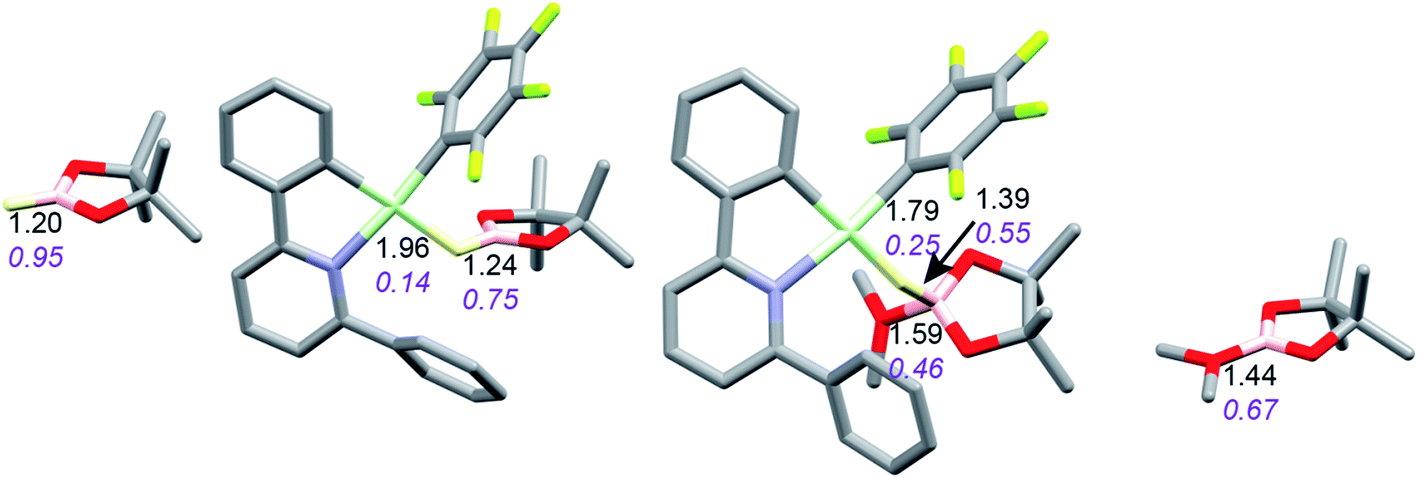 | ||
| Fig. 7 Activation of HBPin by LAu+/OMe2. Bond lengths in Å, Wiberg bond indexes shown in purple and italics. | ||
The reaction with dihydrogen obviously requires a base (here OMe2) to bind the proton released in the heterolytic cleavage of H2. However, one molecule of ether is apparently not enough. The free energy profile (Fig. 8) shows that H2 binding is endergonic by 6.1 kcal mol−1. The coordinated dihydrogen molecule is hardly activated, as is clear from bond lengths and WBIs in Fig. 9. From there, attack by OMe2 at one of the hydrogen atoms of H2 leads to heterolytic cleavage, forming the tightly bound pair [LAuH·HOMe2]+ at +10.5 kcal mol−1 relative to ether-free LAu+. Dissociation into separate LAuH and HOMe2+ would cost another 5.3 kcal mol−1, leading to an effective barrier of 24.7 kcal mol−1 which would correspond to a reaction that is slow at room temperature. More likely, the tightly bound pair [LAuH·HOMe2]+ binds another molecule of ether, forming a less tightly bound combination of LAuH and [H(OMe2)2]+,42 after which separation and trapping of LAuH would lead to the final products. This variation would have an effective barrier of 19.4 kcal mol−1 which would be more compatible with a reaction that is slow at −20 °C. The main difference with the silane and borane variations is that for H2 coordination of the substrate is endergonic and hence contributes to the effective reaction barrier.
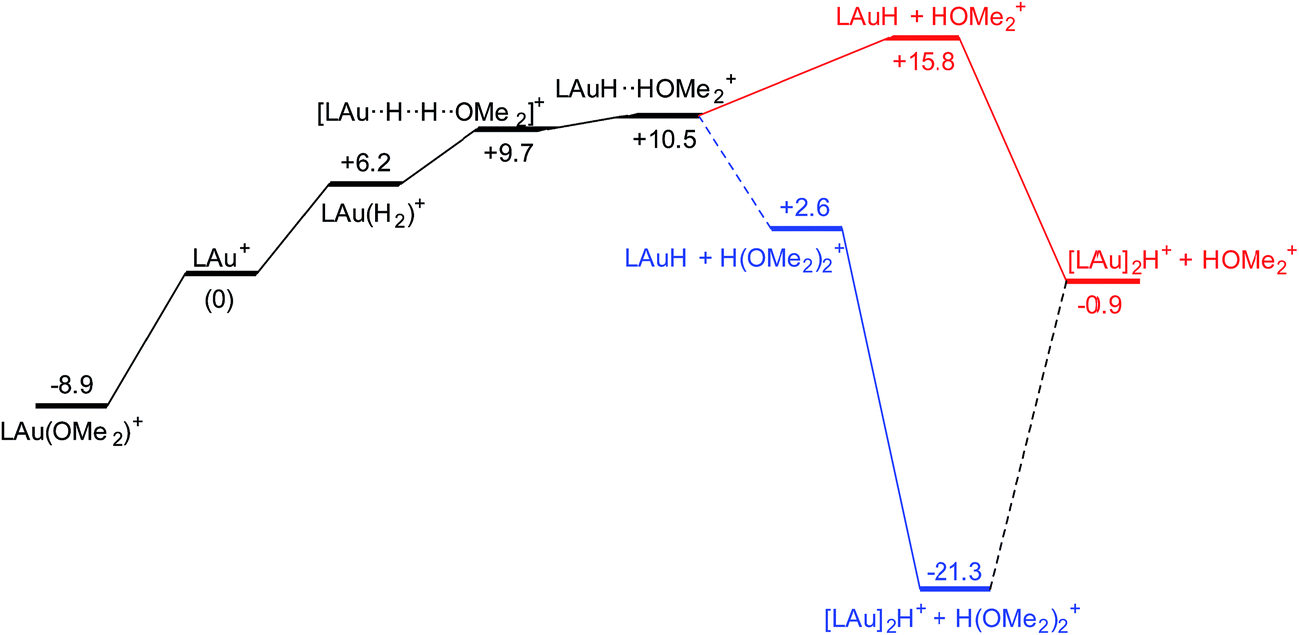 | ||
| Fig. 8 Free energy profile (kcal mol−1) for the reaction of H2 with LAu+ assisted by one (black and red parts) and two (black and blue parts) molecules of ether OMe2. | ||
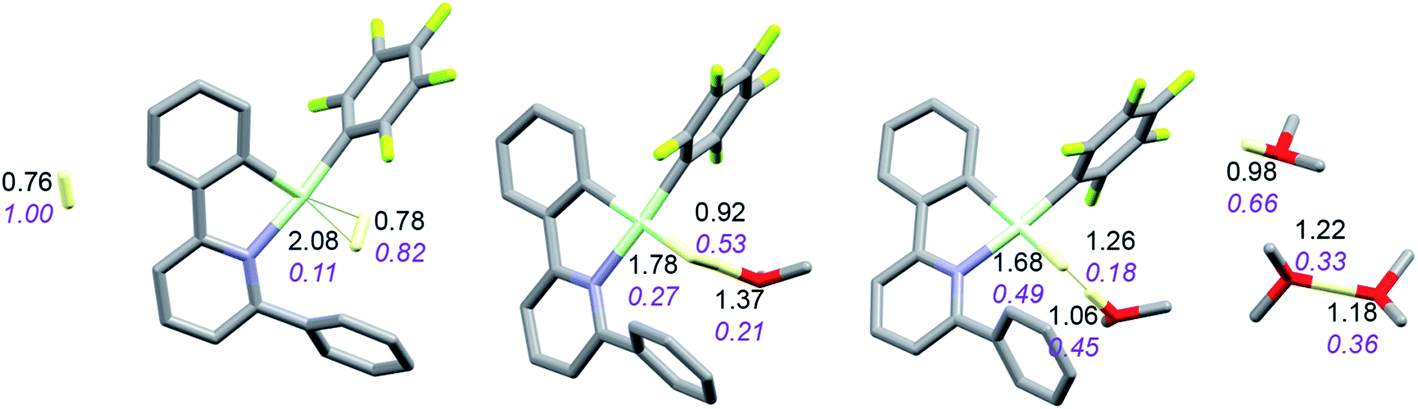 | ||
| Fig. 9 Activation of H2 by LAu+/OMe2. Bond lengths in Å, Wiberg bond indexes shown in purple and italics. | ||
The reaction with dihydropyridine 3,5-dicarboxylate (PyCarH) does not require further assistance by a base: after initial dissociation of ether from the starting complex LAu(OMe2)+, the actual H abstraction step has a barrier of 3.4 kcal mol−1. This leads to an effective activation free energy of 12.3 kcal mol−1, indicating a rather fast reaction. On the other hand, hydride abstraction from toluene or triphenylmethane is thermodynamically not feasible, while abstraction from 1,4-cyclohexadiene (CHDH) in the absence of ether is just on the edge; this reaction is exergonic by only 2.9 kcal mol−1 if LAuH trapping is included, with a calculated barrier of 16.2 kcal mol−1. This is in agreement with the observed slow reaction at room temperature.
To summarize, LAu+ is a strong hydride acceptor, made even more potent by trapping of the initial product LAuH to form the binuclear H-bridged complex [(LAu)2(μ-H)]+. It is comparable in strength to the 3,5-dicarboxylatopyridinium cation, and considerably weaker than the cyclohexadienyl and trityl cations. A common feature of the reactions studied is the surprisingly low barrier for the H transfer step, typically less than 5 kcal mol−1 relative to the substrate complex. These results illustrate how effective gold(III) is in polarizing the H–E bond in preparation for heterolytic cleavage.
Conclusion
Gold(III) complexes have been shown to be able to form σ-complexes with boranes and silanes, species that have long been postulated as the first steps in gold catalysed hydroboration and hydrosilylation reactions. These complexes are sufficiently stable for spectroscopic characterisation. DFT calculations suggest end-on Au–H–E bonding, with binding free energies of the order of 1–5 kcal mol−1. The key to these weak adducts is the use of the gold(III) chelate complex [(C^N–CH)Au(C6F5)]+, which contains a gold–arene π-interaction to the “dangling” aryl substituent which is weak enough to be displaced by H–E, while at the same time the coordinated substrate is protected by this “Pacman”-type ligand structure. In the presence of ether as base heterolytic H–E bond scission is facile. The same holds for the activation of dihydrogen. The observations reported here on gold(III) mediated H2 activation follow the same principle as postulated in earlier computational studies on hydrogen activation by gold(I) compounds,39 where a polar protic solvent like ethanol was shown to be required in the activation process. These results suggest that for gold catalysts in which the active centre is positively polarized to some extent, heterolytic rather than homolytic H–E and H–H bond cleavage processes prevail, at least in the presence of weakly basic solvents or reagents. The range of H-donors for gold could be extended to activated hydrocarbons, where in favourable cases C–H bond activation was shown to be orders of magnitude faster than dihydrogen activation. Overall, these reactions have provided routes to unprecedented types of gold complexes and serve to illustrate the remarkable capacity of gold to polarize and activate substrate H–E bonds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council. M. B. is an ERC Advanced Investigator Award holder (grant no. 338944-GOCAT). We thank S. Woodhouse and Prof. G. G. Wildgoose (UEA) for access to the hydrogen generator.References
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- H. Schmidbaur and A. Schier, Arabian J. Sci. Eng., 2012, 37, 1187–1225 CrossRef CAS.
- H. G. Raubenheimer and H. Schmidbaur, S. Afr. J. Sci., 2011, 107, 459 Search PubMed.
- A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed.
- A. Leyva-Pérez and A. Corma, Angew. Chem., Int. Ed., 2012, 51, 614–635 CrossRef PubMed.
- M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS PubMed.
- J. Miró and C. del Pozo, Chem. Rev., 2016, 116, 11924–11966 CrossRef PubMed.
- M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022–15045 CrossRef CAS PubMed.
- D.-A. Roşca, J. A. Wright and M. Bochmann, Dalton Trans., 2015, 44, 20785–20807 RSC.
- C. Blons, A. Amgoune and D. Bourissou, Dalton Trans., 2018, 47, 10388–10393 RSC.
- P. Claus, Appl. Catal., A, 2005, 291, 222–229 CrossRef CAS.
- L. McEwan, M. Julius, S. Roberts and J. C. Q. Fletcher, Gold Bull., 2010, 43, 298–306 CrossRef CAS.
- M. Pan, A. J. Brush, Z. D. Pozun, H. Chul Ham, W.-Y. Yu, G. Henkelman, G. S. Hwang and C. B. Mullins, Chem. Soc. Rev., 2013, 42, 5002–5013 RSC.
- M. Pan, J. Gong, G. Dong and C. B. Mullins, Acc. Chem. Res., 2014, 47, 750–760 CrossRef CAS PubMed.
- F. Cardenas-Lizana and M. A. Keane, J. Mater. Sci., 2013, 48, 543–564 CrossRef CAS.
- M. A. Keane, M. Li, L. Collado and F. Cárdenas-Lizana, React. Kinet., Mech. Catal., 2018, 125, 25–36 CrossRef CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed.
- A. Corma, P. Serna and H. Garcia, J. Am. Chem. Soc., 2007, 129, 6358–6359 CrossRef CAS PubMed.
- P. Serna, M. Boronat and A. Corma, Top. Catal., 2011, 54, 439–446 CrossRef CAS.
- B. Vilhanova, J. A. van Bokhoven and M. Ranocchiari, Adv. Synth. Catal., 2017, 359, 677–686 CrossRef CAS.
- G. Malta, S. A. Kondrat, S. J. Freakley, C. J. Davies, L. Lu, S. Dawson, A. Thetford, E. K. Gibson, D. J. Morgan, W. Jones, P. P. Wells, P. Johnston, C. R. A. Catlow, C. J. Kiely and G. J. Hutchings, Science, 2017, 355, 1399–1403 CrossRef CAS PubMed.
- M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346, 1498–1501 CrossRef CAS PubMed.
- A. Corma, C. Gonzalez-Arellano, M. Iglesias and F. Sanchez, Angew. Chem., Int. Ed., 2007, 46, 7820–7822 CrossRef CAS PubMed.
- I. Saridakis, M. Kidonakis and M. Stratakis, ChemCatChem, 2018, 10, 980–983 CrossRef CAS.
- M. Kidonakis and M. Stratakis, Org. Lett., 2015, 17, 4538–4541 CrossRef CAS PubMed.
- H. Li, H. Guo, Z. Li, C. Wu, J. Li, C. Zhao, S. Guo, Y. Ding, W. He and Y. Li, Chem. Sci., 2018, 9, 4808–4813 RSC.
- Z. Chen, Q. Zhang, W. Chen, J. Dong, H. Yao, X. Zhang, X. Tong, D. Wang, Q. Peng, C. Chen, W. He and Y. Li, Adv. Mater., 2018, 30, 1704720 CrossRef PubMed.
- R. Bhattacharjee and A. Datta, J. Phys. Chem. C, 2017, 121, 20101–20112 CrossRef CAS.
- H. Lv, J.-H. Zhan, Y.-B. Cai, Y. Yu, B. Wang and J.-L. Zhang, J. Am. Chem. Soc., 2012, 134, 16216–16227 CrossRef CAS PubMed.
- N. Debono, M. Iglesias and F. Sanchez, Adv. Synth. Catal., 2007, 349, 2470–2476 CrossRef CAS.
- Y. Satoh, M. Igarashi, K. Sato and S. Shimada, ACS Catal., 2017, 7, 1836–1840 CrossRef CAS.
- A. Leyva, X. Zhang and A. Corma, Chem. Commun., 2009, 4947–4949 RSC.
- I. P. Silverwood, S. M. Rogers, S. K. Callear, S. F. Parker and C. R. A. Catlow, Chem. Commun., 2016, 52, 533–536 RSC.
- M. Manzoli, A. Chiorino, F. Vindigni and F. Boccuzzi, Catal. Today, 2012, 181, 62–67 CrossRef CAS.
- E. Bus, J. T. Miller and J. A. van Bokhoven, J. Phys. Chem. B, 2005, 109, 14581–14587 CrossRef CAS PubMed.
- A. Corma, M. Boronat, S. González and F. Illas, Chem. Commun., 2007, 3371–3373 RSC.
- B. S. Takale, X. Feng, Y. Lu, M. Bao, T. Jin, T. Minato and Y. Yamamoto, J. Am. Chem. Soc., 2016, 138, 10356–10364 CrossRef CAS PubMed.
- A. Comas-Vives, C. González-Arellano, A. Corma, M. Iglesias, F. Sánchez and G. Ujaque, J. Am. Chem. Soc., 2006, 128, 4756–4765 CrossRef CAS PubMed.
- A. Comas-Vives and G. Ujaque, J. Am. Chem. Soc., 2013, 135, 1295–1305 CrossRef CAS PubMed.
- J. L. Fiorio, N. López and L. M. Rossi, ACS Catal., 2017, 7, 2973–2980 CrossRef CAS.
- J. L. Fiorio, R. V. Gonçalves, E. Teixeira-Neto, M. A. Ortuño, N. López and L. M. Rossi, ACS Catal., 2018, 8, 3516–3524 CrossRef CAS.
- L. Rocchigiani, J. Fernandez-Cestau, P. H. M. Budzelaar and M. Bochmann, Chem. Commun., 2017, 53, 4358–4361 RSC.
- S. J. Lancaster, A. Rodriguez, A. Lara-Sanchez, M. D. Hannant, D. A. Walker, D. L. Hughes and M. Bochmann, Organometallics, 2002, 21, 451–453 CrossRef CAS.
- L. Rocchigiani, J. Fernandez-Cestau, I. Chambrier, P. Hrobarik and M. Bochmann, J. Am. Chem. Soc., 2018, 140, 8287–8302 CrossRef CAS PubMed.
- R. D. Dobrott and W. N. Lipscomb, J. Chem. Phys., 1962, 37, 1779–1784 CrossRef CAS.
- R. K. Hertz, S. Goetze and S. G. Shore, Inorg. Chem., 1979, 18, 2813–2816 CrossRef CAS.
- J. C. Bommer and K. W. Morse, Inorg. Chem., 1980, 19, 587–593 CrossRef CAS.
- M. A. Beckett and P. W. Jones, Synth. React. Inorg. Met.-Org. Chem., 1997, 27, 41–50 CrossRef CAS.
- G. Nuss, G. Saischek, B. N. Harum, M. Volpe, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2011, 50, 12632–12640 CrossRef CAS PubMed.
- N. Carr, M. C. Gimeno, J. E. Goldberg, M. U. Pilotti, F. G. A. Stone and I. Topalğalu, J. Chem. Soc., Dalton Trans., 1990, 2253–2261 RSC.
- J. Y. Corey, Chem. Rev., 2011, 111, 863–1078 CrossRef CAS PubMed.
- M. Joost, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Organometallics, 2013, 32, 898–902 CrossRef CAS.
- L. Rocchigiani, J. Fernandez-Cestau, G. Agonigi, I. Chambrier, P. H. M. Budzelaar and M. Bochmann, Angew. Chem., Int. Ed., 2017, 56, 13861–13865 CrossRef CAS PubMed.
- J.-C. Hierso, Chem. Rev., 2014, 114, 4838–4867 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- L. Rocchigiani, J. Fernandez-Cestau, P. H. M. Budzelaar and M. Bochmann, Chem.–Eur. J., 2018, 24, 8893–8903 CrossRef CAS PubMed.
- T. Ohnishi, S. T. Ohnishi and J. C. Salernoa, Biol. Chem., 2018, 399, 1249–1264 CAS , and cited references.
- B. Bertrand, M. R. M. Williams and M. Bochmann, Chem.–Eur. J., 2018, 24, 11840–11851 CrossRef CAS PubMed , and cited references.
- T. T. Zou, C. T. Lum, C. N. Lok, J. J. Zhang and C. M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, NMR spectroscopic and computational details. See DOI: 10.1039/c8sc05229h |
| This journal is © The Royal Society of Chemistry 2019 |