
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Connective synthesis of 5,5-disubstituted hydantoins by tandem α-amination and α-arylation of silyl ketene acetals†
Rakesh K.
Saunthwal
 ,
Matthew T.
Cornall
,
Roman
Abrams
,
John W.
Ward
* and
Jonathan
Clayden
*
,
Matthew T.
Cornall
,
Roman
Abrams
,
John W.
Ward
* and
Jonathan
Clayden
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: john.ward@liverpool.ac.uk; j.clayden@bristol.ac.uk
First published on 6th February 2019
Abstract
5,5-Disubstituted hydantoins, formally the cyclisation products of quaternary amino acids, were formed connectively from simple ester-derived starting materials by a one-pot tandem method. Amination of the silyl ketene acetal derivative of a methyl ester takes place by silver-catalysed addition to the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of an azocarboxamide, generating a N-amino-N′-aryl urea derivative of a substituted aminoester. Treatment with a base forms an ester enolate which undergoes arylation by intramolecular migration of an aryl ring to the α-position of the ester. The product undergoes ring closure to a hydantoin, which may itself be deprotected and functionalised. Aryl migration is successful with rings of various electronic character and with esters bearing functionalised and unfunctionalised chains, and the products have features in common with several bioactive compounds.
N bond of an azocarboxamide, generating a N-amino-N′-aryl urea derivative of a substituted aminoester. Treatment with a base forms an ester enolate which undergoes arylation by intramolecular migration of an aryl ring to the α-position of the ester. The product undergoes ring closure to a hydantoin, which may itself be deprotected and functionalised. Aryl migration is successful with rings of various electronic character and with esters bearing functionalised and unfunctionalised chains, and the products have features in common with several bioactive compounds.
Introduction
Hydantoin rings, formally the cyclocarbonylation products of amino acids, are found in a number of medicinally significant molecules (Fig. 1).1,2 For example the sodium salts of phenytoin and fosphenytoin have anticonvulsant and antiarrhythmic properties;3a nitrofurantoin is an antibacterial drug;3b,c nilutamide is an androgen receptor antagonist for the treatment of advanced prostate cancer;4 dantrolene is used as a muscle relaxant and to prevent malignant hyperthermia.5 Substituted hydantoins are furthermore valuable intermediates in the synthesis of amino acids using hydantoinases and other related biocatalysts.6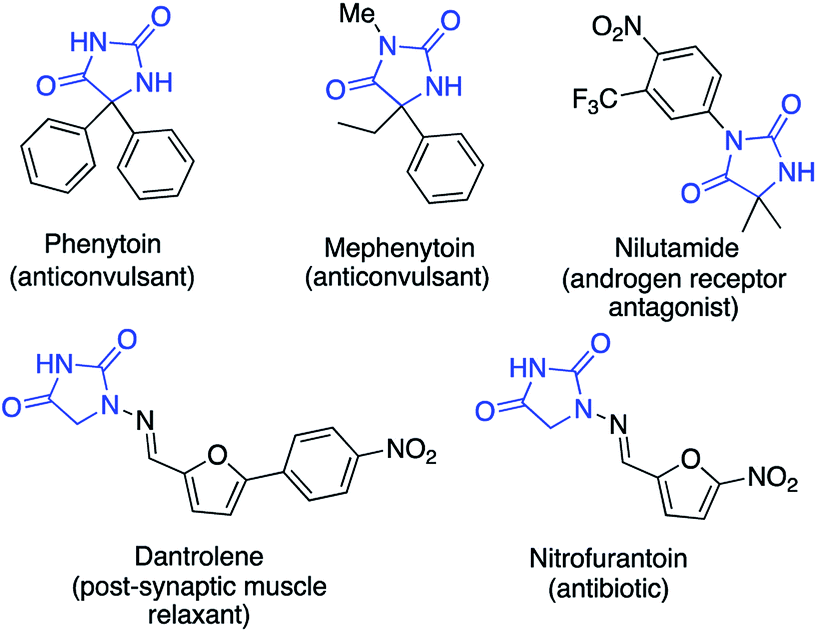 | ||
| Fig. 1 Examples of drugs containing the hydantoin motif. | ||
Methods for the synthesis of substituted hydantoins7 include the classical Urech synthesis,8 and the Bucherer–Bergs9 and Biltz reactions.10 Milder transition metal catalysed approaches have been reported, including the Ugi condensation,11 an aminobarbituric acid-hydantoin rearrangement,12 and reactions of activated carboxylic acids.13 Hydantoins have also been made by α-amination of esters using copper catalysts.14,15
We have shown that intramolecular migration of an aryl ring to the α-position of an amino acid-derived urea can provide a general method for making substituted hydantoins16 in reactions that involve intramolecular nucleophilic aromatic substitution of enolates on even unactivated aromatic rings.17 However, such methods make use of available amino acid starting materials and are less applicable to the synthesis of molecules containing ‘non-proteinogenic’ side chains. We therefore sought to develop a tandem approach from simple precursors in which the α-amination18 of an enolate generates a suitable substrate for a tandem intramolecular arylation,19 leading directly to an α-arylated quaternary hydantoin.20
Our initial plan for a direct route to structurally diverse 5,5-disubstituted hydantoins is illustrated in Scheme 1. We aimed to initiate the hydantoin synthesis with a silver-catalysed regioselective α-amination using an azocarboxamide to generate a urea derivative from which N′-aryl migration to the α-position of the resulting ester followed by ring closure would give a hydantoin.
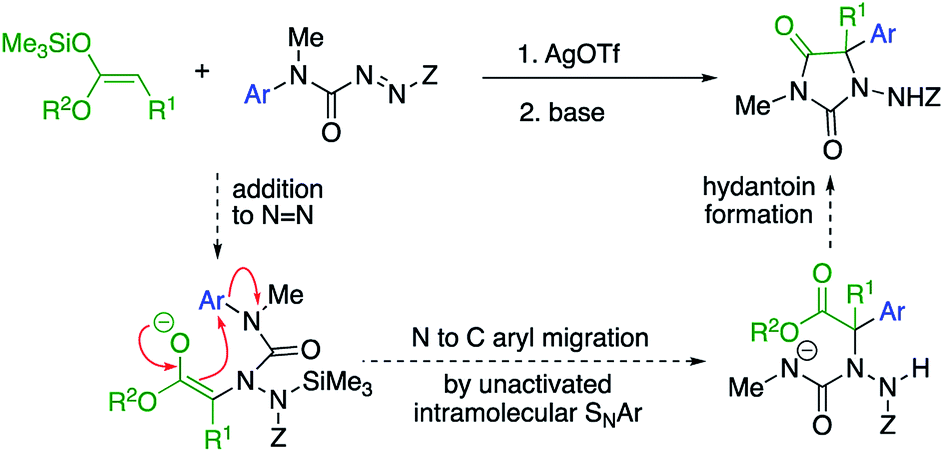 | ||
| Scheme 1 A route to α-arylated quaternary hydantoins by tandem amination-arylation of silyl ketene acetals. Z = electron-withdrawing group. | ||
Three major challenges need to be overcome. Regioselective addition of the silyl ketene acetal to the azocarboxamide must lead to a 2-ureido ester to allow the subsequent aryl migration step. Secondly, the enolate of the product must undergo rearrangement rather than any other alternative reaction (such as substitution or elimination), and finally the product must cyclise to a hydantoin. All these steps ideally should occur in a single, tandem process.
Results and discussion
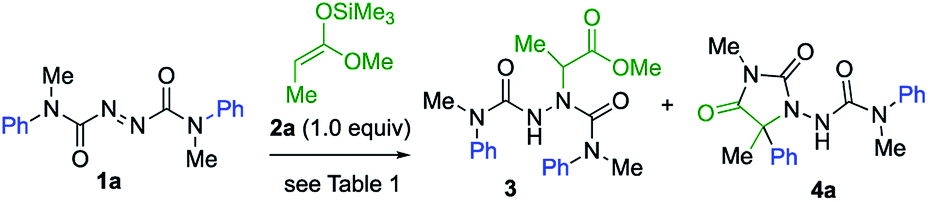
We started by exploring the amination step with a symmetrical azodicarboxamide to allow us to study the viability of the rearrangement while avoiding issues of regioselectivity. Silver-catalysed aminations of silyl ketene acetals were known using azodicarboxylates,21 so azodicarboxamides 1 were made by acylation of hydrazine with N-methyl-N-arylcarbamoyl chloride followed by oxidation with NBS.22 Treatment of a mixture of the azodicarboxamide 1a and the silyl ketene acetal 2a with AgOTf (20 mol%) in dichloromethane gave the addition product 3 in 68% yield (Table 1, entry 1). Reducing the catalyst loading to 10 mol% in THF improved this yield to 80% (entry 2). 3 carries an N′-aryl urea function suitably located for possible aryl transfer to an enolate derivative. We therefore added 2.0 equiv. KHMDS to the reaction mixture in the hope of promoting this intramolecular arylation. The rearranged product 4a was obtained in 60% yield, together with 40% of intermediate 3 (entry 3). Increasing the amount of KHMDS to 3.0 equiv. gave clean product 4a in 75% yield (entry 4).|
|
|||
|---|---|---|---|
| Entry | Reaction conditions | Yieldsb (%) | |
| 3 | 4a | ||
| a Reactions performed using 0.34 mmol of 1a and 0.34 mmol of 2a in 3.4 ml solvent. b Isolated yield. c Similar results obtained with 15 or 20 mol% catalyst. | |||
| 1 | 20 mol% AgOTf, CH2Cl2, −78 → +20 °C, 3 h | 68 | — |
| 2 | 10 mol% AgOTf, THF, −78 → +20 °C, 3 h | 80c | — |
| 3 | (1) 10 mol% AgOTf, THF, −78 → +20 °C, 3 h | 40 | 60 |
| (2) KHMDS (2.0 equiv.), −78 → −40 °C, 3 h | |||
| 4 | (1) 10 mol% AgOTf, THF, −78 → +20 °C, 3 h | 0 | 75 |
| (2) KHMDS (3.0 equiv.), −78 → −40 °C, 3 h | |||
A further series of azodicarboxamides 1b–g were made, and likewise treated with silyl ketene acetals 2a and 2b (Scheme 2). Hydantoin products 4b–f were formed successfully bearing electron donating, electron withdrawing groups, and the reaction was successful even with the pyridyl substituted 1g. Additionally, the structure of the p-tolyl derivative 4b was confirmed by X-ray crystallography.23 The tandem reaction was also successful with the more hindered silyl ketene acetal 2b derived from 3-phenylpropionic acids, generating in one pot hydantoins 4h–j in good yields.
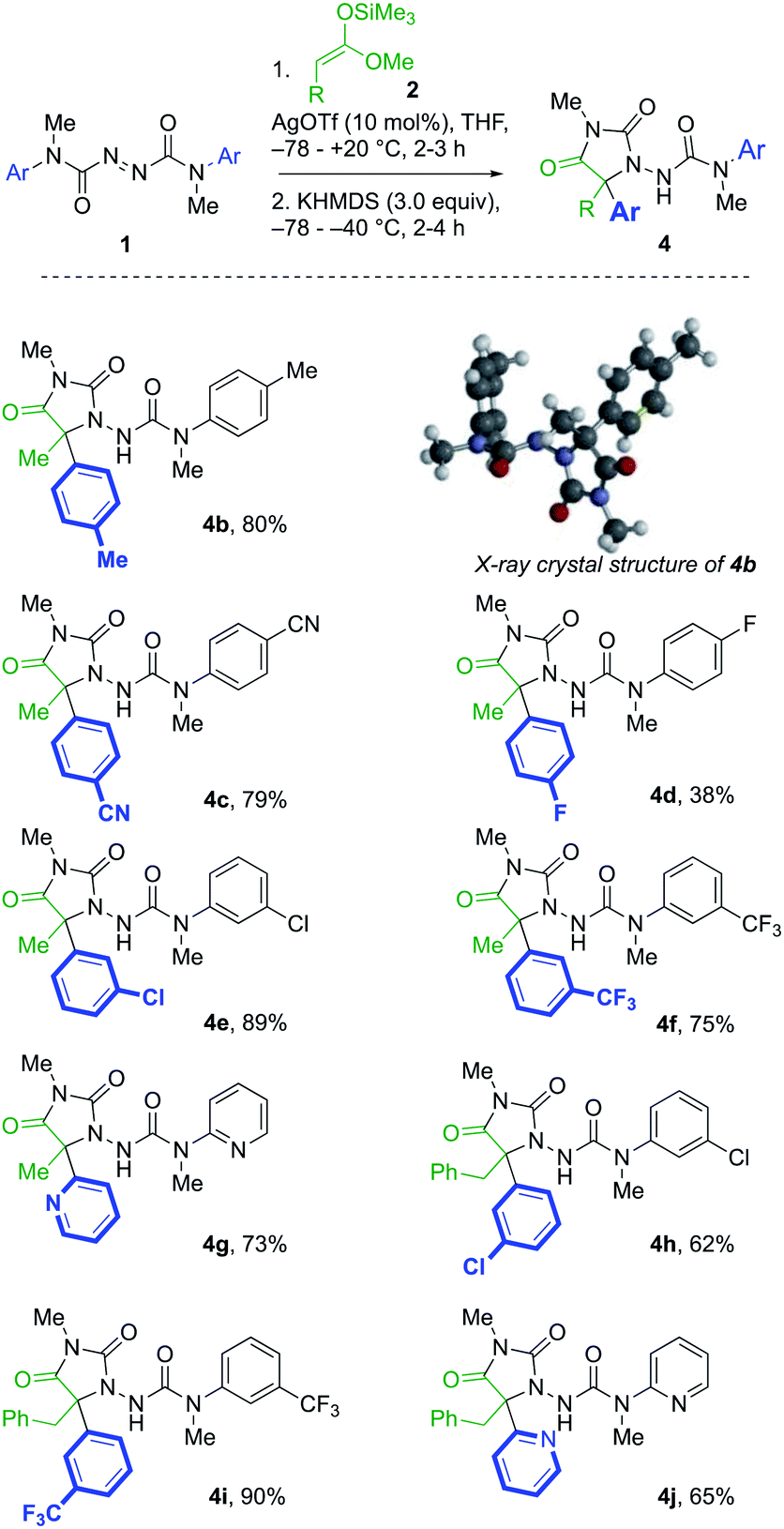 | ||
| Scheme 2 Hydantoin formation from symmetrical azodicarboxamides. | ||
The products 4 all contain a pendent N-aryl urea function derived from the second aryl substituent of the symmetrical starting material, and a greater atom economy would be achieved if an alternative unsymmetrical, mono-N-arylated azocarboxamide was used as the aminating agent. By treating N-methyl-N-tolylcarbamoyl chloride with t-butyl carbazate, and oxidising the product hydrazide with NBS (see ESI†), we were able to form the azocarboxamides 5. Treating silyl ketene acetal 2a with this compound in the presence of 10 mol% AgOTf in CH2Cl2 gave the product 6 in 76% yield, accompanied by less than 5% of the alternative regioisomer (Table 2, entry 1). In THF, the yield of 6 increased to 85% and the amination was fully regioselective (entry 2).
|
|
|||
|---|---|---|---|
| Entry | Reaction conditions | Yieldb (%) | |
| 6 | 7a | ||
| a Reactions performed using 0.36 mmol of 4a, 0.36 mmol of 2a in 3.6 ml solvent. b Isolated yield. c Similar results obtained with 15 or 20 mol% catalyst. | |||
| 1 | 10 mol% AgOTf, CH2Cl2, −78 → +20 °C, 2 h | 76c | — |
| 2 | 10 mol% AgOTf, THF, −78 → +20 °C, 2 h | 85 | — |
| 3 | (1) 10 mol% AgOTf, THF, −78 → +20 °C, 2 h | 40 | 20 |
| (2) KHMDS (2.0 equiv.), −78 → −40 °C, 2 h | |||
| 4 | (1) 10 mol% AgOTf, THF, −78 → +20 °C, 2 h | 0 | 50 |
| (2) KHMDS (3.0 equiv.), −78 → −40 °C, 3 h | |||
| 5 | (1) 10 mol% AgOTf, THF, −78 → +20 °C, 2 h | 0 | 72 |
| (2) KHMDS (3.0 equiv.), −78 → −20 °C, 2 h | |||
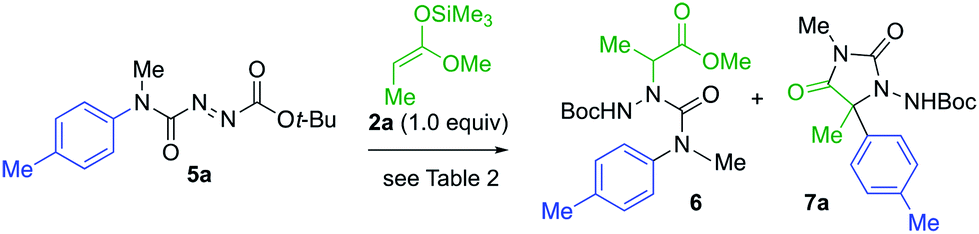
When KHMDS was added directly to the crude reaction mixture containing the amination product, arylation and cyclisation to the N-Boc-protected aminohydantoin 7a (entries 3–5) took place, in parallel with the results seen using the symmetrical aminating agent 1a. With 2.0 equiv. of KHMDS, warming the reaction to −40 °C for 2 h, hydantoin product 7a was formed in 20% yield (entry 3), increasing to 72% yield on warming to −20 °C (entry 5). Other unsymmetrical aminating agents were also explored, including N-benzoyl, N-tert-butyl-carboxamido and N-methyl-N-tert-butyl carboxamido substituted azo compounds. Although intermediate aminated products corresponding to 6 were obtained, treatment with the base led only to decomposition.
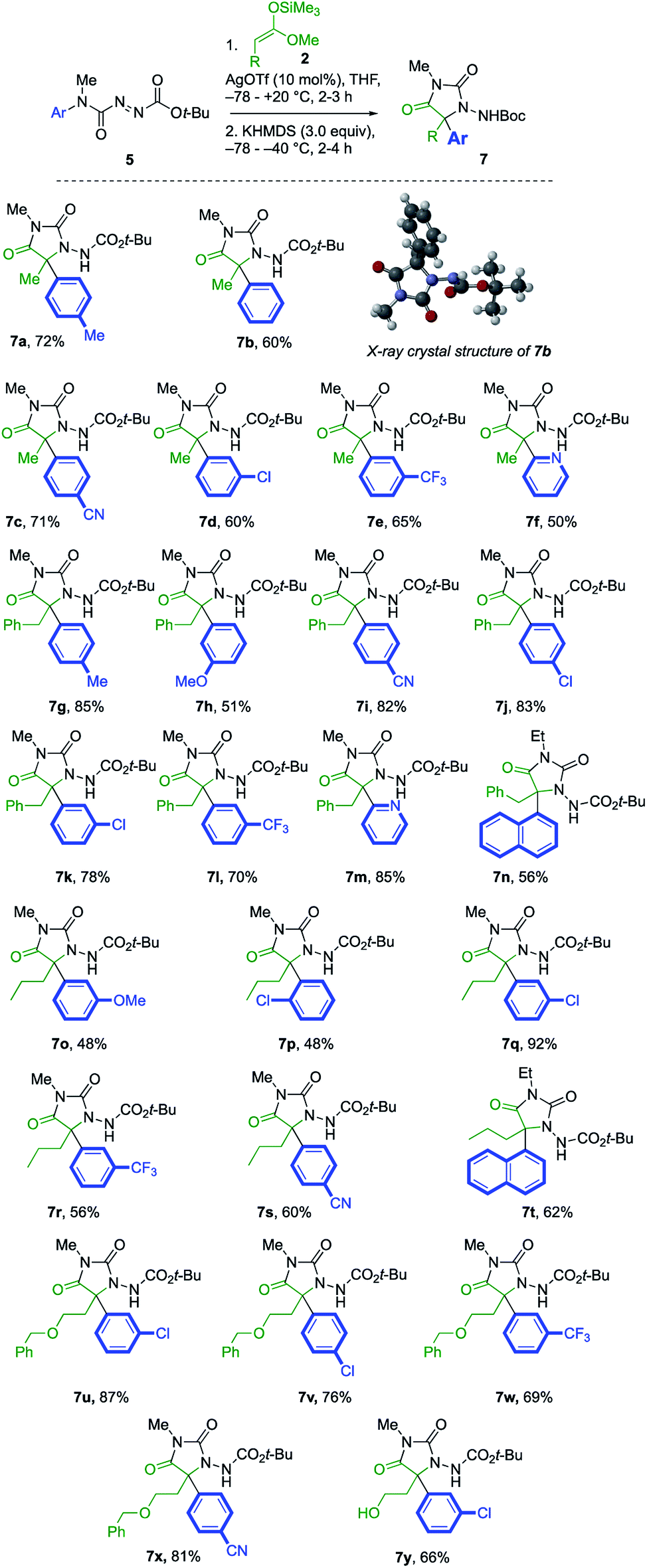
Under these optimised reaction conditions, the generality of the reaction was explored with various azocarboxamides 5 and silyl ketene acetals 2 (Scheme 3). The α-amination of 2a with a range of azocarboxamides 5 was fully regioselective in all cases. With a simple phenyl ring, the product 7b was formed in 60% yield and its structure was confirmed by X-ray crystal structure analysis.23 Electron withdrawing groups p-CN, m-Cl, and m-CF3 were well tolerated, giving the products 7c–7e in 65–71% yields. 2-Pyridyl azocarboxamide 5f likewise performed well and gave hydantoin 7f in 50% yield.
 | ||
| Scheme 3 A general, connective synthesis of protected N-aminohydantoins. | ||
A wider range of alternative silyl ketene acetal partners were explored. 3-Phenylpropionate-derived 2b provided the hydantoins 7g–l by migration of either electron-donating or electron-withdrawing rings in 51–85% yields. With a heteroaryl migrating group, the 2-pyridyl azocarboxamide 5f provided the desired product 7m in 85% yield. The more hindered 1-naphthyl-substituted azocarboxamide 5i formed 7n in 56% yield, and the migration of other aromatic groups substituted at the ortho position, such as the 2-chlorophenyl group of 7p, also gave reduced yields.
The pentanoate-derived precursor 2c likewise yielded the hydantoins 7q–t in the presence of electron-donating, electron-withdrawing and bulky groups, as did the benzyloxy substituted silyl ketene acetal 2d, giving the functionalised hydantoins 7u–x in good to excellent yields. Deprotection of 7u using 10% Pearlman's catalyst in MeOH24 gave 5-hydroxyethyl substituted 7y.
The potential utility of the products was demonstrated by several transformations of the products 7, as illustrated in Scheme 4. Removal of the Boc protecting groups from 7i and 7j proceeded cleanly with 20% trifluoroacetic acid in CH2Cl2. Condensation of the product N-aminohydantoins 8a and 8b with appropriate aromatic aldehydes yielded alkylated analogues of the muscle relaxant dantrolene (9a) and the antibacterial drug nitrofurantoin (9b). Alternatively, the N–N bond of the product could be cleaved to reveal the parent hydantoins 10. Several methods were screened for this transformation, and we found that treatment of a selection of products 7 with sodium nitrite in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetic acid/1 M HCl at 110 °C (ref. 25) gave the hydantoins 10a–c in good yield.
:1 acetic acid/1 M HCl at 110 °C (ref. 25) gave the hydantoins 10a–c in good yield.
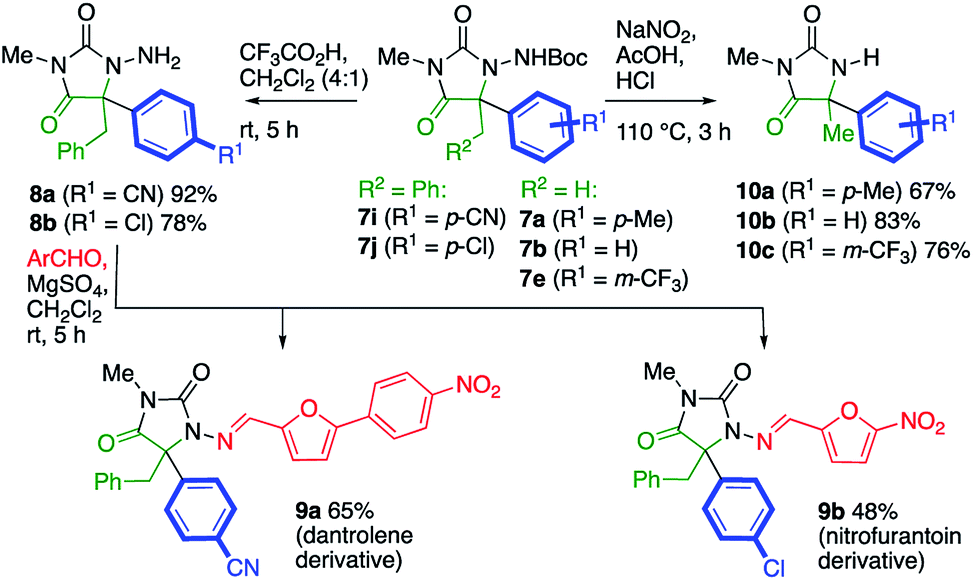 | ||
| Scheme 4 Analogues of bioactive compounds containing the hydantoin motif. | ||
The course of the reaction between model substrates 5c and 2b was studied by in situ infra-red spectroscopy26,16a (see ESI† for full details) and our proposed mechanism for the amination/arylation cascade is presented in Scheme 5a. Silver-catalysed regioselective addition of silyl ketene acetal 2b to the azocarboxamide 5c leads to the silylated addition product A, consistent with the development of CO stretching absorptions at 1745 cm−1 (ester), 1720 cm−1 (carbamate) and 1678 cm−1 (urea). Scheme 5b follows the course of the reaction after addition of KHMDS. A transforms initially into an intermediate which we assign as the enolate B, consistent with the disappearance of the ester CO stretch at 1745 cm−1, and the appearance of peaks we assign to the enolate function at 1640–1660 cm−1 plus a peak at 1604 cm−1 corresponding to the anionic carbamate. The enolate evolves to a species that has CO stretching absorptions at 1764 cm−1 and 1710 cm−1, typical of a hydantoin,16a and retains an anionic carbamate peak at 1606 cm−1. We assign these peaks to species D, the conjugate base of the ultimate product 7i. The rearrangement of B to D presumably passes through an undetectable transient intermediate C that cyclises rapidly to D. Evidence from related reactions suggests that the formation of the new C–C bond and breakage of the old C–N bond during the formation of the proposed intermediate C are to some extent concerted.16e,27
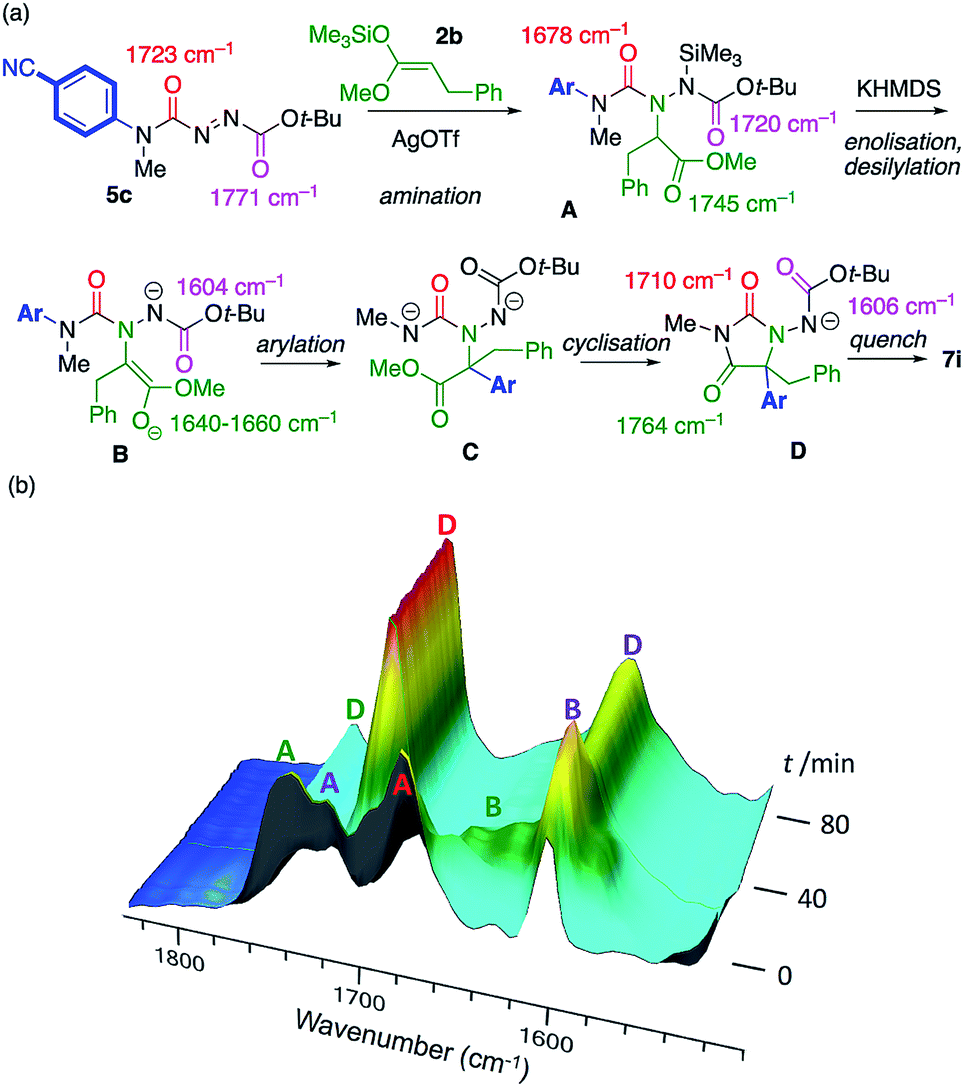 | ||
| Scheme 5 (a) Proposed mechanism of the reaction; (b) Intermediates identified by in situ infra-red spectroscopy. | ||
Conclusions
In conclusion, 5,5-disubstituted hydantoins may be formed by a tandem amination-intramolecular arylation sequence of silyl ketene acetals. The amination entails silver-catalysed regioselective addition to the NN bond of a new class of unsymmetrical azocarboxamides, and the arylation takes place by base-promoted intramolecular N to C migration within the N′-aryl urea linkage that results from the amination step. The hydantoin then forms directly from the product of enolate arylation. In situ infra-red spectroscopy reveals four successive species on the reaction pathway from the amination step to the hydantoin ring closure. The one-pot protocol allowed the connective synthesis of a range of 5,5-disubstituted hydantoins bearing electronically diverse aryl substituents, compounds which have potential applications in the construction of biologically active molecules.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work was supported by the EPSRC and by SERB-India (overseas postdoctoral fellowship to RKS).Notes and references
- (a) C. A. López and G. G. Trigo, Adv. Heterocycl. Chem., 1985, 38, 177–228 CrossRef; (b) W. Kassouf, S. Tanguay and A. G. Aprikian, J. Urol., 2003, 169, 1742–1744 CrossRef CAS PubMed; (c) W. Gao and J. T. Dalton, Drug Discovery Today, 2007, 12, 241–248 CrossRef CAS PubMed; (d) N. Cachet, G. Genta-Jouve, E. L. Regalado, R. Mokrini, P. Amade, G. Culioli and O. P. Thomas, J. Nat. Prod., 2009, 72, 1612–1615 CrossRef CAS PubMed.
- (a) M. Dhanawat, A. G. Banerjee and S. K. Shrivastava, Med. Chem. Res., 2012, 21, 2807–2822 CrossRef CAS; (b) L. Grosse, S. Pâquet, P. Caron, L. Fazli, P. S. Rennie, A. Bélanger and O. Barbier, Cancer Res., 2013, 73, 6963–6971 CrossRef CAS PubMed; (c) Z. Iqbal, S. Ali, J. Iqbal, Q. Abbas, I. Z. Qureshi and S. Hameed, Bioorg. Med. Chem. Lett., 2013, 23, 488–491 CrossRef CAS PubMed; (d) J. Marton, J. Enisz, S. Hosztafi and T. Timar, J. Agric. Food Chem., 1993, 41, 148–152 CrossRef CAS.
- (a) M. A. Rogawski and W. Löscher, Nat. Rev. Neurosci., 2004, 5, 553–564 CrossRef CAS PubMed; (b) W. Liu, C. Zhao, Y. Zhang, S. Lu, J. Liu and R. Xi, J. Agric. Food Chem., 2007, 55, 6829–6834 CrossRef CAS PubMed; (c) Q. Wang, Y.-C. Liu, Y.-J. Chen, W. Jiang, J.-L. Shi, Y. Xiao and M. Zhang, Anal. Methods, 2014, 6, 4414–4420 RSC.
- (a) M. Moguilewsky, C. Bertagna and M. Hucher, J. Steroid Biochem., 1987, 27, 871–875 CrossRef CAS PubMed; (b) W. Kassouf, S. Tanguay and A. G. Aprikian, J. Urol., 2003, 169, 1742–1744 CrossRef CAS PubMed.
- (a) S. Murasawa, K. Iuchi, S. Sato, T. Noguchi-Yachide, M. Sodeoka, T. Yokomatsu, K. Dodo, Y. Hashimoto and H. Aoyama, Bioorg. Med. Chem., 2012, 20, 6384–6393 CrossRef CAS PubMed; (b) K. Kumata, M. Ogawa, M. Takei, M. Fujinaga, Y. Yoshida, N. Nengaki, T. Fukumura, K. Suzuki and M.-R. Zhang, Bioorg. Med. Chem., 2012, 20, 305–310 CrossRef CAS PubMed.
- (a) S. G. Burton and R. A. Dorrington, Tetrahedron: Asymmetry, 2004, 15, 2737–2741 CrossRef CAS; (b) J. Altenbuchner, M. Siemann-Herzberg and C. Syldatk, Curr. Opin. Biotechnol., 2001, 12, 559–563 CrossRef CAS PubMed.
- L. Konnert, F. Lamaty, J. Martinez and E. Colacino, Chem. Rev., 2017, 117, 13757–13809 CrossRef CAS PubMed.
- (a) W. T. Read, J. Am. Chem. Soc., 1922, 44, 1746–1755 CrossRef CAS; (b) E. Ware, Chem. Rev., 1950, 46, 403–470 CrossRef CAS PubMed.
- (a) H. T. Bucherer and V. A. Lieb, J. Prakt. Chem., 1934, 141, 5–43 CrossRef CAS; (b) C. Montagne and M. Shipman, Synlett, 2006, 17, 2203–2206 Search PubMed; (c) V. O. Knizhnikov, Z. V. Voitenko, V. B. Golovko and M. V. Gorichko, Tetrahedron: Asymmetry, 2012, 23, 1080–1083 CrossRef CAS.
- (a) H. Biltz, Ber. Dtsch. Chem. Ges., 1908, 41, 1379–1393 CrossRef CAS; (b) L. Konnert, B. Reneaud, R. M. de Figueiredo, J.-M. Campagne, F. Lamaty, J. Martinez and E. Colacino, J. Org. Chem., 2014, 79, 10132–10142 CrossRef CAS PubMed.
- (a) C. Hulme, L. Ma, J. J. Romano, G. Morton, S.-Y. Tang, M.-P. Cherrier, S. Choi, J. Salvino and R. Labaudiniere, Tetrahedron Lett., 2000, 41, 1889–1893 CrossRef CAS; (b) M. Sanudo, M. Garcia-Valverde, S. Marcaccini and T. A. Torroba, Tetrahedron, 2012, 68, 2621–2629 CrossRef CAS; (c) J. M. Ignacio, S. Macho, S. Marcaccini, R. Pepino and T. Torroba, Synlett, 2005, 3051–3054 CAS.
- M. Meusel, A. Ambrożak, T. K. Hecker and M. Gütschow, J. Org. Chem., 2003, 68, 4684–4692 CrossRef CAS PubMed.
- (a) A. Volonterio, C. R. de Arellano and M. Zanda, J. Org. Chem., 2005, 70, 2161–2170 CrossRef CAS PubMed; (b) F. Olimpieri, A. Volonterio and M. Zanda, Synlett, 2008, 3016–3020 CAS; (c) O. A. Attanasi, L. De Crescentini, G. Favi, S. Nicolini, F. R. Perrulli and S. Santeusanio, Org. Lett., 2011, 13, 353–355 CrossRef CAS PubMed; (d) F. Olimpieri, M. C. Bellucci, T. Marcelli and A. Volonterio, Org. Biomol. Chem., 2012, 10, 9538–9555 RSC.
- B. Zhao, H. Du and Y. Shi, J. Am. Chem. Soc., 2008, 130, 7220–7221 CrossRef CAS PubMed.
- J. Song, Z. J. Zhang, S. S. Chen, T. Fan and L. Z. Gong, J. Am. Chem. Soc., 2018, 140, 3177–3180 CrossRef CAS PubMed.
- (a) R. C. Atkinson, D. J. Leonard, J. Maury, D. Castagnolo, N. Volz and J. Clayden, Chem. Commun., 2013, 49, 9734–9736 RSC; (b) M. B. Tait, S. Butterworth and J. Clayden, Org. Lett., 2015, 17, 1236–1239 CrossRef CAS PubMed; (c) F. Fernández-Nieto, J. M. Roselló, S. Lenoir, S. Hardy and J. Clayden, Org. Lett., 2015, 17, 3838–3841 CrossRef PubMed; (d) J. Maury and J. Clayden, J. Org. Chem., 2015, 80, 10757–10768 CrossRef CAS PubMed; (e) D. J. Leonard, J. W. Ward and J. Clayden, Nature, 2018, 562, 105–109 CrossRef CAS PubMed.
- R. C. Atkinson, F. Fernández-Nieto, J. M. Roselló and J. Clayden, Angew. Chem., Int. Ed., 2015, 54, 8961–8965 CrossRef CAS PubMed.
- (a) K. Juhl and K. A. Jørgensen, J. Am. Chem. Soc., 2002, 124, 2420–2421 CrossRef CAS PubMed; (b) J. M. Janey, Angew. Chem., Int. Ed., 2005, 44, 4292–4300 CrossRef CAS PubMed; (c) D. Sandoval, A. V. Samoshin and J. R. de Alaniz, Org. Lett., 2015, 17, 4514–4517 CrossRef PubMed; (d) D. A. Evans, T. C. Britton, R. L. Dorow and J. F. Dellaria, J. Am. Chem. Soc., 1986, 108, 6395–6397 CrossRef CAS.
- Z. Huang, Z. Liu and J. Zhou, J. Am. Chem. Soc., 2011, 133, 15882–15885 CrossRef CAS PubMed.
- K. Tomohara, T. Yoshimura, R. Hyakutake, P. Yang and T. Kawabata, J. Am. Chem. Soc., 2013, 135, 13294–13297 CrossRef CAS.
- Y. Yamashita, H. Ishitani and S. Kobayashi, Can. J. Chem., 2000, 78, 666–672 CrossRef CAS.
- R. Guo, K.-N. Li, B. Liu, H.-J. Zhu, Y.-M. Fana and L.-Z. Gong, Chem. Commun., 2014, 50, 5451–5454 RSC.
- CCDC 1867365 and 1867366 (4b and 7b) contain the supplementary crystallographic data for this paper.†.
- F. Soucy, L. Grenier, M. L. Behnke, A. T. Destree, T. A. McCormack and L. Plamondon, J. Am. Chem. Soc., 1999, 121, 9967–9976 CrossRef CAS.
- H. Vogt, S. Vanderheiden and S. Bräse, Chem. Commun., 2003, 19, 2448–2449 RSC.
- (a) D. Stead, G. Carbone, P. O'Brien, K. R. Campos, I. Coldham and A. Sanderson, J. Am. Chem. Soc., 2010, 132, 7260–7261 CrossRef CAS PubMed; (b) J. M. Roselll, S. Hachisu and J. Clayden, Angew. Chem., Int. Ed., 2017, 56, 10750–10754 CrossRef PubMed.
- (a) S. J. Zuend, M. P. Coughlin, M. P. Lalonde and E. N. Jacobsen, Nature, 2009, 461, 968–970 CrossRef CAS PubMed; (b) E. E. Kwan, Y. Zeng, H. A. Besser and E. N. Jacobsen, Nat. Chem., 2018, 10, 917–923 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1867365 and 1867366. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05263h |
| This journal is © The Royal Society of Chemistry 2019 |