
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegiodivergent hydrosilylation, hydrogenation, [2π + 2π]-cycloaddition and C–H borylation using counterion activated earth-abundant metal catalysis†
Riaz
Agahi‡
a,
Amy J.
Challinor‡
a,
Joanne
Dunne
a,
Jamie H.
Docherty
a,
Neil B.
Carter
b and
Stephen P.
Thomas
 *a
*a
aEaStCHEM School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: stephen.thomas@ed.ac.uk; Fax: +44 (0)131 650 6543
bSyngenta, Jealott's Hill International Research Centre, Bracknell, Berkshire RG42 6EX, UK
First published on 8th April 2019
Abstract
The widespread adoption of earth-abundant metal catalysis lags behind that of the second- and third-row transition metals due to the often challenging practical requirements needed to generate the active low oxidation-state catalysts. Here we report the development of a single endogenous activation protocol across five reaction classes using both iron- and cobalt pre-catalysts. This simple catalytic manifold uses commercially available, bench-stable iron- or cobalt tetrafluoroborate salts to perform regiodivergent alkene and alkyne hydrosilylation, 1,3-diene hydrosilylation, hydrogenation, [2π + 2π]-cycloaddition and C–H borylation. The activation protocol proceeds by fluoride dissociation from the counterion, in situ formation of a hydridic activator and generation of a low oxidation-state catalyst.
Introduction
The ubiquity of catalytic protocols using precious metals such as platinum, palladium and rhodium can be ascribed to the highly robust and reliable nature of these methods and widespread commercial availability of the catalyst precursors. Operational simplicity has made reaction screening and optimisation routine using these metals. However, beyond high value applications the low natural abundance, volatile cost and toxicity of these metals remains problematic. Earth-abundant transition metals offer an alternative, sustainable platform for catalysis, particularly for bulk and dispersive technologies, but are yet to achieve widespread adoption even though excellent catalytic activity has been achieved in exemplar industrial reactions.1 Thus, the operational simplicity of earth-abundant-metal-catalysed reactions must be addressed to enable the widespread use and development of these powerful methodologies.The global silicone industry is forecast to be worth $18.3 billion in 2021 and finds applications in areas as diverse as soft materials, cosmetics and food additives.2 Alkene and alkyne hydrosilylation reactions underpin this industry and the homogenous nature of these processes results in the loss of over 5 tonnes of platinum annually.3 Thus a transition to earth-abundant metal catalysis would be beneficial both environmentally and economically. Seminal studies using isolated, low oxidation-state iron- and cobalt pre-catalysts have shown the potential of these metals for alkene hydrosilylation,4 and in situ activation of metal(II/III) pre-catalysts using organometallic reagents1b,5 has decreased the operational barrier to use. Methods have also been developed using bench-stable reductants such as alkoxide reagents6 or amines7 (Scheme 1a). However, an additional, external reagent is still required for pre-catalyst activation. Iron- and cobalt carboxylate salts have been shown to act as pre-catalysts for alkene hydrosilylation that do not require an external activator (Scheme 1b).8 Similarly, Huang showed that a tridentate PNN–cobalt(II) dichloride pre-catalyst could be activated thermally for alkene hydrosilylation.5b However, these are limited to a single reaction class and the carboxylate counterions are pre-catalyst specific.
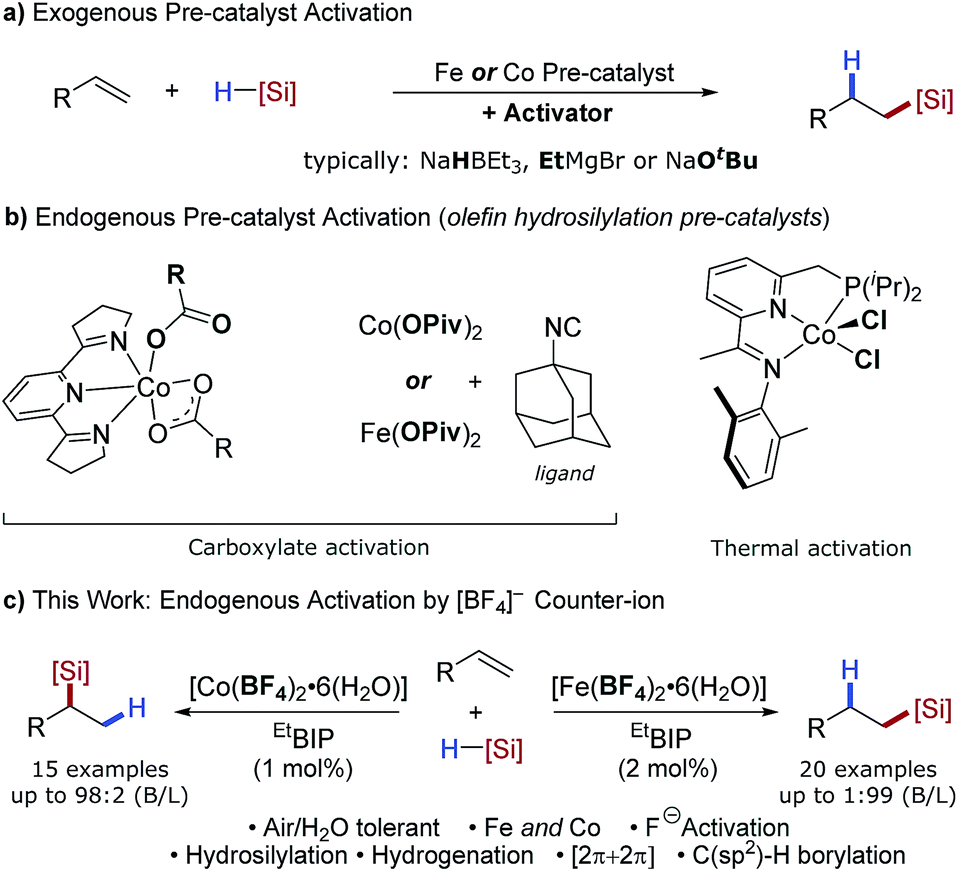 | ||
| Scheme 1 Overview of prior alkene hydrosilylation reactions using iron- and cobalt catalysts. (a) Pre-catalyst activation using additives. (b) Catalyst activity facilitated by carboxylate ligands or thermal activation. (c) This work: catalyst activation through use of weakly coordinating tetrafluoroborate counterions. | ||
The alkoxide activation of iron- and cobalt pre-catalysts (Scheme 1a) was proposed to proceed by reaction of the alkoxide and silane to form a hydridic silicon ‘ate’ complex which reduces the pre-catalyst by a hydride transfer.6a In contrast, the carboxylate and thermal activation methods were proposed to proceed by a σ-bond metathesis reaction between the metal carboxylate and silane (Scheme 1b).8 We postulated that using a counterion which is known to dissociate a nucleophile would allow an activation method that combined the operational simplicity of carboxylate activation and the broad scope of alkoxide activation. The tetrafluoroborate counterion is known to dissociate to BF3 and fluoride.9 The fluoride could react with the silane to give a hydridic silicon ‘ate’ complex,10 and activate a pre-catalyst by hydride transfer (Scheme 1c).11
Results and discussion
In order to establish the utility of the tetrafluoroborate counterion for activation of iron(II) pre-catalysts we selected alkene hydrosilylation as a model reaction. Baseline reactivity was determined by the use of iron pre-catalysts bearing strongly coordinating chloride anions [EtBIPFeCl2] (Table 1, entry 1) and weakly coordinating triflate anions [EtBIPFe(OTf)2] (entry 2). Both reactions showed no catalytic activity. The tetrafluoroborate pre-catalyst, formed in situ by the reaction of the commercially available hydrate salt Fe(BF4)2·6H2O with bisiminopyridine ligand (EtBIP), showed excellent catalytic activity without an external activator, giving the linear silane product 2a in excellent yield and regioselectivity (Table 1, entry 3). Lower catalyst loadings showed good, but reduced, reactivity in the production of 2a (Table 1, entries 4–5). Using iron(II) tetrafluoroborate, with less hindered bisiminopyridine ligands (HBIP, MeBIP, MesBIP) led to low reactivity, with only the N-mesityl ligand giving good yield, with similar linear:branched selectivity to that observed with EtBIP, while the more hindered N-isopropyl ligand gave only trace reactivity (Table 1, entries 6–9). Control reactions showed the need for both iron and ligand to achieve catalysis (see ESI, Table SI 1.1†).|
|
||||
|---|---|---|---|---|
| Entry | [M] | Loading (mol%) | Ligand | Yield (%) (2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a) :3a) |
| a Reaction conditions: 1-octene (1.00 equiv.), phenylsilane (1.10 equiv.) and metal tetrafluoroborate (n mol%), THF (1 M), r.t., 1 h. Yields determined by 1H NMR spectroscopy of the crude reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. | ||||
| 1 | FeCl2 | 2 | EtBIP | 0 |
| 2 | Fe(OTf)2 | 2 | EtBIP | 0 |
| 3 | Fe(BF 4 ) 2 ·6H 2 O | 2 | Et BIP |
87 (93:7) |
| 4 | Fe(BF4)2·6H2O | 0.5 | EtBIP | 67 |
| 5 | Fe(BF4)2·6H2O | 1 | EtBIP | 82 |
| 6 | Fe(BF4)2·6H2O | 2 | HBIP | 0 |
| 7 | Fe(BF4)2·6H2O | 2 | MeBIP | Trace |
| 8 | Fe(BF4)2·6H2O | 2 | MesBIP | 78 (>95:5) |
| 9 | Fe(BF4)2·6H2O | 2 | iPrBIP | Trace |
| 10 | Co(BF4)2·6H2O | 2 | HBIP | 84 (86:14) |
| 11 | Co(BF4)2·6H2O | 2 | MeBIP | 72 (4:96) |
| 12 | Co(BF4)2·6H2O | 2 | MesBIP | 68 (3:97) |
| 13 | Co(BF4)2·6H2O | 2 | EtBIP | 82 (5:95) |
| 14 | Co(BF4)2·6H2O | 2 | iPrBIP | 31 (16:84) |
| 15 | Co(BF 4 ) 2 ·6H 2 O | 1 | Et BIP |
90 (8:92) |
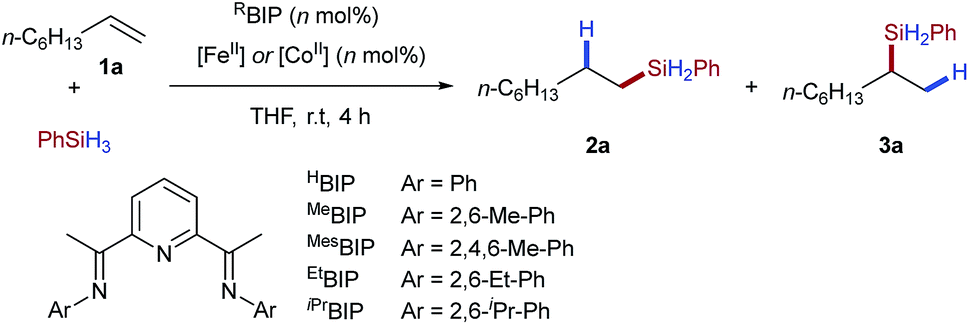
Having established the tetrafluoroborate activation for iron-catalysed hydrosilylation, we next attempted to apply the same protocol to cobalt-catalysed alkene hydrosilylation. Using commercially available cobalt(II) tetrafluoroborate and the N-dimethylphenyl bisiminopyridine ligand gave the linear silane 3a in excellent yield and good regioselectivity (Table 1, entry 11); the opposite to that observed under iron catalysis. Variation of the ligand N-aryl substituent showed that all cobalt catalysed systems selectively gave the branched silane, with the more hindered catalysts giving reduced yields of silane 3a (Table 1, entries 10–14). Notably, and to the best of our knowledge, this is the first example of a regiodivergent alkene hydrosilylation where high levels of regioselectivity are observed using an identical ligand and the same reaction conditions, but only varying the metal used.5b Use of a lower catalyst loading of 1 mol% gave the branched silane product in excellent yield and selectivity (Table 1, entry 15).
Having established optimal conditions for olefin hydrosilylation, we set out to investigate the scope and limitations of these reactions (Scheme 2). A variety of alkyl- and alkoxysilane reagents were successfully used for alkene hydrosilylation with both iron- (anti-Markovnikov) and cobalt (Markovnikov) pre-catalysts providing the linear- 2a–p and branched alkyl silanes 3a–l in excellent yield and regioselectivity, respectively. Iron-catalysed hydrosilylation of 1-octene with a tertiary silane (triethoxysilane) gave the linear silane 2d with a turnover number of 425 and a turnover frequency of 5100 h−1. In the cobalt-catalysed system, using only 110 ppm cobalt with phenylsilane gave the branched silane 3a with an overall turnover number of 5940, and turnover frequency of 2970 h−1. As all reaction components are air- and moisture stable the reactions can be set up without the need for specialist equipment. Therefore the iron- and cobalt catalysed hydrosilylation of 1-octene 1a was carried out in air with only a limited loss of catalyst activity and regioselectivity (Scheme 2, 2a and 3a). Alkyl- and aryl substituted alkenes underwent hydrosilylation in excellent yield and regioselectivity for both the iron- 2e–p and cobalt-catalysed 3d–l systems. The iron-catalysed system was found to tolerate electron-withdrawing substituents 2k and substituted alkenes such as β-pinene 2m, norbornene 2n, α-methylstyrene 2o and limonene 2p without detriment to yield or regioselectivity. The lower oxophilicity of cobalt was exemplified by chemoselective alkene hydrosilylation in the presence of ketone 3i, ester 3j, epoxy 3k and amido 3l functionalities. Divergent diastereoselectivity was observed for the hydrosilylation of terminal alkynes6a with cobalt catalysis preferentially giving the (E)-alkenylsilane 3m and the iron-catalysed system giving the (Z)-alkenylsilane 2q.
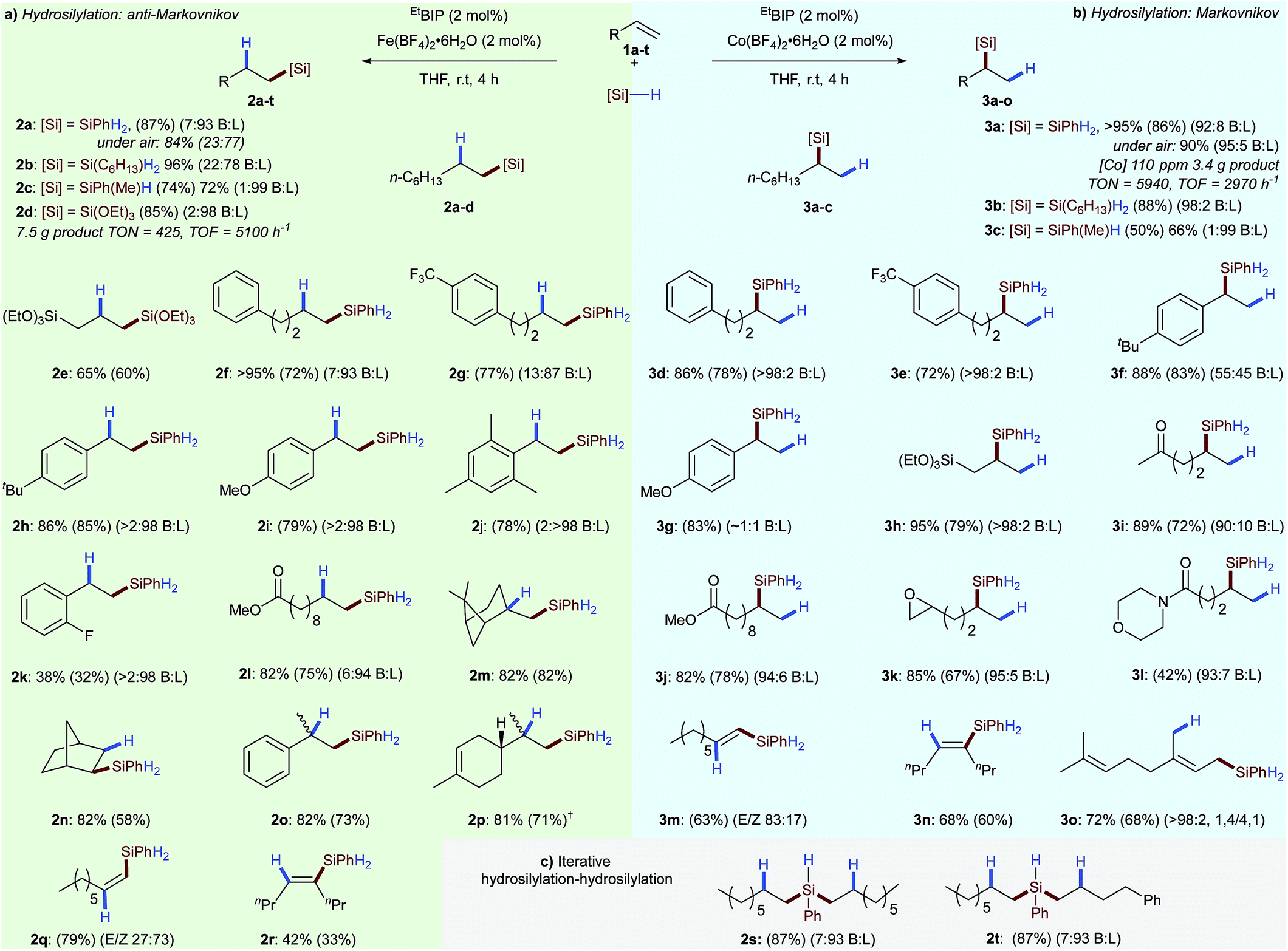 | ||
| Scheme 2 Scope for iron- and cobalt-catalysed hydrosilylation reactions enabled by tetrafluoroborate activation. (a) Reaction conditions: olefin, PhSiH3 (1.1 eq.), EtBIP (2 mol%) and Fe(BF4)2·6H2O (2 mol%), THF, r.t., 4 h. †MesBIP (2 mol%) used. (b) Reaction conditions: olefin, PhSiH3 (1.1 eq.), EtBIP (2 mol%) and Co(BF4)2·6H2O (2 mol%), THF, r.t., 4 h. (c) Reaction conditions: alkene (1 equiv.), PhSiH3 (1 equiv.) Fe(BF4)2·6H2O (2 mol%), EtBIP (2 mol%), THF, r.t., 30 min then a second alkene (1 equiv.) added, 3 h. Yields determined by 1H NMR spectroscopy of the crude reaction mixture using 1,3,5-trimethoxybenzene as an internal standard, isolated yields in parenthesis. | ||
Both the iron and cobalt catalysts gave the (E)-alkenylsilane, the product of syn addition, in the hydrosilylation of 4-octyne 2r and 3n, respectively. Hydrosilylation of the 1,3-diene myrcene proceeded with 1,4-selectivity 3o.
The potential of the developed tetrafluoroborate activated hydrosilylations for polymer crosslinking and late-stage functionalisation was demonstrated by iterative homo- 2s and hetero- 2t bis-hydrosilylations using two alkenes. Beyond showing that the tetrafluoroborate activation strategy can be applied across a number of unique ligand classes and metal salts, these results also demonstrate how, as a tool for reaction screening, this method can provide a facile method for uncovering new and contrasting reactivity.
To assess the generality of tetrafluoroborate iron- and cobalt-salts for hydrosilylation we opted to perform alkene hydrosilylation reactions with ligands distinct from bisiminopyridine. We initially selected bis-phosphine ligands, xantphos and dppf, as these had been used previously by Ge for cobalt-catalysed alkene hydrosilylation.12 Use of both of these ligands in combination with Co(BF4)2·6H2O effectively catalysed the hydrosilylation of 1-octene 1a with phenylsilane to generate linear silane 2a in excellent yield as a single exclusive regioisomer (Scheme 3a). Adamantyl isocyanide as a ligand additionally proved effective for both iron- and cobalt-catalysed alkene hydrosilylation to generate 2u and 2v respectively (Scheme 3b).13 The bidentate iminopyridine ligand, used previously by Ritter for 1,4-hydrosilylation of 1,3-dienes,14 was effective using Fe(BF4)2·6H2O to give allylic silane 2w (Scheme 3c).
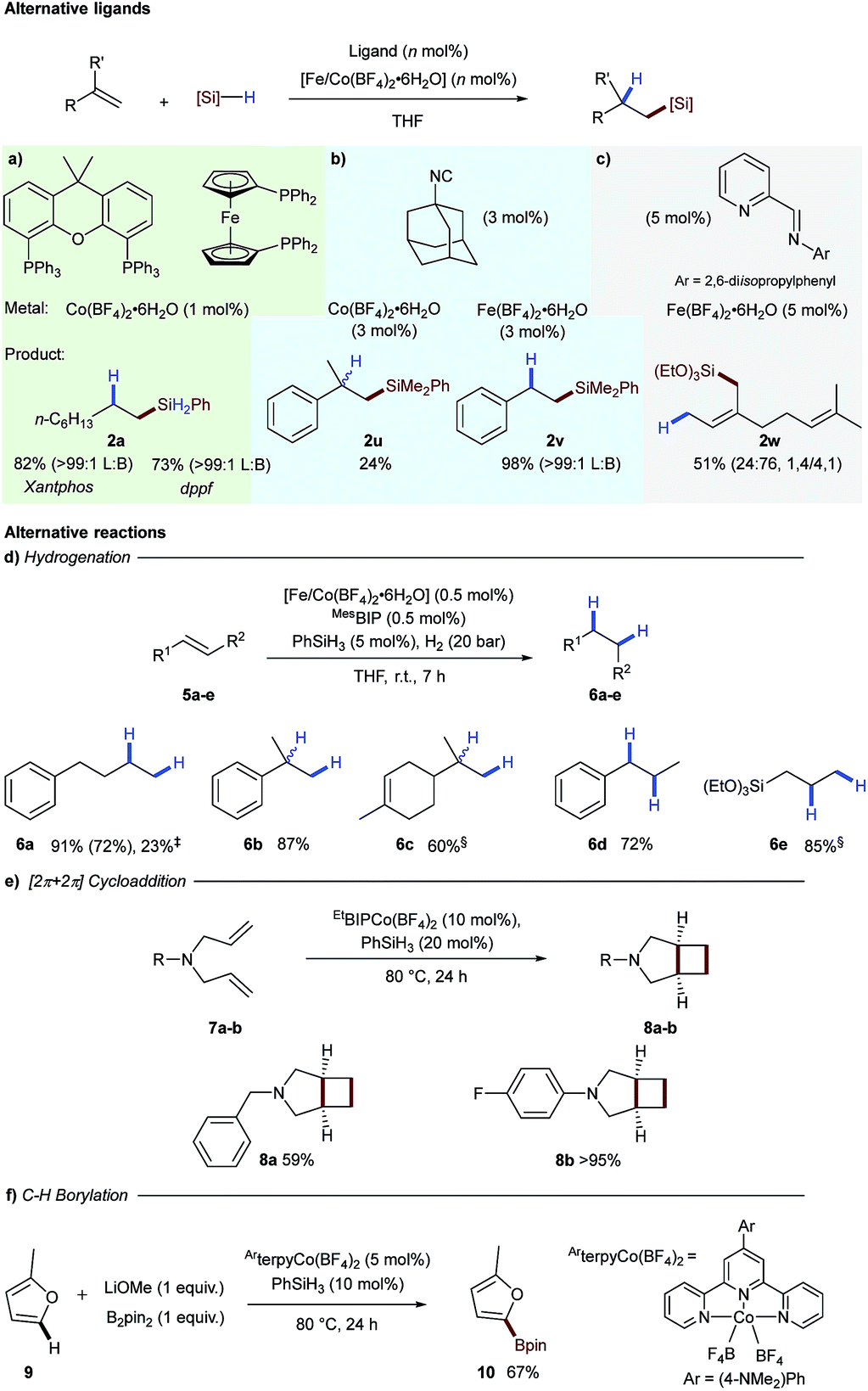 | ||
| Scheme 3 Application of tetrafluoroborate activation to other ligand classes for alkene hydrosilylation (top). Reaction conditions: (a) xantphos or dppf (1 mol%), Co(BF4)2·6H2O (1 mol%), 1-octene (1 mmol), phenylsilane (1.1 mmol), THF (2 M), r.t., 4 h. (b) Adamantyl isocyanide (9 mol% [Co] or 6 mol% [Fe]), metal tetrafluoroborate (3 mol%), α-methylstyrene ([Co], 1 mmol) or styrene ([Fe], 1 mmol), phenyldimethylsilane (1.3 mmol), THF (2 M), 80 °C, 3 h. (c) iPrIP (5 mol%), Fe(BF4)2·6H2O (5 mol%), myrcene (1 mmol), triethoxysilane (1.2 mmol), THF (2 M), r.t., 16 h. (d) Alkene (1 equiv.), Co(BF4)2·6H2O (0.5 mol%), MesBIP (1 mol%), PhSiH3 (5 mol%), H2 (20 bar), r.t., 7 h. ‡Alkene (1 equiv.), Fe(BF4)2·6H2O (2 mol%), MesBIP (2 mol%), PhSiH3 (5 mol%), H2 (20 bar), r.t., 7 h. §Co(BF4)2·6H2O (1 mol%) and MesBIP (1 mol%). (e) 1,6-Diene (1 equiv.), EtBIPCo(BF4)2·6H2O (10 mol%), PhSiH3 (20 mol%), 80 °C, 24 h. (f) Arene (15 equiv.), (4-NMe2-Ph-terpy)Co(BF4)2 (5 mol%), PhSiH3 (20 mol%), LiOMe (1 equiv.), B2pin2 (1 equiv.), 80 °C, 24 h. | ||
As we presumed the active hydrosilylation catalyst was a low oxidation-state metal species, we next explored the use of the tetrafluoroborate activation as a general platform to access these species. We postulated that mixing the tetrafluoroborate pre-catalysts with a substoichiometric amount of silane reagent would give a generic low oxidation-state catalyst that would be applicable to transformations beyond hydrosilylation. This would negate the need to isolate a catalyst with limited stability or use pyrophoric reagents in low oxidation-state iron- and cobalt catalysis.6a
The first reaction tested was hydrogenation of alkenes, an industrially important transformation.15,16 Using 0.5 mol% of the iron- or cobalt tetrafluoroborate salts and the MesBIP ligand in combination with substoichiometric phenylsilane gave an active catalyst for alkene reduction in both cases (Scheme 3d). 4-Phenyl-1-butene underwent hydrogenation to the alkene 6a in good yield under cobalt catalysis and reduced yield with the analogous iron system. The cobalt-catalysed hydrogenation was found to be successful for 1,1-disubstituted alkenes 6b and 6c, 1,2-disubstituted alkenes 6d and allyl silane 6e (Scheme 3d).
Another example of a reaction which has been catalysed by low oxidation-state species is the intramolecular [2π + 2π]-cycloaddition of 1,6-dienes to give [3.2.0] bicyclic systems.17 In this case, N-benzyl-N,N-diallylamine 7a was converted into N-benzyl-3-azabicyclo[3.2.0]heptane 8a in good yield and N-4-fluorophenyl-N,N-diallyl amine 7b cyclised to give bicyclic 8b in excellent yield (Scheme 3e). A number of procedures have been developed for cobalt-catalysed C–H borylation, and cobalt(I) boryl complexes have been proposed to be the key catalytic intermediate.18 In order to apply tetrafluoroborate activation to a range of mechanistically distinct reactions, C–H borylation of 2-methylfuran 9 was carried out using a cobalt-terpyridine tetrafluoroborate pre-catalyst, to give the boronic ester 10 in 67% yield (Scheme 3f).
The facile activation observed using this protocol was thought to result from pre-catalyst reduction by an in situ formed hydridic silicon ‘ate’; formed by reaction of the silane reagent with fluoride dissociated from the counterion. This putative ‘ate’ complex then transfers hydride to the pre-catalyst, facilitating reductive elimination of dihydrogen (Scheme 4a).4b Silane reagents have been shown to be hydridic reagents in the presence of suitable nucleophiles, such as fluoride, and this has been applied in the reduction of carbonyls.10 To examine whether similar reactivity could be obtained in this case, n-butylammonium tetrafluoroborate (TBABF4) was reacted with phenylsilane in the presence of 4-fluorobenzaldehyde 10 to give the primary alcohol reduction product 11 in excellent yield (Scheme 4b). This underlines the possibility of pre-catalyst reduction by a hydridic silicon ‘ate’ complex6a formed by reaction of fluoride and phenylsilane.11 Although the exact nature of the active catalyst is not known, the reaction was tested in the presence of radical inhibitors and was unaffected (Scheme 4c and Table SI 10†), suggesting that reduction occurs by a two-electron mechanism.4b
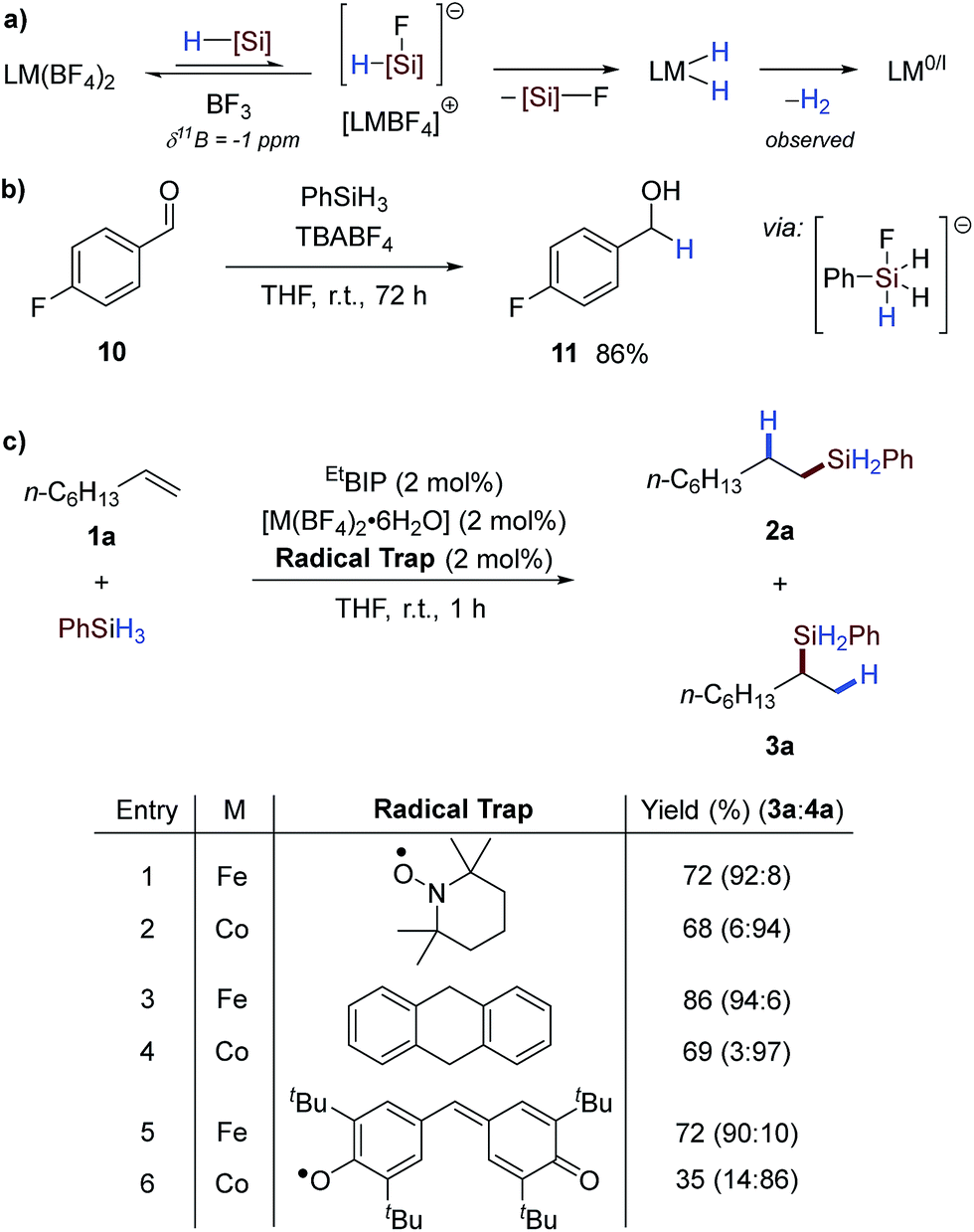 | ||
| Scheme 4 Proposed activation mechanism and mechanistic studies. (a) Metal tetrafluoroborate pre-catalyst activation strategy from reaction with silane reagents to generate a low oxidation-state active catalyst. (b) Interaction of tetrabutylammonium tetrafluoroborate and phenylsilane for the reduction of 4-fluorobenzaldehyde, suggestive of hydride formation. (c) Attempted radical inhibition experiments with radical trapping reagents. | ||
Conclusions
A procedure for the regiodivergent hydrosilylation of olefins, a highly valuable industrial reaction, has been developed using iron- and cobalt tetrafluoroborate catalysts without the need for an external activator or the use of isolated low oxidation-state complexes. This has been used as an activation platform to access the low oxidation-state catalysts in a range of iron- and cobalt-catalysed reactions including hydrosilylation, 1,3-diene hydrosilylation, alkene hydrogenation, [2π + 2π]-cycloaddition and C–H borylation. The developed tetrafluoroborate activation represents a versatile platform for activation, and serves as a generic strategy for accessing low oxidation-state reactivity with both iron and cobalt. It is hoped that this work will streamline the discovery of new reactivity, development of novel synthetic methodology and, ultimately, the replacement of precious metals with their earth-abundant counterparts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RA and SPT thank the Royal Society for funding a PhD studentship. AJC and SPT thank Syngenta for part funding a PhD studentship. SPT thanks the Royal Society for a University Research Fellowship. JD and SPT thank Prof. M. Shaver for useful discussions. All thank the University of Edinburgh, School of Chemistry and Technical Staff for generous support.Notes and references
- (a) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed; (b) X. Du and Z. Huang, ACS Catal., 2017, 7, 1227–1243 CrossRef CAS; (c) R. I. Khusnutdinov, A. R. Bayguzina and U. M. Dzhemilev, Russ. J. Org. Chem., 2012, 48, 309–348 CrossRef CAS; (d) J. R. Carney, B. R. Dillon and S. P. Thomas, Eur. J. Org. Chem., 2016, 23, 3912–3929 CrossRef; (e) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS PubMed; (f) A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed.
- Global Silicones Market by Market, Product and Country, 4th edn, Summary, 1.
- A. J. Holwell, Platinum Met. Rev., 2008, 52, 243–246 CrossRef.
- (a) A. M. Tondreau, C. C. H. Atienza, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis and P. J. Chirik, Science, 2012, 335, 567–570 CrossRef CAS PubMed; (b) S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794–13807 CrossRef CAS PubMed.
- (a) W. Y. Chu, R. Gilbert-Wilson, T. B. Rauchfuss, M. Van Gastel and F. Neese, Organometallics, 2016, 35, 2900–2914 CrossRef CAS; (b) X. Du, Y. Zhang, D. Peng and Z. Huang, Angew. Chem., Int. Ed., 2016, 55, 6671–6675 CrossRef CAS PubMed; (c) M. D. Greenhalgh, D. J. Frank and S. P. Thomas, Adv. Synth. Catal., 2014, 356, 584–590 CrossRef CAS; (d) X. Jia and Z. Huang, Nat. Chem., 2016, 8, 157–161 CrossRef CAS PubMed; (e) J. Guo and Z. Lu, Angew. Chem., Int. Ed., 2016, 55, 10835–10838 CrossRef CAS PubMed; (f) C. Chen, M. B. Hecht, A. Kavara, W. W. Brennessel, B. Q. Mercado, D. J. Weix and P. L. Holland, J. Am. Chem. Soc., 2015, 137, 13244–13247 CrossRef CAS PubMed; (g) M. D. Greenhalgh, A. S. Jones and S. P. Thomas, ChemCatChem, 2015, 7, 190–222 CrossRef CAS; (h) J. Sun and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef CAS; (i) J. Guo and Z. Lu, Angew. Chem., Int. Ed., 2016, 55, 10835–10838 CrossRef CAS PubMed.
- (a) J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017, 9, 595–600 CrossRef CAS PubMed; (b) I. Buslov, S. C. Keller and X. Hu, Org. Lett., 2016, 18, 1928–1931 CrossRef CAS PubMed; (c) J. Peng, J. H. Docherty, A. P. Dominey and S. P. Thomas, Chem. Commun., 2017, 53, 4726–4729 RSC; (d) J. L. Boyer and A. K. Roy, US pat. 0343311 A1, 2014.
- A. J. Challinor, M. Calin, G. S. Nichol, N. B. Carter and S. P. Thomas, Adv. Synth. Catal., 2016, 358, 2404–2409 CrossRef CAS.
- (a) D. Noda, A. Tahara, Y. Sunada and H. Nagashima, J. Am. Chem. Soc., 2016, 138, 2480–2483 CrossRef CAS PubMed; (b) A. Sanagawa and H. Nagashima, Organometallics, 2018, 17, 2859–2871 CrossRef; (c) C. H. Schuster, T. Diao, I. Pappas and P. J. Chirik, ACS Catal., 2016, 6, 2632–2636 CrossRef CAS; (d) G. Wu, U. Chakraborty and A. Jacobi von Wangelin, Chem. Commun., 2018, 54, 12322–12325 RSC; (e) H. L. Sang, S. Yu and S. Ge, Chem. Sci., 2018, 9, 973–978 RSC.
- (a) A. J. Cresswell, S. G. Davies, P. M. Roberts and J. E. Thomson, Chem. Rev., 2015, 115, 566–611 CrossRef CAS PubMed; (b) C. A. Wamser, J. Am. Chem. Soc., 1948, 70, 1209–1215 CrossRef CAS; (c) M. G. Freire, C. M. S. S. Neves, I. M. Manucho, J. A. P. Coutinho and A. M. Fernandes, J. Phys. Chem. A, 2010, 114, 3744–3749 CrossRef CAS PubMed; (d) R. Agahi, A. J. Challinor, N. B. Carter and S. P. Thomas, Org. Lett., 2019, 21, 993–997 CrossRef CAS PubMed.
- (a) J. Boyer, R. Corriu, R. Perz, M. Poirier and C. Reye, Synthesis, 1981, 7, 558–559 CrossRef; (b) M. Das and D. F. O'Shea, Tetrahedron, 2013, 69, 6448–6460 CrossRef CAS.
- J. M. Larsson and K. J. Szabó, J. Am. Chem. Soc., 2013, 135, 443–455 CrossRef CAS PubMed.
- C. Wang, W. J. Teo and S. Ge, ACS Catal., 2017, 7, 855–863 CrossRef CAS.
- D. Noda, A. Tahara, Y. Sunada and H. Nagashima, J. Am. Chem. Soc., 2016, 138, 2480–2483 CrossRef CAS PubMed.
- J. Y. Wu, B. N. Stanzl and T. Ritter, J. Am. Chem. Soc., 2010, 132, 13214–13216 CrossRef CAS PubMed.
- (a) J. G. de Vries and C. J. Elsevier, The Handbook of Homogeneous Hydrogenation, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2006 CrossRef; (b) P. Etayo and A. Vidal-Ferran, Chem. Soc. Rev., 2013, 42, 728–754 RSC; (c) C. Pettinari, F. Marchetti and D. Martini, in Comprehensive Coordination Chemistry II, Elsevier, 2003, pp. 75–139 Search PubMed.
- For examples of olefin hydrogenation using iron and cobalt see: (a) P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687–1695 CrossRef CAS PubMed; (b) D. Gärtner, A. Welther, B. R. Rad, R. Wolf and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2014, 53, 3722–3726 CrossRef PubMed; (c) L. J. Murphy, M. J. Ferguson, R. McDonald, M. D. Lumsden and L. Turculet, Organometallics, 2018, 37, 4814–4826 CrossRef CAS; (d) A. J. MacNair, M.-M. Tran, J. E. Nelson, G. U. Sloan, A. Ironmonger and S. P. Thomas, Org. Biomol. Chem., 2014, 12, 5082–5088 RSC; (e) D. J. Frank, L. Guiet, A. Käslin, E. Murphy and S. P. Thomas, RSC Adv., 2013, 3, 25698 RSC; (f) T. N. Gieshoff, U. Chakraborty, M. Villa and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2017, 56, 3585–3589 CrossRef CAS.
- (a) J. M. Hoyt, V. A. Schmidt, A. M. Tondreau and P. J. Chirik, Science, 2013, 775, 772–775 Search PubMed; (b) V. A. Schmidt, J. M. Hoyt, G. W. Margulieux and P. J. Chirik, J. Am. Chem. Soc., 2015, 137, 7903–7914 CrossRef CAS PubMed.
- (a) J. V. Obligacion, M. J. Bezdek and P. J. Chirik, J. Am. Chem. Soc., 2017, 139, 2825–2832 CrossRef CAS PubMed; (b) J. V. Obligacion and P. J. Chirik, ACS Catal., 2017, 7, 4366–4371 CrossRef CAS PubMed; (c) N. G. Léonard, M. J. Bezdek and P. J. Chirik, Organometallics, 2017, 36, 142–150 CrossRef; (d) J. V. Obligacion, H. Zhong and P. J. Chirik, Isr. J. Chem., 2017, 57, 1032–1036 CrossRef CAS PubMed; (e) W. N. Palmer, J. V. Obligacion, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 766–769 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05391j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |