DOI:
10.1039/C8SC05543B
(Edge Article)
Chem. Sci., 2019,
10, 2875-2881
A rational quest for selectivity through precise ligand-positioning in tandem DNA-catalysed Friedel–Crafts alkylation/asymmetric protonation†
Received
11th December 2018
, Accepted 22nd January 2019
First published on 24th January 2019
Abstract
Covalent anchorage of a metallic co-factor to a DNA-based architecture is merely the only way to ensure an accurate positioning of a catalytic site within the chiral micro-environment offered by the DNA double helix. Ultimately, it also allows a fine-tuning of the catalytic pocket through simple synthetic modifications of the DNA sequence. Here, we report highly selective copper(II)-catalysed asymmetric Friedel–Crafts conjugate addition/enantioselective protonation, which is due to a careful positioning of a bipyridine ligand within a DNA framework. Most importantly, this study unveils specific structural features that account for an optimal chirality transfer from the duplex to the Friedel–Crafts adducts.
Introduction
In the last decade, a large variety of artificial metalloenzymes based on various macromolecular architectures have been designed and engineered by chemists resulting in several particularly effective catalytic processes.1 While the first protein-based artificial metalloenzymes date back to the 1970s,2 it was only in 2005 that Roelfes and Feringa3 described the very first DNA-based artificial metalloenzyme, where the well-defined DNA structure provides a unique chiral microenvironment able to accommodate a transition metal complex which in turn catalysed Diels–Alder cycloaddition with high levels of enantiodiscrimination. Since then, DNA-based chiral amplifications have been applied to a wide range of Cu(II)-catalysed reactions including Friedel–Crafts alkylation,4 Michael addition,5syn-hydration,6 and fluorination reactions.7 Among all the ligands tested, 2,2′-dimethyl-4,4′-bipyridine (dmbipy) was quickly identified as one of the most promising, which led multiple groups around the world including ours to evaluate the influence on the selectivity of various anchorage strategies by which the ligand can be incorporated in the DNA scaffold. While supramolecular interactions allow a straightforward self-assembly of the catalyst and thus a rapid screening/optimization of the reaction conditions, they unfortunately do not enable a precise positioning of the metallic cofactor into the DNA duplex.1 In contrast, the covalent attachment of a metallic cofactor allows the construction of finely tuned DNA-based catalysts with a clear knowledge of the surrounding environment, thus allowing a more straightforward rationalization of the catalytic efficacy of a given bio-hybrid system. Interestingly, while several groups have embarked on this route,8 none have taken into consideration the fundamental influence of the positioning of the metal-chelating ligand. This is all the more surprising that it is well known that DNA base modifications at the 5-position of the pyrimidines tend to place the substituents in the major groove of the double helix, while 2′-modifications place the substituents inside the minor groove.9 These fundamental structural differences prompted us to further investigate this matter with the aim of developing a strong mechanistic rational and eventually designing the ultimate bio-hybrid catalyst. The asymmetric Friedel–Crafts conjugate addition/enantioselective protonation of α-substituted α,β-unsaturated acyl imidazoles appeared to us as the perfect model reaction to study since the selectivities reported in the field were rather low and, most importantly, highly substrate dependent.4c Indeed, the challenge in this reaction is that the chirality is not introduced during the conjugate addition step but rather during the protonation of the highly reactive pro-chiral enolate intermediate. As a general trend, the enantioselectivity in such reactions is usually obtained using a chiral enolate in conjunction with an achiral proton source or by using an achiral enolate in combination with a chiral protonating agent.10 In both cases however, exclusion of water is crucial in order to prevent any non-selective protonation. To the best of our knowledge only two examples of tandem Friedel–Crafts conjugate addition/asymmetric protonation in water have been reported in the literature so far. The first one was reported by Luo and Cheng in 2011 and featured the conjugate addition of various indoles on α-substituted acroleins through chiral enamine catalysis.11 The second one was reported more recently by Roelfes and co-workers and involved the conjugate addition of a variety of indoles on 2-methyl-1-(thiazol-2-yl)prop-2-en-1-one catalysed by DNA in conjunction with a Cu–dmbipy metallic cofactor.4c Interestingly, while the presence of DNA was found to accelerate the reaction at an exceptional rate (up to 990-fold), the enantioselectivities were nonetheless highly dependent on the nature of the indole. Moreover, this study unveiled a close connexion between the selectivity obtained and the binding affinity of the Cu(II)–dmbipy complex, the enone and the indole with DNA.4c This indispensable affinity balance between all the species involved inspired us to evaluate the effect of a covalent anchorage of the metallic co-factor onto the DNA scaffold and ultimately ascertain the influence of the grooves on the enantioselectivity.
Results and discussion
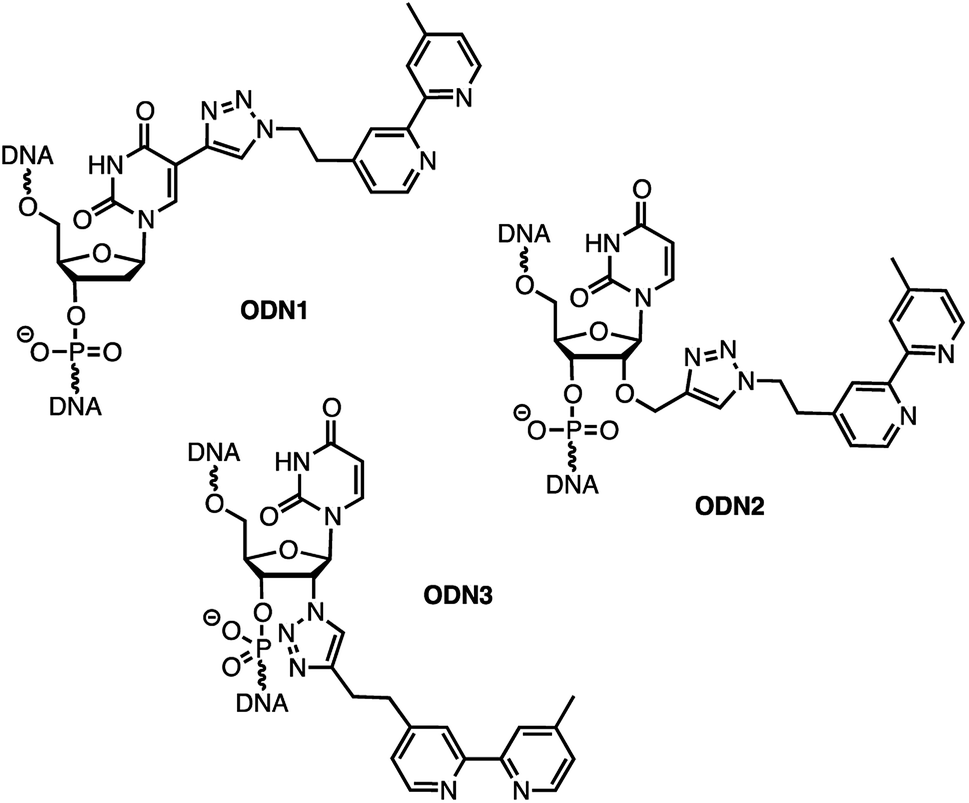
In order to analyse the influence of the positioning of the metallic co-factor on both the reactivity and the selectivity, we chose to synthesize ODN1, ODN2 and ODN3, which incorporate a bipyridine ligand either in the major groove (ODN1) or in the minor groove (ODN2 and ODN3) once hybridised with a common complementary strand ODN4 (Scheme 1).
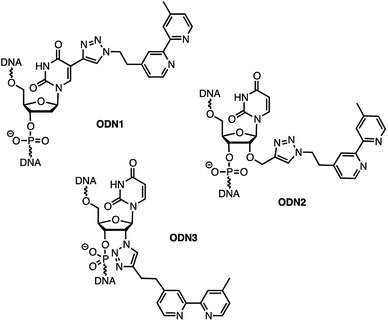 |
| | Scheme 1 Structures of bipyridine-functionalised ODNs. | |
The usual methods to introduce modifications into oligonucleotides involve either the synthesis of appropriately modified phosphoramidite building blocks or the post-synthetic conjugation of a defined reactive group. This latter strategy is usually preferred as it is easier to handle, gives better yields and allows higher degrees of modularity.12 As rate acceleration and improved enantioselectivities have been previously demonstrated with G-rich self-complementary dodecamers,13 we designed a duplex 5′-GCCAGCXGACCG-3′/5′-CGGTCAGCTGGC-3′ which incorporates a unique modification (X) at residue 7 of the sense strand. Commercially available phosphoramidite derivatives of 2′-deoxy-5-ethynyluridine 7 and 2′-O-propargyluridine 10 were used to prepare CPG-bound oligonucleotides 8 and 11 respectively (DMT-off). The azido-dmbipy partner 4, on the other hand, was synthesized in three steps starting from commercially available 4,4′-dimethyl-2,2′-bipyridine 1. Hence, monolithiation of 1 using LDA followed by the addition of an excess of paraformaldehyde afforded the corresponding hydroxymethyl derivative 2,14 which was eventually subjected to tosylation [TsCl, DIEA, DCM, rt, and 57%] and azidation [NaN3, DMF, 0 °C, and 85%] to afford the desired azide 4 (Scheme 2A). A Cu(I)-catalysed azide–alkyne cycloaddition (CuAAC) reaction was then carried out on a solid support by mixing the representative ODN with azide 4 (2 equiv.) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O/dioxane mixture in the presence of a freshly prepared aqueous solution of CuSO4·5H2O (1 equiv.), sodium ascorbate (5 equiv.) and THPTA (3 equiv.), and by heating the mixture for 75 min at 55 °C under microwave irradiation (Scheme 2B and C).15 The resulting CPG supports were then filtered and washed with a saturated solution of EDTA. After treatment with aqueous ammonia, ODN1 and ODN2 were purified by preparative HPLC and finally desalted.
:1 H2O/dioxane mixture in the presence of a freshly prepared aqueous solution of CuSO4·5H2O (1 equiv.), sodium ascorbate (5 equiv.) and THPTA (3 equiv.), and by heating the mixture for 75 min at 55 °C under microwave irradiation (Scheme 2B and C).15 The resulting CPG supports were then filtered and washed with a saturated solution of EDTA. After treatment with aqueous ammonia, ODN1 and ODN2 were purified by preparative HPLC and finally desalted.
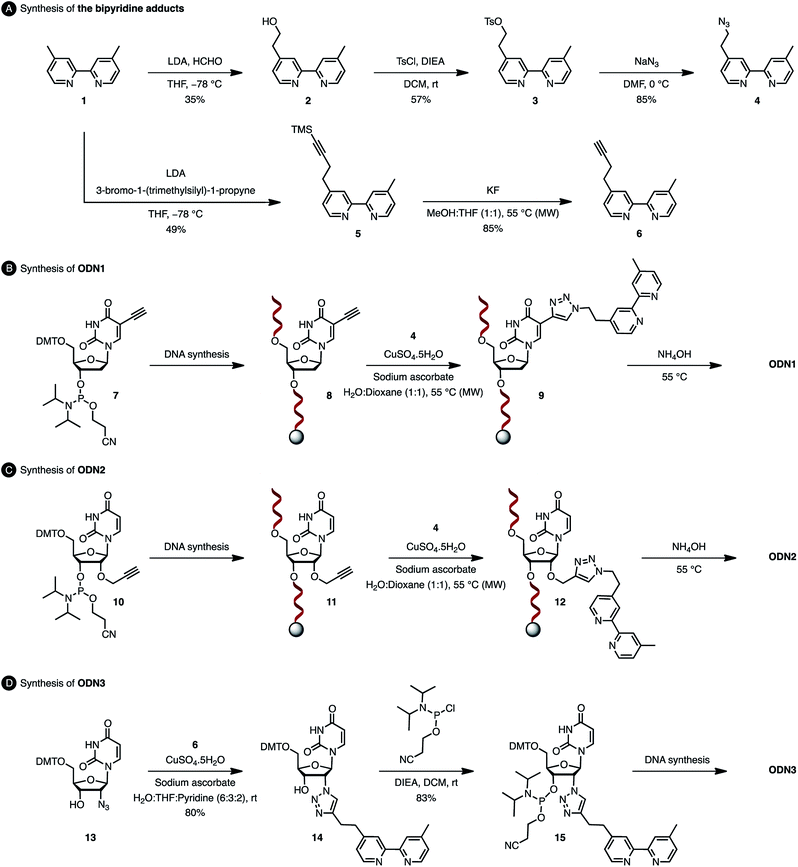 |
| | Scheme 2 Synthesis of oligonucleotides ODN1, ODN2 and ODN3. | |
In order to probe the influence of the flexibility of the ligand inside the minor groove, we also synthesized ODN3, which bears a triazole moiety directly attached onto the 2′-position at residue 7 of the sense strand. This modification required the use of 2′-azido uridine which is not compatible with standard phosphoramidite chemistry because of the inherent reactivity of the phosphorus(III) species in the presence of azides.16 Although the post-synthetic labelling of 2′-azido modified RNA has been described by Micura and co-workers by combining both phosphotriester and phosphoramidite chemistries,17 we decided to embark on the chemical synthesis of the appropriately modified phosphoramidite 15 (Scheme 2D). In this context, the monolithiation of 1 using LDA at −78 °C followed by the addition of bromo-1-(trimethylsilyl)-1-propyne (1 equiv.) afforded 5, which was ultimately deprotected with potassium fluoride under microwave irradiation. The resulting terminal alkyne 6 was then reacted with 5′-dimethoxytrityl-2′-azido-2′-deoxyrudine 13 in the presence of CuSO4·5H2O and sodium ascorbate [H2O/THF/pyridine (6:3:2), 30 min, and rt] to afford triazole derivative 14, which was subsequently converted to the corresponding phosphoramidite 15 using 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (1.3 equiv.). The requisite oligonucleotide ODN3 was then assembled through solid-phase synthesis, purified by HPLC and desalted. ODN1, ODN2 and ODN3 were eventually hybridised with complementary ODN4 (5′-CGG TCA GCT GGC-3′) and evaluated in the context of the tandem Friedel–Crafts conjugate addition/asymmetric protonation reaction.
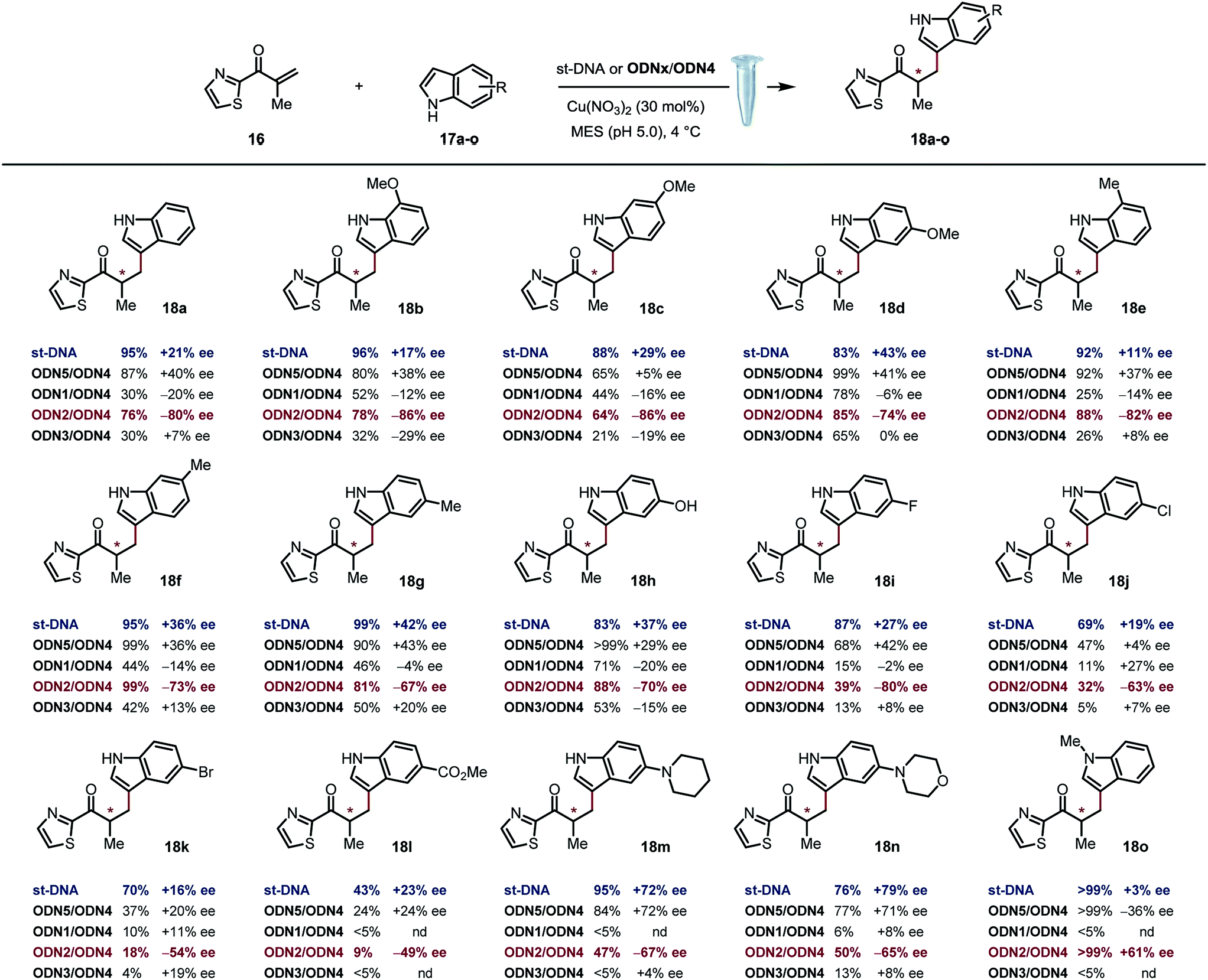
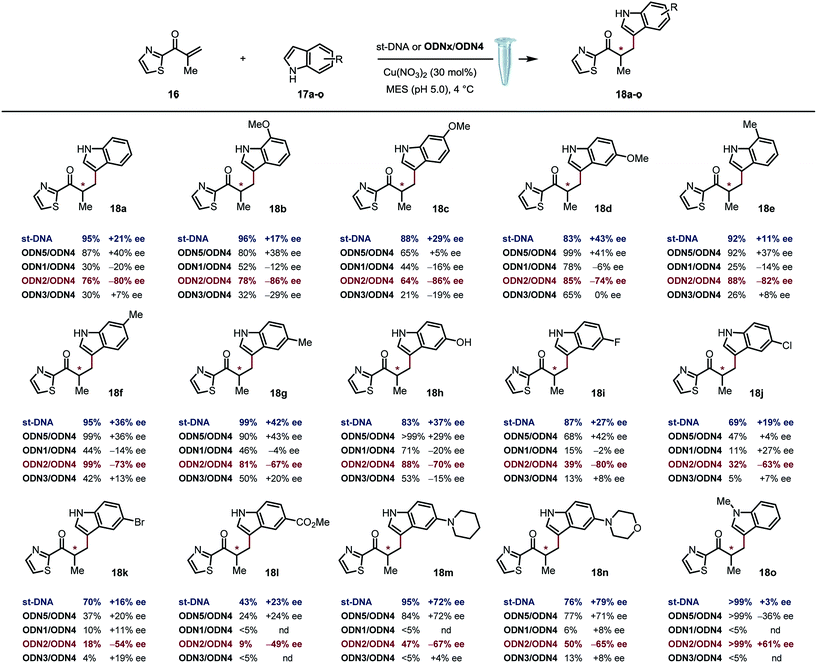
Based on previously optimized conditions, we decided to run the reactions using a 1:1 ratio of 2-methyl-1-(thiazol-2-yl)prop-2-en-1-one and indole, 30 mol% of Cu(NO3)2 and a 0.4 mM solution of ODNx/ODN4 in a MES buffer (pH 5.0) at 4 °C for three days.18 The supramolecular approach using the non-covalently linked Cu–dmbipy complex in the presence of either st-DNA (2 mM base pair) or the non-modified duplex ODN5/ODN4 (5′-GCCAGCTGACCG-3′/5′-CGGTCAGCTGGC-3′) was also evaluated for comparison purposes. A large variety of indoles differing in their substitution pattern were assessed (Table 1). Three major trends could be identified from these reactions: (1) good to excellent conversions were observed albeit lower that the ones obtained using the supramolecular approach,4c (2) the non-covalent approach using st-DNA or ODN5/ODN4 in the presence of dmbipy afforded very similar ee values, and (3) the best selectivities were obtained with the covalently modified sequences, particularly ODN2/ODN4, which clearly outperformed the supramolecular approach in terms of enantioselectivity. In contrast, ODN1/ODN4 (major groove) and ODN3/ODN4 (minor groove) could not clearly differentiate between the two faces of the pro-chiral enolate, thus suggesting that the positioning of the ligand in either groove did not seem to have a significant effect on the selectivity of the reactions. Moreover, the catalytic efficacy of our lead duplex appeared to be far less dependent on the nature of the indole than st-DNA as showcased by the good to excellent ee values obtained with the various indoles tested. Finally, an interesting inversion of selectivity was observed when using ODN2/ODN4 compared to any of the non-covalent approaches (ODN5/ODN4 or st-DNA) and this trend was confirmed for pretty much all the indoles tested independently of their substitution pattern. Hence, the use of 1H-indole led to the corresponding saturated ketone in −80% ee with ODN2/ODN4 while only +21% ee was obtained with st-DNA (Table 1, 18a). Likewise, the 5-, 6- and 7-methoxy-substituted indoles afforded ee values ranging from 74 to 86% (Table 1, 18b–d) with ODN2/ODN4 while the same reactions run in the presence of st-DNA afforded only moderate selectivities (up to +43% ee). A similar trend was also observed with the 5-, 6- and 7-methyl-substituted indoles as well as the 5-hydroxy indole, which afforded ee values ranging between −67 and −82%, while the enantioselectivities did not exceed +43% ee when using the supramolecular approach (Table 1, 18e–h). ODN2/ODN4 also proved to be superior to st-DNA when indoles bearing an electron-withdrawing group at the 5 position were used. This was the case with 5-fluoro indole (−80% ee vs. +27% ee, 18i), 5-chloro indole (−63% ee vs. +19% ee, 18j), 5-bromo indole (−54% ee vs. +16% ee, 18k) and methyl indole-5-carboxylate (−49% ee vs. +23% ee, 18l). ODN2/ODN4 displayed a similar selectivity to st-DNA when indoles bearing a tertiary amine at the 5 position such as a piperidine (−67% ee vs. +72% ee, 18m) or a morpholine (−65% ee vs. +79% ee, 18n) were used. Protonation of the amine in the reaction media was advanced to explain the higher ee values obtained with st-DNA;4c however in view of our results, this does not seem to be the case in our covalent approach. Finally, the use of N-methyl indole led to the corresponding product in only +3% ee with st-DNA, while ODN2/ODN4 afforded up to +61% ee under otherwise identical conditions (Table 1, 18o). It is worth pointing out as well that this was actually the only example that did not lead to an inversion of the selectivity thus confirming the importance of specific interactions between the substrate and the oligonucleotides.
Table 1 Scope of the a tandem Friedel–Crafts conjugate addition/asymmetric protonation reactiona,b
|
Conditions with st-DNA: st-DNA [2 mM in Milli-Q H2O (29 μL)], 200 mM MES buffer solution (10 μL, pH 5.0), 1.0 mM of Cu(NO3)2–dmbipy in Milli-Q H2O solution (33 μL, 30 mol%), 0.05 M solution of freshly prepared enone in DMSO (2.0 μL, 1 equiv.), 0.05 M solution of indole in DMSO (2.0 μL, 1 equiv.), 3 d, and 4 °C. Conditions with ODNx/ODN4: ODNx/ODN4 (40 mol%), 200 mM MES buffer solution (10 μL, pH 5.0), 1.0 mM of Cu(NO3)2 in Milli-Q H2O solution (30 μL, 30 mol%), 0.05 M solution of freshly prepared enone in DMSO (2.0 μL, 1 equiv.), 0.05 M solution of indole in DMSO (2.0 μL, 1 equiv.), 3 d, and 4 °C.
Conversion and ee values were determined by High Pressure Liquid Chromatography (HPLC) analysis. ODNx/ODN4: 5′-GCCAGCXGACCG-3′/5′-CGGTCAGCTGGC-3′.
|
|
Discussion
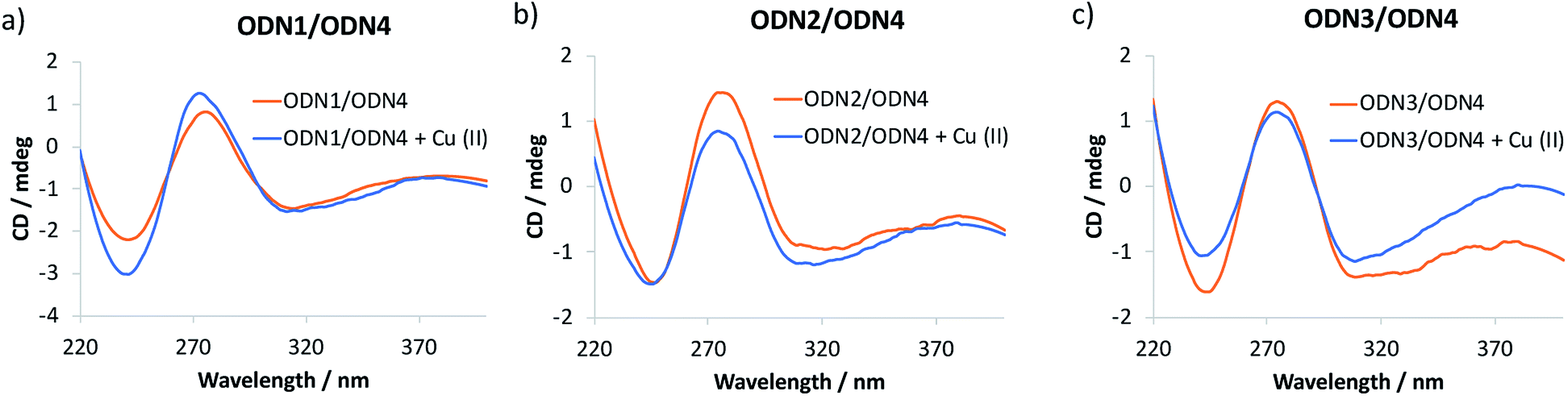
The catalytic performance of ODN2/ODN4 compared to either ODN1/ODN4 or ODN3/ODN4 or the non-covalent approach is quite remarkable and leads to unprecedented levels of enantioselectivities for tandem Friedel–Crafts conjugate addition/asymmetric protonation in water. In order to rationalize the differences observed, especially when the bipyridine ligand is positioned in the major groove or in the minor groove through a 2′-triazolyl linker, we recorded circular dichroism (CD) spectra of the different modified duplexes in the absence and presence of Cu(II) (Fig. 1). For all three duplexes, the CD spectra were very similar and exhibited a typical B-type CD spectrum characterized by a negative band around 240 nm and a positive band around 260 nm. The attachment of the dmbipy ligand through a triazoyl linker either at the 5-position of the thymidine or at the 2′-position of the 2′-deoxyuridine did not seem to induce any significant change in the spectrum compared to that of the non-modified duplex ODN4/ODN5. Similarly, addition of Cu(II) ions did not induce substantial ICD (Fig. 1). According to the CD spectra, these observations indicate that the covalent attachment of the bipyridine ligand has no major influence on the folding of these duplexes. ODN2/ODN4 is the only duplex that shows a small decrease in the intensity of the positive band after addition of Cu(II) ions thus revealing a small change in the interaction of the ligand with the DNA helix (Fig. 1b). This small structural change may be the key factor that accounts for the higher enantioselectivities observed with ODN2/ODN4 compared to all the other modified duplexes. To further investigate these systems, the influence of the covalent modification was analysed by thermal denaturation studies.
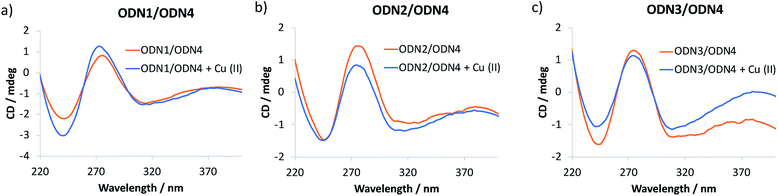 |
| | Fig. 1 CD spectra of (a) ODN1/ODN4, (b) ODN2/ODN4 and (c) ODN3/ODN4 duplexes folding in the absence and in the presence of Cu(II) ions. | |
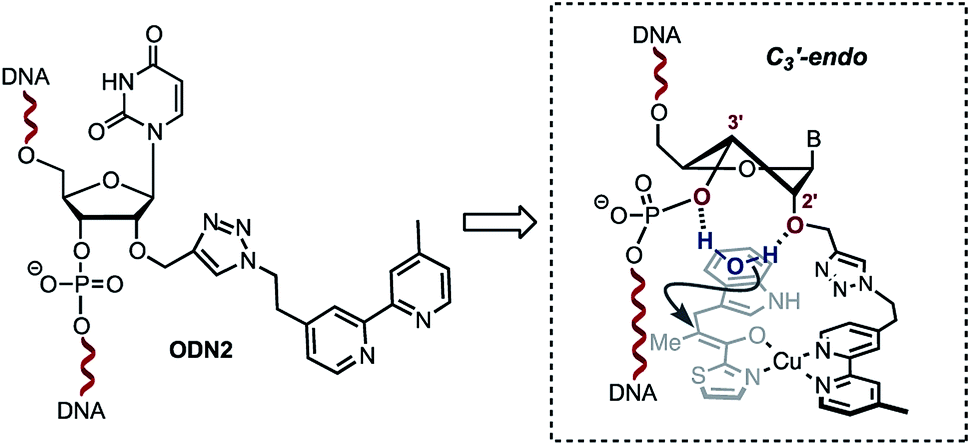
A single sigmoidal transition was obtained in all cases. Compared with the non-modified duplex ODN4/ODN5, the presence of the bipyridine linker destabilised the duplex ranging from ΔTm = −5 °C for ODN1/ODN4 and ODN2/ODN4 to ΔTm = −11 °C for ODN3/ODN4. This strong destabilization might also affect the catalytic efficiency observed with ODN3/ODN4. Interestingly, no significant effect was observed upon addition of copper(II) ions (Table 2). The steric restrictions imposed by the ligand and the substrate/DNA-binding interactions might be the main parameter affecting the stereochemical outcome of the reaction (Table 1, 18g–j, comparing ODN2/ODN4 with ODN3/ODN4). In terms of the catalytic activity, the only difference between ODN2/ODN4 and ODN3/ODN4 is the attachment of the triazolyl linker. Interestingly however, we observed no influence of the groove on the selectivity outcome as only one duplex which happened to position the ligand in the minor groove gave successful results. The structural difference was related to the attachment of the triazolyl linker on ODN2/ODN4, which was longer and more flexible. Structural changes caused by modification of the sugar at the 2′-position have been extensively investigated. In particular, it was shown that 2′-O-modifications, like the ones found in ODN2, deviate the conformational equilibrium of the sugar toward the C3′-endo (North) pucker in order to prevent steric clash between the phosphate backbone and the neighboring nucleobases.19–22 This puckering generates locally an A-form geometry into the helix, causing the distance between two adjacent bases to be reduced, a characteristic of RNA duplexes. Interestingly, 2′-azido groups have also been shown to induce a 3′-endo sugar puckering conformation.23 However, examination of these modifications revealed that the 2′-azido group mainly interacts with the adjacent 3′-phosphate group,23 whereas in the case of 2′-O-modifications, the 2′- and 3′-oxygen atoms as well as the 2′-O-substituents provide a stable cavity which can potentially coordinate to a molecule of water.22,24 We believe that this structural feature combined with the local A-form geometry adopted by the duplex accounts for the high enantioselectivities observed with ODN2/ODN4. Indeed, the ability of the 2′ and 3′ oxygen atoms in the 2′-O-substituted RNA residues to coordinate to a molecule of water, which was demonstrated by Egli and co-workers,22,24 prompted us to propose the hypothesis that the configuration adopted by the ODN2/ODN4 duplex is likely to sequester a molecule of H2O inside the cavity through H-bond interactions with the 2′-O present in ODN2 and the 3′-O of the 3′-phosphate group (Scheme 3). In contrast, this organized coordination of H2O is most probably lacking in both ODN1/ODN4 and ODN3/ODN4. This plausible mechanism by which a molecule of water sequestered in an organized cavity can readily protonate a highly reactive pro-chiral enolate intermediate is likely to differ from the mechanism taking place in the non-covalent approach reported by Roelfes and co-workers.4c This may actually explain why the selectivities obtained in our case are not dependent on the electronics of the indoles.
Table 2 Melting temperatures (Tm) of covalently modified duplexes in the absence (−) and presence (+) of Cu(II) ions
| Entry |
Duplex |
Cu(NO3)2 |
T
m
(°C) |
|
Melting temperatures are obtained from the maxima of the first derivatives of the melting curve (A260 vs. temperature) recorded in a buffer containing 1 M NaCl and 10 mM sodium cacodylate. Curve fits data were averaged from fits of three denaturation curves.
|
| 1 |
ODN4/ODN5 |
− |
56.9 |
| 2 |
ODN1/ODN4 |
− |
52.1 |
| 3 |
ODN1/ODN4 |
+ |
53.2 |
| 4 |
ODN2/ODN4 |
− |
52.1 |
| 5 |
ODN2/ODN4 |
+ |
49.8 |
| 6 |
ODN3/ODN4 |
− |
46.1 |
| 7 |
ODN3/ODN4 |
+ |
44.9 |
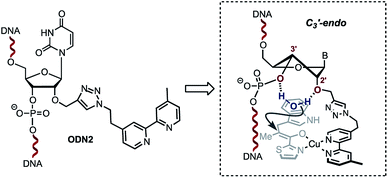 |
| | Scheme 3 Plausible sugar puckering conformation in ODN2. The 2′-modification is likely to favour the C3′-endo conformation positioning a water molecule between the 2′-O and the 3′-phosphate. | |
Conclusions
Through this study, we were able to devise a particularly effective bio-hybrid catalyst capable of achieving unprecedented levels of enantioselectivity in the challenging copper(II)-catalysed asymmetric Friedel–Crafts conjugate addition/enantioselective protonation of α-substituted α,β-unsaturated enones. Interestingly, our 2′-modified bipyridine-containing duplex ODN2/ODN4 proved not only to be highly effective but also highly versatile affording excellent levels of enantioselectivity with minimum substrate dependency thus setting a new benchmark in the field. Most importantly, by closely examining the structure of the duplex and that of the catalytic pocket, we suggested that the high selectivities could be induced by both the formation of a local A-type helix around the metallic co-factor and the sequestration of a molecule of water between the hydroxymethyltriazole arm and the 3′-phosphate which can readily protonate the highly reactive enolate intermediate. Finally, we believe that this study demonstrates the value of the covalent anchorage approach, which allows a fine tuning of the chiral micro-environment around the metallic co-factor and thus allows designing highly selective catalytic systems for specific transformations.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Agence Nationale de la Recherche (D-CYSIV project; ANR-2015-CE29-0021-01) and Queen Mary University of London.
Notes and references
-
(a) F. Schwizer, Y. Okamoto, T. Heinisch, Y. F. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Kohler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142 CrossRef CAS PubMed;
(b) N. Duchemin, I. Heath-Apostolopoulos, M. Smietana and S. Arseniyadis, Org. Biomol. Chem., 2017, 15, 7072 RSC;
(c) I. Drienovska and G. Roelfes, Isr. J. Chem., 2015, 55, 21 CrossRef CAS;
(d) M. Smietana and S. Arseniyadis, Chimia, 2018, 72, 630 CrossRef CAS PubMed.
-
(a) K. Yamamura and E. T. Kaiser, J. Chem. Soc., Chem. Commun., 1976, 830 RSC;
(b) M. E. Wilson and G. M. Whitesides, J. Am. Chem. Soc., 1978, 100, 306 CrossRef CAS.
- G. Roelfes and B. L. Feringa, Angew. Chem., Int. Ed., 2005, 44, 3230 CrossRef CAS.
-
(a) A. J. Boersma, B. L. Feringa and G. Roelfes, Angew. Chem., Int. Ed., 2009, 48, 3346 CrossRef CAS PubMed;
(b) S. Park, K. Ikehata, R. Watabe, Y. Hidaka, A. Rajendran and H. Sugiyama, Chem. Commun., 2012, 48, 10398 RSC;
(c) A. Garcia-Fernandez, R. P. Megens, L. Villarino and G. Roelfes, J. Am. Chem. Soc., 2016, 138, 16308 CrossRef CAS PubMed;
(d) K. Amirbekyan, N. Duchemin, E. Benedetti, R. Joseph, A. Colon, S. A. Markarian, L. Bethge, S. Vonhoff, S. Klussmann, J. Cossy, J. J. Vasseur, S. Arseniyadis and M. Smietana, ACS Catal., 2016, 6, 3096 CrossRef CAS.
-
(a) D. Coquiere, B. L. Feringa and G. Roelfes, Angew. Chem., Int. Ed., 2007, 46, 9308 CrossRef CAS PubMed;
(b) Y. Li, C. Wang, G. Jia, S. Lu and C. Li, Tetrahedron, 2013, 69, 6585 CrossRef CAS.
- A. J. Boersma, D. Coquiere, D. Geerdink, F. Rosati, B. L. Feringa and G. Roelfes, Nat. Chem., 2010, 2, 991 CrossRef CAS PubMed.
-
(a) N. Shibata, H. Yasui, S. Nakamura and T. Toru, Synlett, 2007, 1153 CrossRef CAS;
(b) E. Benedetti, N. Duchemin, L. Bethge, S. Vonhoff, S. Klussmann, J.-J. Vasseur, J. Cossy, M. Smietana and S. Arseniyadis, Chem. Commun., 2015, 51, 6076 RSC;
(c) J. Wang, E. Benedetti, L. Bethge, S. Vonhoff, S. Klussmann, J.-J. Vasseur, J. Cossy, M. Smietana and S. Arseniyadis, Angew. Chem., Int. Ed., 2013, 52, 11546 CrossRef CAS PubMed;
(d) A. J. Boersma, R. P. Megens, B. L. Feringa and G. Roelfes, Chem. Soc. Rev., 2010, 39, 2083 RSC;
(e) S. Park and H. Sugiyama, Molecules, 2012, 17, 12792 CrossRef CAS PubMed;
(f) A. Rioz-Martinez and G. Roelfes, Curr. Opin. Chem. Biol., 2015, 25, 80 CrossRef CAS PubMed;
(g) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752 CrossRef CAS PubMed.
-
(a) U. Jakobsen, K. Rohr and S. Vogel, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 1419 CrossRef CAS PubMed;
(b) L. Ropartz, N. J. Meeuwenoord, G. A. van der Marel, P. van Leeuwen, A. M. Z. Slawin and P. C. J. Kamer, Chem. Commun., 2007, 1556 RSC;
(c) M. Caprioara, R. Fiammengo, M. Engeser and A. Jaschke, Chem.–Eur. J., 2007, 13, 2089 CrossRef CAS PubMed;
(d) P. Fournier, R. Fiammengo and A. Jaeschke, Angew. Chem., Int. Ed., 2009, 48, 4426 CrossRef CAS PubMed;
(e) N. S. Oltra and G. Roelfes, Chem. Commun., 2008, 6039 RSC;
(f) L. Gjonaj and G. Roelfes, ChemCatChem, 2013, 5, 1718 CrossRef CAS;
(g) S. Dey, C. L. Ruhl and A. Jaschke, Chem.–Eur. J., 2017, 23, 12162 CrossRef CAS PubMed;
(h) D. Dey and A. Jaschke, Angew. Chem., Int. Ed., 2015, 54, 11279 CrossRef PubMed;
(i) S. Park, L. Zheng, S. Kumakiri, S. Sakashita, H. Otomo, K. Ikehata and H. Sugiyama, ACS Catal., 2014, 4, 4070 CrossRef CAS.
-
(a) J. Matyasovsky, P. Perlikova, V. Malnuit, R. Pohl and M. Hocek, Angew. Chem., Int. Ed., 2016, 55, 15856 CrossRef CAS PubMed;
(b) M. Kalek, A. S. Madsen and J. Wengel, J. Am. Chem. Soc., 2007, 129, 9392 CrossRef CAS PubMed;
(c) N. K. Andersen, H. Døssing, F. Jensen, B. Vester and P. Nielsen, J. Org. Chem., 2011, 76, 6177 CrossRef CAS PubMed.
-
(a) J. P. Phelan and J. A. Ellman, Beilstein J. Org. Chem., 2016, 12, 1203 CrossRef CAS PubMed;
(b) N. Fu, L. Zhang and S. Luo, Org. Biomol. Chem., 2018, 16, 510 RSC.
- N. K. Fu, L. Zhang, J. Y. Li, S. Z. Luo and J. P. Cheng, Angew. Chem., Int. Ed., 2011, 50, 11451 CrossRef CAS PubMed.
-
(a) P. M. E. Gramlich, C. T. Wirges, A. Manetto and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 8350 CrossRef CAS PubMed;
(b) A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388 RSC.
- A. J. Boersma, J. E. Klijn, B. L. Feringa and G. Roelfes, J. Am. Chem. Soc., 2008, 130, 11783 CrossRef CAS PubMed.
- Z. Lu, T. Ladrak, O. Roubeau, J. van der Toorn, S. J. Teat, C. Massera, P. Gamez and J. Reedijk, Dalton Trans., 2009, 3559 RSC.
- C. Bouillon, A. Meyer, S. Vidal, A. Jochum, Y. Chevolot, J.-P. Cloarec, J.-P. Praly, J.-J. Vasseur and F. Morvan, J. Org. Chem., 2006, 71, 4700 CrossRef CAS PubMed.
-
(a) T. Wada, A. Mochizuki, S. Higashiya, H. Tsuruoka, S. Kawahara, M. Ishikawa and M. Sekine, Tetrahedron Lett., 2001, 42, 9215 CrossRef CAS;
(b) A. M. Jawalekar, N. Meeuwenoord, J. G. O. Cremers, H. S. Overkleeft, G. A. van der Marel, F. P. J. T. Rutjes and F. L. van Delft, J. Org. Chem., 2008, 73, 287 CrossRef CAS PubMed.
-
(a) M. Aigner, M. Hartl, K. Fauster, J. Steger, K. Bister and R. Micura, ChemBioChem, 2011, 12, 47 CrossRef CAS PubMed;
(b) K. Fauster, M. Hartl, T. Santner, M. Aigner, C. Kreutz, K. Bister, E. Ennifar and R. Micura, ACS Chem. Biol., 2012, 7, 581 CrossRef CAS PubMed.
- N. Duchemin, E. Benedetti, L. Bethge, S. Vonhoff, S. Klussmann, J. J. Vasseur, J. Cossy, M. Smietana and S. Arseniyadis, Chem. Commun., 2016, 52, 8604 RSC.
- M. Ikehara, Heterocycles, 1984, 21, 75 CrossRef CAS.
- E. A. Lesnik, C. J. Guinosso, A. M. Kawasaki, H. Sasmor, M. Zounes, L. L. Cummins, D. J. Ecker, P. D. Cook and S. M. Freier, Biochemistry, 1993, 32, 7832 CrossRef CAS PubMed.
- S. Freier, Nucleic Acids Res., 1997, 25, 4429 CrossRef CAS PubMed.
- M. Egli and P. S. Pallan, Chem. Biodiversity, 2010, 7, 60 CrossRef CAS PubMed.
- M. Ikehara, Y. Takatsuka and S. Uesugi, Chem. Pharm. Bull., 1979, 27, 1830 CrossRef CAS.
- V. Tereshko, S. Portmann, E. C. Tay, P. Martin, F. Natt, K.-H. Altmann and M. Egli, Biochemistry, 1998, 37, 10626 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional experimental results. See DOI: 10.1039/c8sc05543b |
| ‡ These authors have contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Stellios
Arseniyadis
a,
Stellios
Arseniyadis