
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA versatile catalyst system for enantioselective synthesis of 2-substituted 1,4-benzodioxanes†
Eugene
Chong
 *a,
Bo
Qu
*a,
Yongda
Zhang
*a,
Zachary P.
Cannone
a,
Joyce C.
Leung
a,
Sergei
Tcyrulnikov
b,
Khoa D.
Nguyen
a,
Nizar
Haddad
a,
Soumik
Biswas
a,
Xiaowen
Hou
a,
Katarzyna
Kaczanowska
c,
Michał
Chwalba
c,
Andrzej
Tracz
c,
Stefan
Czarnocki
c,
Jinhua J.
Song
a,
Marisa C.
Kozlowski
*b and
Chris H.
Senanayake
ad
*a,
Bo
Qu
*a,
Yongda
Zhang
*a,
Zachary P.
Cannone
a,
Joyce C.
Leung
a,
Sergei
Tcyrulnikov
b,
Khoa D.
Nguyen
a,
Nizar
Haddad
a,
Soumik
Biswas
a,
Xiaowen
Hou
a,
Katarzyna
Kaczanowska
c,
Michał
Chwalba
c,
Andrzej
Tracz
c,
Stefan
Czarnocki
c,
Jinhua J.
Song
a,
Marisa C.
Kozlowski
*b and
Chris H.
Senanayake
ad
aChemical Development, Boehringer Ingelheim Pharmaceuticals, Inc., 900 Ridgebury Road, Ridgefield, CT 06877, USA. E-mail: eugene.chong@boehringer-ingelheim.com; bo.qu@boehringer-ingelheim.com; yongda.zhang@boehringer-ingelheim.com
bDepartment of Chemistry, University of Pennsylvania Philadelphia, PA 19104, USA. E-mail: marisa@sas.upenn.edu
cApeiron Synthesis S.A. Wroclaw Technology Park ul, Duńska 9, 54-427 Wrocław, Poland
dAstatech BioPharmaceutical Corporation, 488 Kelin West Road, Wenjiang Dist., Chengdu, Sichuan 611130, P. R. China
First published on 13th March 2019
Abstract
We report the synthesis of enantiomerically enriched 1,4-benzodioxanes containing alkyl, aryl, heteroaryl, and/or carbonyl substituents at the 2-position. The starting 1,4-benzodioxines were readily synthesized via ring closing metathesis using an efficient nitro-Grela catalyst at ppm levels. Excellent enantioselectivities of up to 99:1 er were obtained by using the versatile catalyst system [Ir(cod)Cl]2/BIDIME-dimer in the asymmetric hydrogenation of 2-substituted 1,4-benzodioxines. Furthermore, DFT calculations reveal that the selectivity of the process is controlled by the protonation step; and coordinating groups on the substrate may alter the interaction with the catalyst, resulting in a change in the facial selectivity.
Enantiomerically pure 1,4-benzodioxane derivatives are widely found in biologically active compounds.1 They possess interesting biological activities such as α-adrenergic blocking,2 antigastric,3 spasmolytic,4 antipsychotic,5 anxiolytic,6 and hepatoprotective properties7 (see Fig. 1 for selected examples). As a result, their enantioselective syntheses have attracted great attention in the past decades.
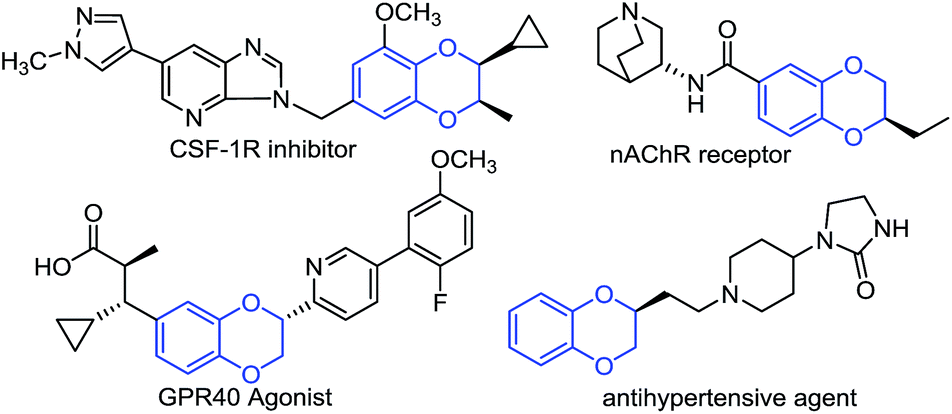 | ||
| Fig. 1 Examples of chiral 1,4-benzodioxane containing molecules. | ||
Whereas early efforts to synthesize enantiomerically enriched 2,3-dihydro-1,4-benzodioxine containing compounds focused primarily on the use of chiral building blocks8 or enzymatic kinetic resolution of carboxylic acids and derivatives,9 recent investigations have increasingly focused on exploiting chiral catalysts. Highly enantioselective processes have been developed including Pd-catalyzed asymmetric intramolecular O-arylative coupling reactions10 and asymmetric intramolecular alkene aryloxyarylation reactions.11 Furthermore, direct asymmetric hydrogenation of the 1,4-benzodioxine derivatives has been realized recently via Ir-catalyzed reduction of 2-aryl-1,4-benzodioxines12 and Rh-catalyzed reduction of 2-carboxyl-1,4-benzodioxines,13 each of which has limited substrate scope. We aimed to discover a catalyst system that is effective and enantioselective for valuable alkyl and heteroaryl substituted products. Herein, we report a versatile [Ir(cod)Cl]2/BIDIME-dimer catalyst system for enantioselective synthesis of a wide range of 2-substituted 1,4-benzodioxanes containing polar directing carbonyl groups as well as largely unfunctionalized groups including alkyls, aryls and heteroaryls. Furthermore, a mechanistic rationale for the reversal of facial selectivity between different substrates is provided based on DFT calculations.
We commenced our studies by preparing the requisite starting materials, 2-substituted 1,4-benzodioxines. We proposed to construct the key 1,4-benzodioxines fragment using a ring closing metathesis (RCM) strategy after alkylation of catechol derivatives (Scheme 1). RCM on substrates containing vinylic ethers is known to be problematic.14 High catalyst loadings of Grubb's second-generation catalyst (5–8 mol%) were reported for the synthesis of 1,4-benzodioxines,15 which hindered the practicality of this otherwise powerful transformation. Furthermore, any coordinating group on the substrate would negatively impact the efficiency of the metathesis reaction. In searching for an effective catalyst, we hypothesized that Grela's nitro-aryl RCM catalysts would facilitate the initial metathesis event due to the labile and less Lewis basic nature of the isopropoxy donor. Gratifyingly, the bismesityl-substituted dichloro nitro-Grela catalyst is very reactive even for ortho-methyl ester substrate 3b. As a result, compounds 4a and 4b were prepared using 150 or 300 ppm of the catalyst in >80% yields (Scheme 1).
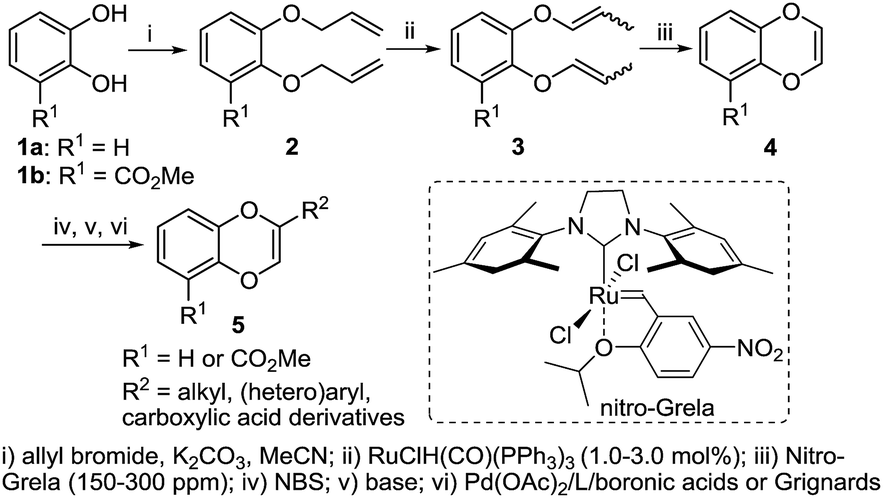 | ||
| Scheme 1 Synthesis of 1,4-benzodioxines. | ||
With a highly efficient synthesis of the 2-substituted 1,4-benzodioxines in hand, enantioselective reduction was tested. The initial focus was on the asymmetric hydrogenation of 2-cyclopropyl-substituted substrate 5a (Table 1). Given that a general, highly enantioselective method for 2-alkyl-substituted 1,4-benzodioxanes has not been reported to date, we theorized that the small size of the cyclopropyl substituent would be a good test case for this purpose. More than 20 commercially available bidentate phosphine ligands were screened16 (representative examples are shown in Table 1, entries 1–8); however, all of the screened ligands provided moderate enantioselectivities (<90:10 er), and the reaction did not reach full conversion even with addition of acetic acid (AcOH), which was reported to increase the reactivity of the catalyst for iridium-catalyzed hydrogenation.17 We hypothesized that the chiral bis-dihydrobenzooxaphosphole (BIBOP) type ligands (L1–L5) could be used to advantage for this transformation, as they have the potential to provide a deep chiral pocket as well as steric tunability for enantioinduction.19 By increasing the size of the substituent on the BIBOP ligands from hydrogen to aryl, a general increase in enantioselectivity and conversion was observed (Table 1, entries 9–13). Notably, 9-anthracenyl-substituted ligand L4 (WingPhos) provided 95:5 er; and 2,6-dimethoxyphenyl-substituted ligand L5 (BIDIME-dimer) provided the highest enantioselectivity (97:3 er). Importantly, side products from cyclopropyl ring opening or ester hydrolysis were not observed under the reaction conditions and product 6a was isolated in 98% yield using the BIDIME-dimer ligand.
|
|
|||
|---|---|---|---|
| Entry | Ligand | Convb (%) | erc |
| a Reaction conditions:185a (0.1 mmol), [Ir(cod)Cl]2 (0.001 mmol), ligand (0.003 mmol), THF (0.2 mL), MeOH (0.2 mL), AcOH (4 mmol), H2 (600 psi), 50 °C for 24 h. b Conversion was determined by HPLC. c Enantiomeric ratio was determined by chiral SFC. d Isolated yield. | |||
| 1 | (R)-Ph-Garphos | 79 | 76:24 |
| 2 | (R)-3,5-tBu-MeOBIPHEP | 93 | 81:19 |
| 3 | (S)-SEGPHOS | 67 | 16:84 |
| 4 | (R)-BINAP | 92 | 81:19 |
| 5 | (R)-C3-TunePhos | 83 | 77:23 |
| 6 | (S)-Phanephos | 91 | 88:12 |
| 7 | (1R,1′R,2S,2′S)-DuanPhos | 63 | 41:59 |
| 8 | (R)-Josiphos SL-J009-1 | 75 | 56:44 |
| 9 | L1 | 68 | 47:53 |
| 10 | L2 | 94 | 59:41 |
| 11 | L3 | >98 | 66:34 |
| 12 | L4 | >98 | 95:5 |
| 13 | L5 | >98 (98)d | 97:3 |
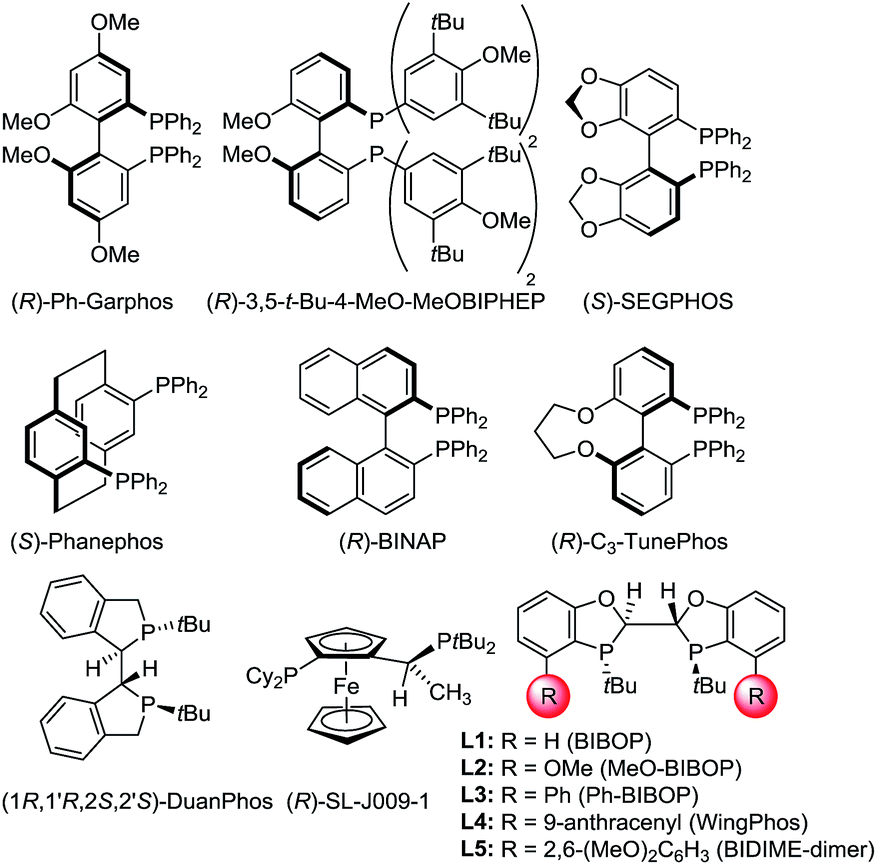
|
|||
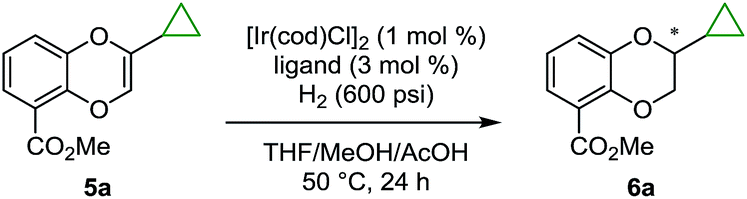
Having identified BIDIME-dimer as the optimal ligand, other 2-alkyl-substituted 1,4-benzodioxines were examined in the Ir-catalyzed asymmetric hydrogenation reaction (Scheme 2). The cyclopropyl analogue without the methyl ester substituent at the 5-position was hydrogenated to afford 6b with comparable enantioselectivity (94:6 er). Other cycloalkyl substituents ranging from 4- to 7-membered-rings also performed well in the reaction to give the corresponding 1,4-benzodioxanes (6c–6e) with very good enantioselectivities. Additionally, 2-aryl-substituted 1,4-benzodioxanes 6f and 6g were synthesized with excellent enantioselectivities of 98:2 er and >99:1 er, respectively. The synthesis of 6f (98:2 er) is a significant improvement over the recently reported IrBARF/In-BiphPHOX system (91.5:8.5 er) (BARF = tetrakis(3,5-bis(trifluoromethyl) phenyl)borate).12 Furthermore, gram scale reduction of 5f was demonstrated with 0.1 mol% [Ir(cod)Cl]2. Quantitative yield (98%) and the same enantiomeric ratio (98:2) were obtained. Even for the challenging ethyl substituted 6h, much higher enantioselectivity was obtained (90:10 er vs. 74:26 er12).
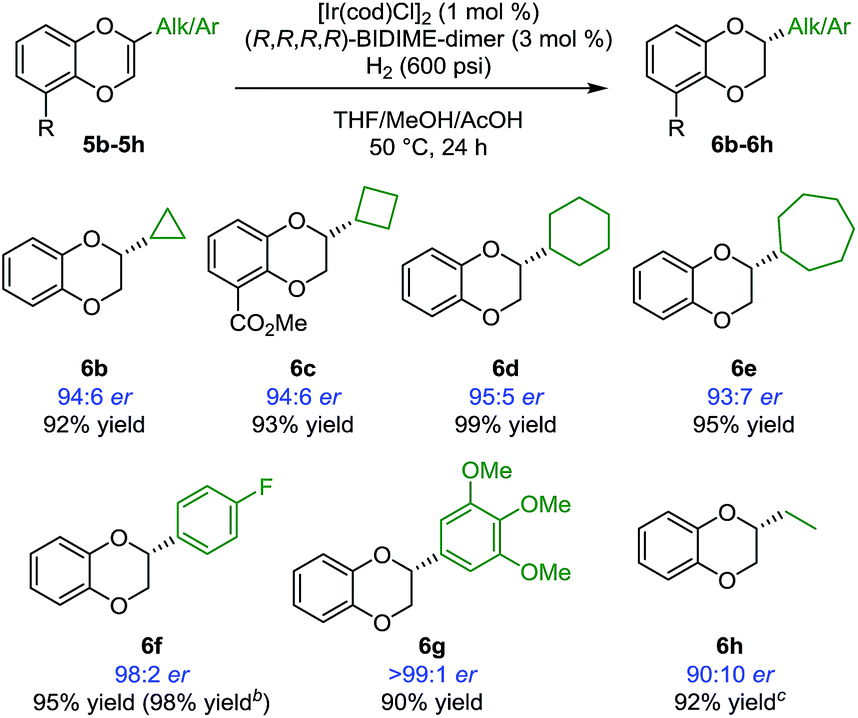 | ||
| Scheme 2 Asymmetric hydrogenation of 2-alkyl and 2-aryl-substituted 1,4-benzodioxinesa. aReaction conditions: 5 (0.2 mmol), [Ire(cod)Cl]2 (0.002 mmol), (R,R,R,R)-BIDIME-dimer L5 (0.006 mmol), THF (0.3 mL), MeOH (0.3 mL), AcOH (8 mmol, 40 equiv.), H2 (600 psi), 50 °C for 24 h. Isolated yield. Enantiomeric ratio was determined by chiral SFC or HPLC. b5f (5.0 mmol, 1.14 g), [Ir(cod)Cl]2 (0.005 mmol), L5 (0.015 mmol), THF (6 mL), MeOH (6 mL), AcOH (150 mmol, 30 equiv.), H2 (600 psi), 70 °C for 24 h. c25 °C. | ||
In most reductions of largely unfunctionalized alkenes, non-coordinating anions such as BARF are needed to enhance the coordination of the iridium catalyst to the substrate. As a result, nitrogen-containing heterocycles are generally not compatible with Ir-BARF catalyst systems.12,20 With the excellent enantioselectivities observed in our initial studies, we envisioned that [Ir(cod)Cl]2/BIDIME-dimer catalyst system would allow selective alkene coordination vs. heterocycle coordination due to the lower Lewis acidity of the iridium and the deep binding pocket. If so, a number of products containing heterocyclic substituents of pharmaceutical relevance would be accessible in an expeditious manner. Indeed, a variety of benzodioxines with heteroaryl substituents were reduced under the reaction conditions (Scheme 3). 1,3-Benzodioxoles 6i and 6j were obtained in 98:2 er and 94:6 er, respectively. Benzothiophene 6k was generated in 99:1 er. Excellent enantioselectivities (>99:1 er) were obtained with more sterically demanding dibenzofuran (6l) and dibenzothiophene (6m) substituents. Importantly, many 1,4-benzodioxanes containing N-heterocyclic substituents at the 2-position (6n–6x) can be prepared with consistently excellent enantioselectivities (≥98:2 er). These cases include heterocycles containing two adjacent heteroatoms such as pyrazole (6n), indazole (6o), and isoxazole (6p). Furthermore, a variety of substrates containing pyridine functionality (5q–5v) underwent asymmetric hydrogenation successfully. Substrates containing challenging pyrimidine derivatives (6w and 6x) also performed well in the reaction.
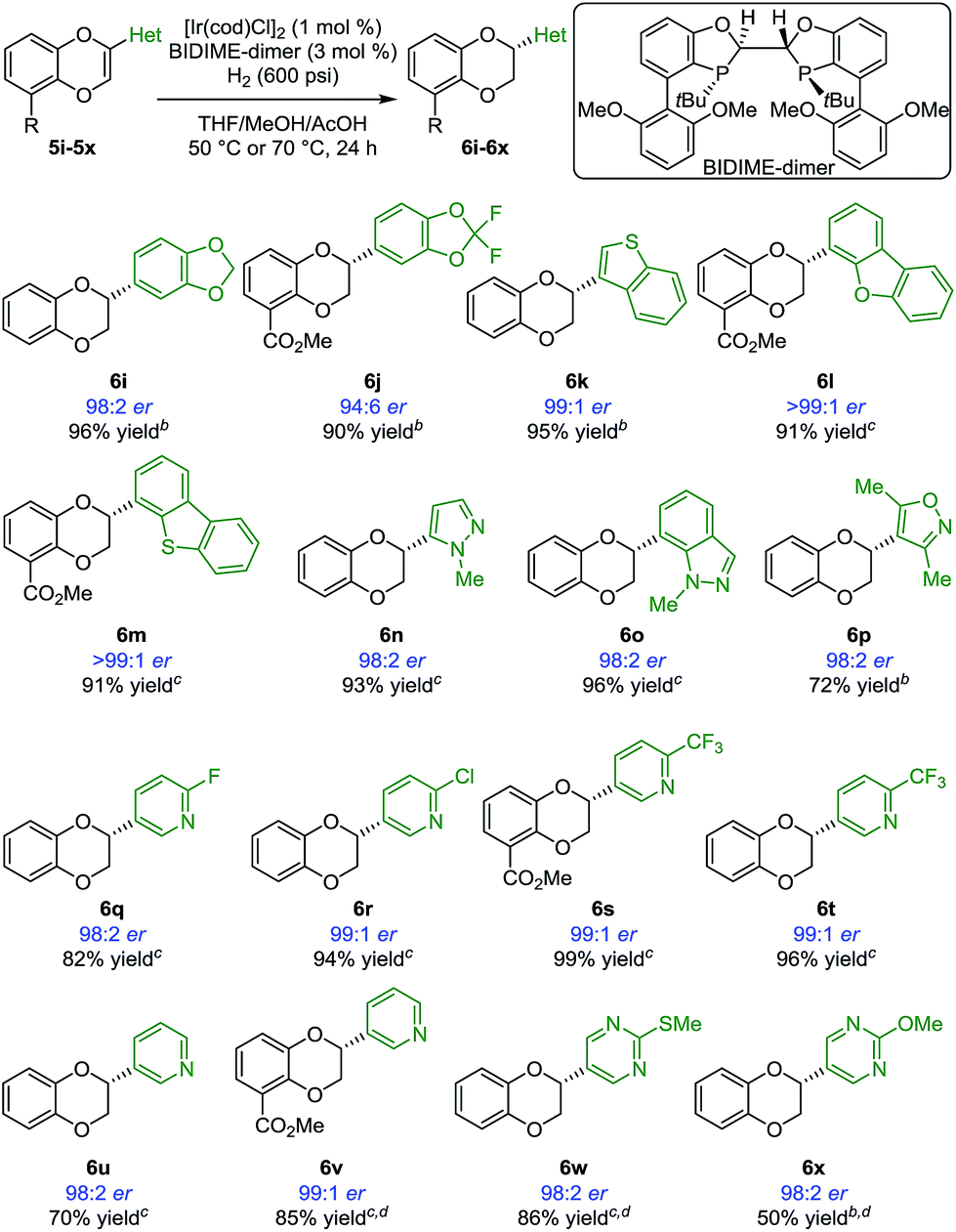 | ||
| Scheme 3 Scope of Ir-catalyzed asymmetric hydrogenation of 2-heterocycle-substituted 1,4-benzodioxinesa. aUnless noted, standard reaction conditions: substrate 5 (0.2 mmol), [Ir(cod)Cl]2 (0.002 mmol), (R,R,R,R)-BIDIME-dimer L5 (0.006 mmol), THF (0.3 mL), MeOH (0.3 mL), AcOH (8 mmol), H2 (600 psi), 50 or 70 °C for 24 h. Isolated yield. Enantiomeric ratio was determined by chiral SFC or HPLC. b50 °C. c70 °C. d[Ir(cod)Cl]2 (2 mol%), BIDIME-dimer L5 (6 mol%). | ||
In addition to heterocyclic substituents, the [Ir(cod)Cl]2/BIDIME-dimer catalyst system is able to furnish chiral 1,4-benzodioxanes containing polar coordinating carbonyl derivatives21 (Scheme 4). Excellent enantioselectivity of >99:1 er was obtained for the double methyl ester containing compound 6y. 2-Methyl ester containing substrate 6z was obtained in 95:5 er. A variety of amide derivatives incorporating pyrrolidine 6aa (93:7 er), morpholine 6ab (98:2 er), piperidine 6ac (97:3 er), piperazine 6ad (98:2 er), and tetrahydroisoquinoline 6ae (97:3 er) were also generated with high enantioselectivity. Overall, these results establish the broad applicability of the IrCl/BIDIME-dimer catalyst system for enantioselective synthesis of 2-substituted 1,4-benzodioxanes with a range of both coordinating and non-coordinating substituents. Interestingly, the opposite chirality was observed when a polar carbonyl group was incorporated at the 2-position. To understand this unexpected result and gain insight into mechanism of stereoinduction, we initiated a computational study of this transformation.
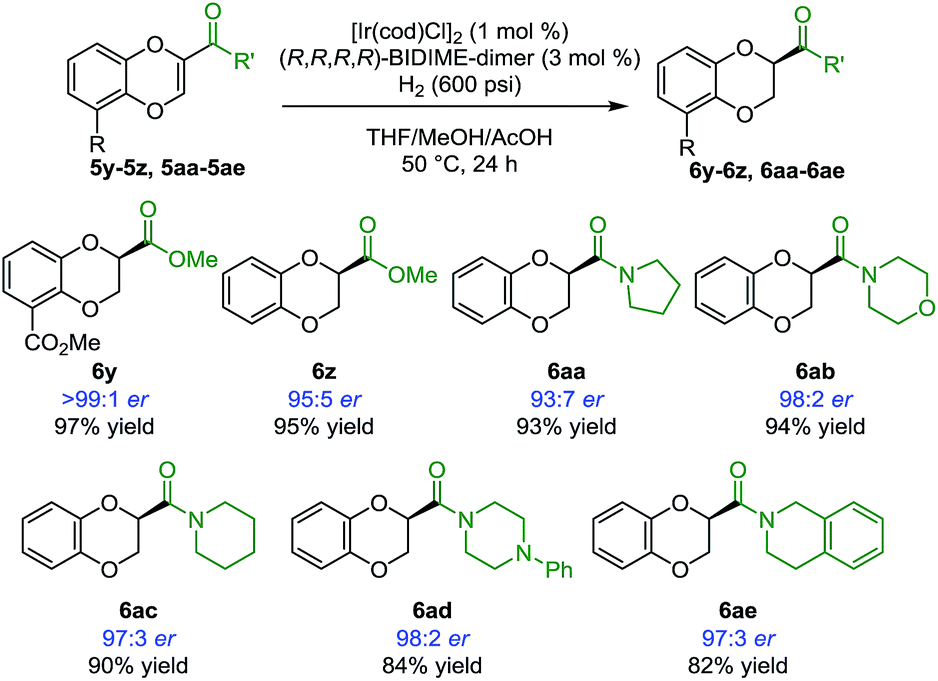 | ||
| Scheme 4 Scope of Ir-catalyzed asymmetric hydrogenation of 1,4-benzodioxinesa. a Reaction conditions: 5 (0.2 moll), [Ir(cod)Cl]2 (0.002 mmol), (R,R,R,R)-BIDIME-dimer L5 (0.006 mmol), THF (0.3 mL), MeOH (0.3 mL), AcOH (8 mmol), H2 (600 psi), 50 °C for 24 h. Isolated yield. Enantiomeric ratio was determined by chiral SFC or HPLC. | ||
Based on the previous studies of related Ir-catalyzed hydrogenations, reduction was expected to occur by sequential protonation-hydride transfer mechanism (Fig. 2).22
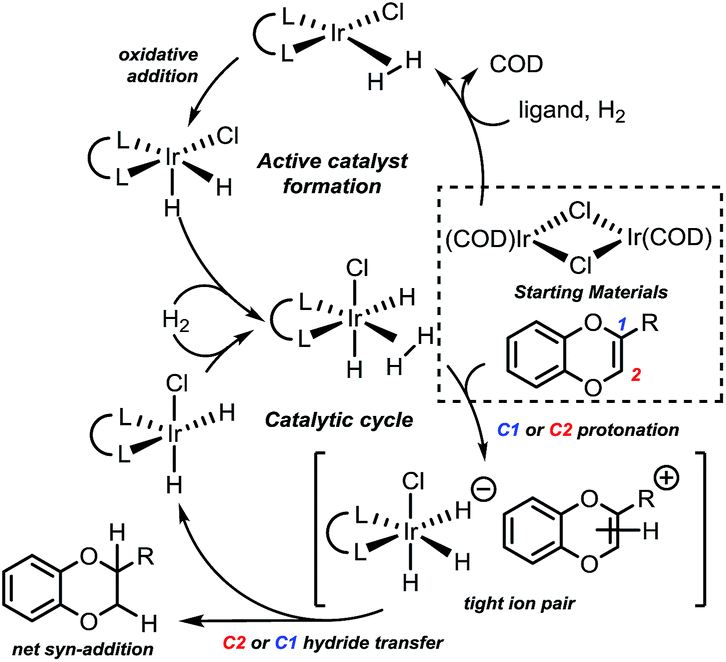 | ||
| Fig. 2 Proposed hydrogenation mechanism. See ESI† for the computational analysis of the active catalyst formation. Computational study performed for R = Me and R = CO2Me. | ||
In such a mechanism, protonation or hydride transfer can be enantiodetermining. Calculations of this system indicate that the energy of the separated ion pair is much higher than that of the protonation transition state (by ∼25 kcal mol−1) or the subsequent hydride transfer transition state. Since transfer of the iridium hydride to the opposite face via the separated ion pair is not favorable energetically, the calculations support the Ir catalyst establishing a facial preference during the protonation stage. To understand the origin of this facile preference and the enantioselectivity observed, we further analyzed the protonation step computationally. We found that protonation of both alkyl and carboxymethyl substituted benzodioxines proceeds as a Markovnikov process (C2 protonation on Fig. 2. See ESI† for details). We confirmed this regioselectivity for the reaction on both faces of the benzodioxine. This feature of the mechanism allows us to use the same model for both electron-poor and electron-rich R substituents.
As the reaction takes place in a polar medium, ionization of or loss of dihydrogen from the Ir complexes is highly likely23 (Fig. 3). For the non-coordinating substrates, we propose protonation via a ‘mode A’ mechanism, where substrate reacts with a neutral or cationic Ir complex. Both neutral and cationic mode A mechanisms provide very similar ligand–substrate interactions that determine the stereochemical outcome of the reaction, allowing us to consider only one of these mechanisms in our study.
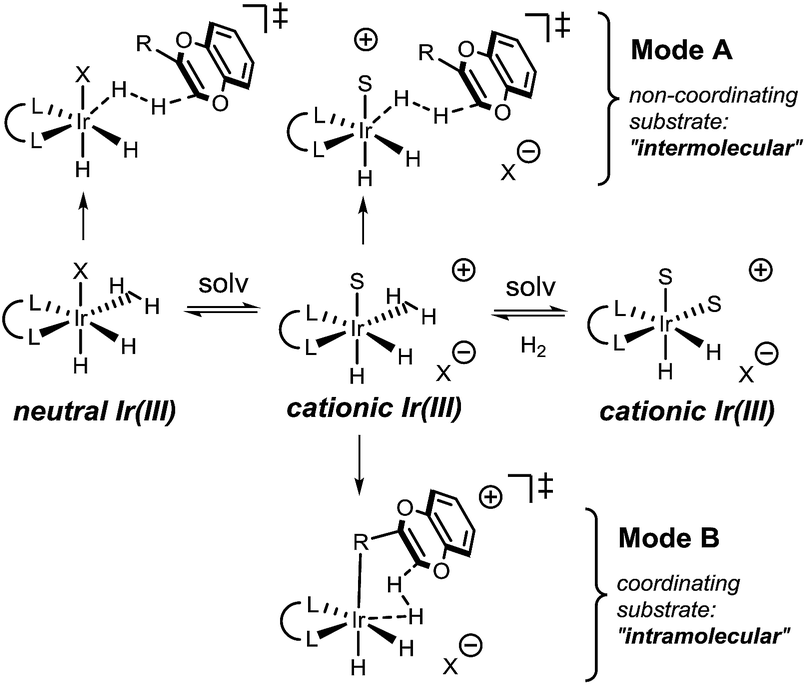 | ||
| Fig. 3 Possible solvation equilibrium and proposed protonation transition states for coordinating and non-coordinating substrates. | ||
For the coordinating substrate we envision an intramolecular process where the substrate coordinates the metal center prior to protonation (mode B). Such an arrangement will cause the subsequent reaction with dihydrogen to be more entropically favorable than in mode A. We propose that different arrangements of the mode A and mode B transition states alter the selectivity determining ligand–substrate interactions, thereby affecting the facial preference of the protonation. To test this hypothesis, the reactions of methyl and carboxymethyl substituted benzodioxines were analyzed. Computational analysis of the mode A-protonation of the methyl substrate indicated that reaction takes place preferentially on the face A of the starting material (see Fig. 4).
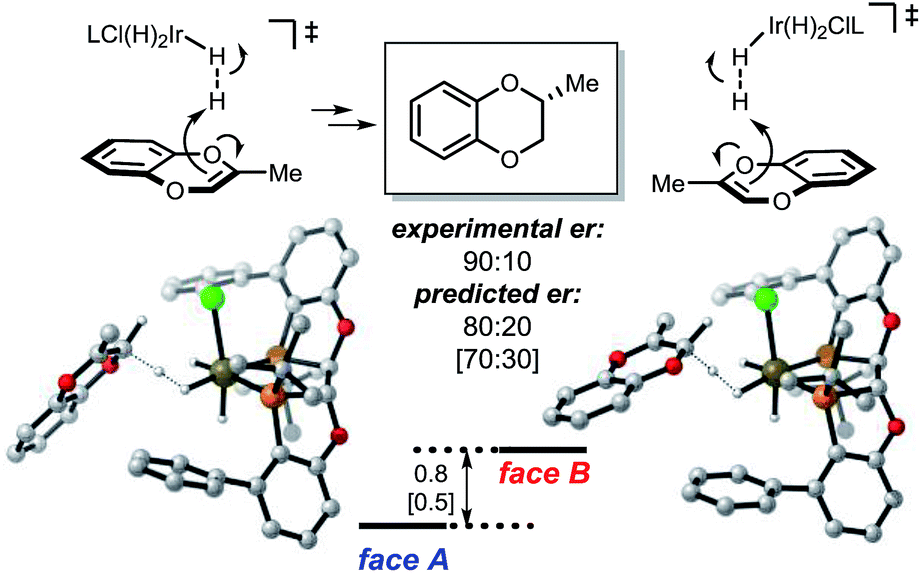 | ||
| Fig. 4 Energetics of the mode A protonation of the Me-substituted benzodioxine. Selectivity-determining conformations are shown. Free energy and enthalpy (in brackets) gaps are calculated using PBE-D2/6-311+G(d,p), Ir:LANL2DZ(f), IEFPCM-MeOH//PBE/6-31G(d), Ir:LANL2DZ.24 Values are in kcal mol−1. Ligand L3 was used for the calculation to minimize conformational isomers; the experimental value was taken from 6h. | ||
In contrast, the protonation of the methoxycarbonyl substituted benzodioxine via mode B favors proton delivery to face B, resulting in the opposite selectivity (Fig. 5).
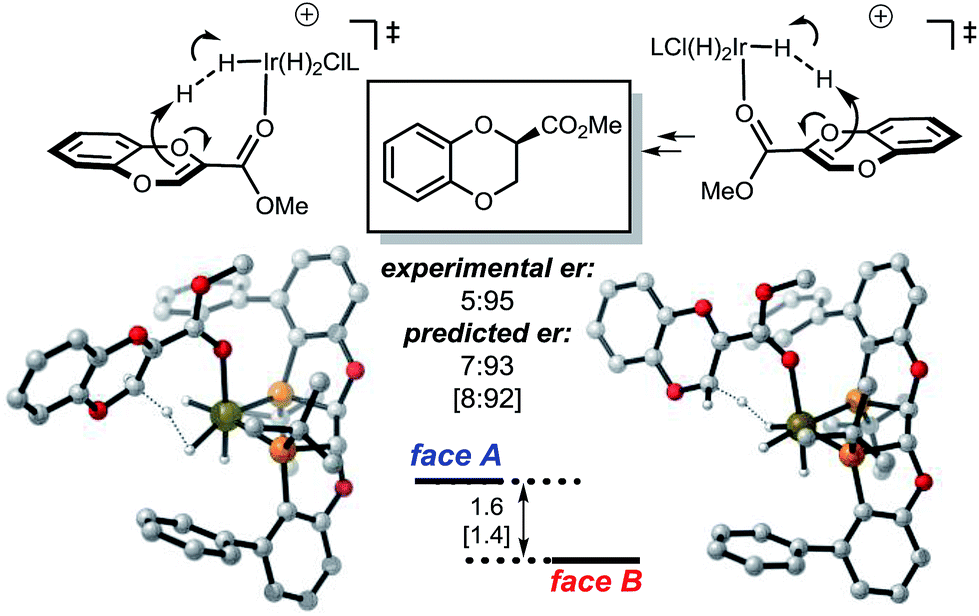 | ||
| Fig. 5 Energetics of the mode B protonation of the CO2Me-substituted benzodioxine. Selectivity-determining conformations are shown. Free energy and enthalpy (in brackets) gaps are calculated using PBE-D2/6-311+G(d,p), Ir:LANL2DZ(f), IEFPCM-MeOH//PBE/6-31G(d), Ir:LANL2DZ. Values are in kcal mol−1. Ligand L3 was used for the calculation to minimize conformational isomers; the experimental value was taken from 6z. | ||
The selectivity of the methyl substrate appears to be dictated by an unfavorable steric interaction between the substrate and the aryl portion of the ligand (Fig. 6, mode A). The computationally predicted selectivity is in good agreement with experimental results (Fig. 4, computed 80:20 vs. experimental 90:10). In contrast, control of the stereoselectivity with the methyl ester is provided by the stabilizing electronic interactions (Fig. 6, mode B). Computationally predicted values for enantiomeric ratio of this substrate are also in very good agreement with experimentally obtained result (Fig. 5, computed 7:93 vs. experimental 5:95).
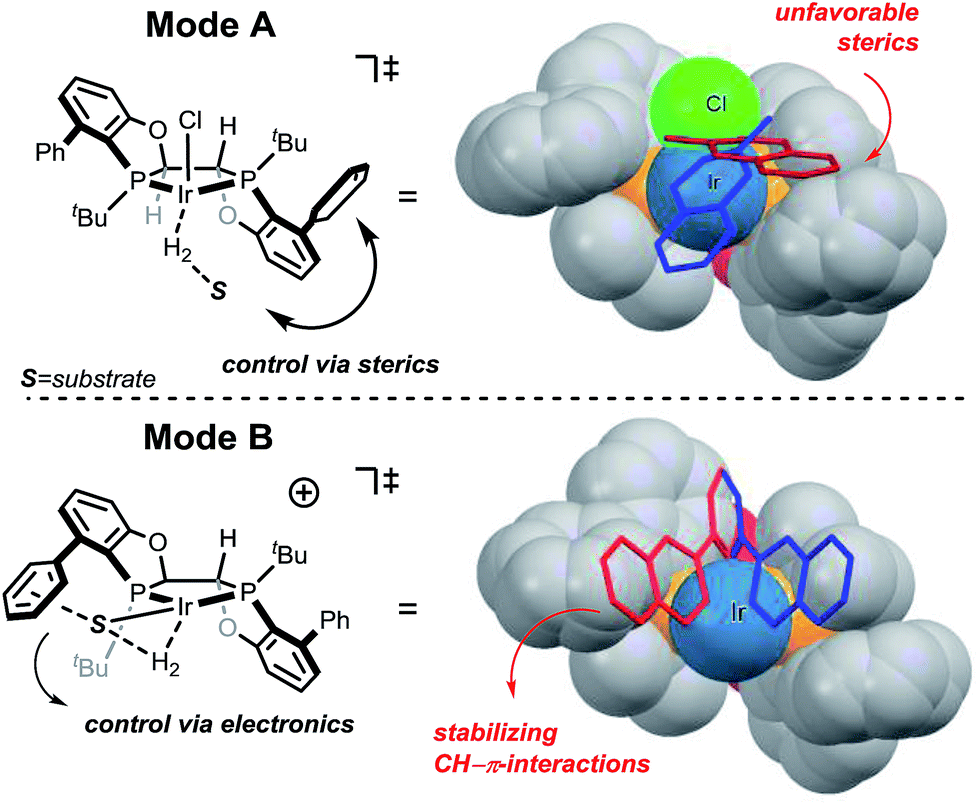 | ||
| Fig. 6 Stereochemical models for mode A and mode B protonations. Substrate approaches for face A and face B attacks are shown in blue and red respectively. | ||
The selectivity basis for the mode B approach is outlined in greater detail in Fig. 7. From the transition state geometry, the structural motif contributing to stabilization is a combination of slipped overlapping π–π and sp2CH–π interactions,25 which are reported to be relatively strong (∼2.5 kcal mol−1).26 In our case, imperfect positioning of the CH unit relative to the aromatic system (3.13 Å vs. typical 2.73 Å and 136° vs. 148° for typical interactions)27 results in an overall weaker energetic effect (1.5 kcal mol−1 in this case). This interaction is absent in the transition state arising from the other facial approach (face A in Fig. 5). Even though this electronic interaction is not optimal, the resultant stabilization is sufficient to drive the differentiation of these diastereomeric transition states leading to high selectivity.
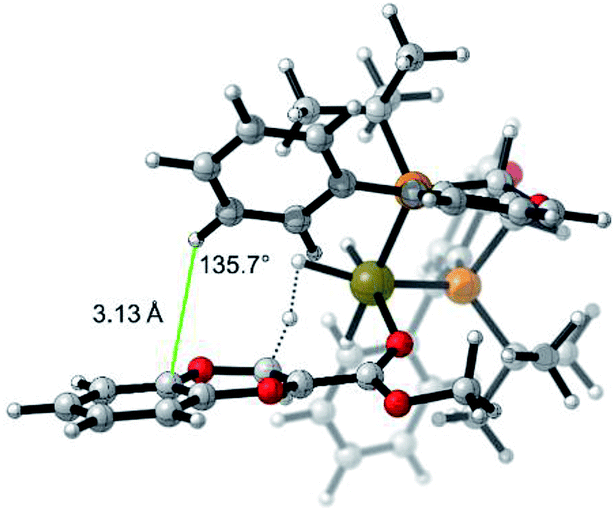 | ||
| Fig. 7 Model of lowest energy mode B protonation for the 2-methoxycarbonyl-1,4-benzodioxine showing electronic stabilizing interactions (π–π and sp2CH–π interactions).25 | ||
In summary, we report the discovery of a highly enantioselective (>99:1 er) and versatile IrCl/BIDIME-dimer catalyst system for asymmetric hydrogenation reactions to prepare a wide range of chiral 1,4-benzodioxanes including those with alkyl, aryl, heteroaryl, and carbonyl groups at the 2-position. The route commences with a key RCM step to construct the ring of the requisite 1,4-benzodioxines with ppm level catalyst loadings using the nitro-Grela catalyst. Overall, this route represents a general and atom-economical catalytic approach to construct synthetically challenging chiral benzodioxanes. Computational studies suggest that enantioselectivity of the hydrogenation process is controlled by an outer sphere protonation step. Depending on the nature of the substrate, however, coordination to the metal center can intervene creating an inner sphere approach in the corresponding transition states. This specific coordination is a powerful way of altering the spatial orientation of the substrate during the protonation, providing the means to affect the absolute and relative stereochemical outcome of the reaction.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the NIH (GM087605 M. C. K.) and Boehringer Ingelheim Pharmaceuticals for financial support. Computational support was provided by XSEDE (TG-CHE120052). We also thank Dr. Fenghe Qiu, Scott Pennino and Keith McKellop for HRMS analyses and Dr. Dongyue Xin for structural confirmation of 6ae.Notes and references
- (a) L. I. Pilkington and D. Barker, Nat. Prod. Rep., 2015, 32, 1369 RSC; (b) A. Abeywardane; M. J. Burke; T. M. Kirrane; M. R. Netherton; A. K. Padyana; L. L. Smith Keenan; H. Takahashi; M. R. Turner; Q. Zhang; Q. Zhang, Benzodioxane Inhibitors of Leukotrien Production, International Patent WO 2012125598 A1, September 9, 2012; (c) R. Zhou, G. Luo and A. G. Ewing, J. Neurosci., 1994, 14, 2402 CrossRef CAS PubMed; (d) A. Arnoldi, A. Bassoli, L. Merlini and E. Ragg, J. Chem. Soc., Perkin Trans. 1, 1993, 1359 RSC; (e) H. Hikino, Y. Kiso, H. Wagner and M. Fiebig, Planta Med., 1984, 50, 248 CrossRef CAS PubMed; (f) L.-G. Zhuang, O. Seligmann, K. Jurcic and H. Wagner, Planta Med., 1982, 45, 172 CrossRef CAS.
- S. D. Patel, W. M. Habeski, H. Min, J. S. Zhang, R. Roof, B. Snyder, G. Bora, B. Campbell, C. Li, D. Hidayetoglu, D. S. Johnson, A. Chaudhry, M. E. Charlton and N. M. Kablaoui, Bioorg. Med. Chem. Lett., 2008, 18, 5689 CrossRef CAS PubMed.
- T. Tomiyama, S. Wakabayashi and M. Yokota, J. Med. Chem., 1989, 32, 1988 CrossRef CAS PubMed.
- R. Ertan and H. Göker, FABAD J. Pharm. Sci., 1987, 12, 152 CAS.
- M. F. Hibert, M. W. Gittos, D. N. Middlemiss, A. K. Mir and J. R. Fozard, J. Med. Chem., 1988, 31, 1087 CrossRef CAS PubMed.
- A. K. Mir, M. Hilbert, M. D. Trickleband, D. N. Middlemiss, E. J. Kidd and J. R. Fozard, Eur. J. Pharmacol., 1988, 149, 107 CrossRef CAS PubMed.
- G. Vogel, W. Trost, R. Braatz, K. P. Odenthal, G. Brusewitz, H. Antweiler and R. Seeger, Arzneim.-Forsch., 1975, 25, 179 CAS.
- (a) A. Rouf, M. A. Aga, B. Kumar and S. C. Taneja, Tetrahedron Lett., 2013, 54, 6420 CrossRef CAS; (b) S. I. Kuwabe, K. E. Torraca and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 12202 CrossRef CAS PubMed; (c) E. Valoti, M. Pallavicini, L. Villa and D. Pezzetta, J. Org. Chem., 2001, 66, 1018 CrossRef CAS PubMed; (d) W. L. Nelson and J. E. Wennerstrom, J. Chem. Soc., Chem. Commun., 1976, 921 RSC.
- (a) A. Rouf, P. Gupta, M. A. Aga, B. Kumar, A. Chaubey, R. Parshad and S. C. Taneja, Tetrahedron: Asymmetry, 2012, 23, 1615 CrossRef CAS; (b) S. M. Kasture, R. Varma, U. K. Kalkote, S. Nene and B. D. Kulkarni, Biochem. Eng. J., 2005, 27, 66 CrossRef CAS; (c) T. Sakai, K. Hayashi, F. Yano, M. Takami, M. Ino, T. Korenaga and T. Ema, Bull. Chem. Soc. Jpn., 2003, 76, 1441 CrossRef CAS; (d) S. Antus, A. Gottsegen, J. Kajtar, T. Kovacs, T. S. Toth and H. Wagner, Tetrahedron: Asymmetry, 1993, 4, 339 CrossRef CAS.
- (a) J. Shi, T. Wang, Y. Huang, X. Zhang, Y. Wu and Q. Cai, Org. Lett., 2015, 17, 840 CrossRef CAS PubMed; (b) W. Yang, J. Yan, Y. Long, S. Zhang, J. Liu, Y. Zeng and Q. Cai, Org. Lett., 2013, 15, 6022 CrossRef CAS PubMed.
- N. Hu, K. Li, Z. Wang and W. Tang, Angew. Chem., Int. Ed., 2016, 55, 5044 CrossRef CAS PubMed.
- Y. Wang, J. Xia, G. Yang and W. Zhang, Tetrahedron, 2018, 74, 477 CrossRef CAS.
- X. Yin, Y. Huang, Z. Chen, Y. Hu, L. Tao, Q. Zhao, X.-Q. Dong and X. Zhang, Org. Lett., 2018, 20, 4173 CrossRef CAS PubMed.
- A. K. Chatterjee, J. P. Morgan, M. Scholl and R. H. Grubbs, J. Am. Chem. Soc., 2000, 122, 3783 CrossRef CAS.
- G. L. Morgans, E. L. Ngidi, L. G. Madeley, S. D. Khanye, J. P. Michael, C. B. de Koning and W. A. L. van Otterlo, Tetrahedron, 2009, 65, 10650 CrossRef CAS.
- See the ESI.†.
- (a) B. Hans-Ulrich, Adv. Synth. Catal., 2002, 344, 17 CrossRef; (b) K. Makino, Y. Hiroki and Y. Hamada, J. Am. Chem. Soc., 2005, 127, 5784 CrossRef CAS PubMed.
- Solvent mixture of MeOH:THF (1:1) was selected for all the substrates for solubility enhancement. AcOH was applied at 30–40 equiv to the starting material for increased catalyst activity. For substrate 5a, a methoxy incorporation impurity was generated when the reaction was run in the absence of AcOH, see the ESI† for details..
- (a) G. Liu, X. Liu, Z. Cai, G. Jiao, G. Xu and W. Tang, Angew. Chem., Int. Ed., 2013, 52, 4235 CrossRef CAS PubMed; (b) R. S. Luo, K. Li, Y. Hu and W. Tang, Adv. Synth. Catal., 2013, 355, 1297 CrossRef CAS; (c) W. Tang, B. Qu, A. G. Capacci, S. Rodriguez, X. Wei, N. Haddad, B. Narayanan, S. Ma, N. Grinberg, N. K. Yee, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2010, 12, 176 CrossRef CAS PubMed.
- B. Qu, L. P. Samankumara, S. Ma, K. R. Fandrick, J.-N. Desrosiers, S. Rodriguez, Z. Li, N. Haddad, Z. S. Han, K. McKellop, S. Pennino, N. Grinberg, N. C. Gonnella, J. J. Song and C. H. Senanayake, Angew. Chem., Int. Ed., 2014, 53, 14428 CrossRef CAS PubMed.
- During preparation of this manuscript, hydrogenation of the similar substrates was described by Zhang's group using rhodium catalysis: see ref. 13.
- (a) B. Qu, H. P. R. Mangunuru, S. Tcyrulnikov, D. Rivalti, O. V. Zatolochnaya, D. Kurouski, S. Radomkit, S. Biswas, S. Karyakarte, K. R. Fandrick, J. D. Sieber, S. Rodriguez, J.-N. Desrosiers, N. Haddad, K. McKellop, S. Pennino, H. Lee, N. K. Yee, J. J. Song, M. C. Kozlowski and C. H. Senanayake, Org. Lett., 2018, 20, 1333 CrossRef CAS PubMed; (b) X. Wei, B. Qu, X. Zeng, J. Savoie, K. R. Fandrick, J.-N. Desrosiers, S. Tcyrulnikov, M. A. Marsini, F. G. Buono, Z. Li, B.-S. Yang, W. Tang, N. Haddad, O. Gutierrez, J. Wang, H. Lee, S. Ma, S. Campbell, J. C. Lorenz, M. Eckhardt, F. Himmelsbach, S. Peters, N. D. Patel, Z. Tan, N. K. Yee, J. J. Song, F. Roschangar, M. C. Kozlowski and C. H. Senanayake, J. Am. Chem. Soc., 2016, 138, 15473 CrossRef CAS PubMed; (c) H. Zhou, Z. Li, Z. Wang, T. Wang, L. Xu, Y. He, Q.-H. Fan, J. Pan, L. Gu and A. S. C. Chan, Angew. Chem., Int. Ed., 2008, 47, 8464 CrossRef CAS PubMed; (d) K. H. Hopmann and A. Bayer, Organometallics, 2011, 30, 2483 CrossRef CAS.
- (a) M.-L. Li, S. Yang, X.-C. Su, H.-L. Wu, L.-L. Yang, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 541 CrossRef CAS PubMed; (b) G. E. Dobereiner, A. Nova, N. D. Schley, N. Hazari, S. J. Miller, O. Eisenstein and R. H. Crabtree, J. Am. Chem. Soc., 2011, 133, 7547 CrossRef CAS PubMed.
- K. H. Hopmann, Organometallics, 2016, 35, 3795 CrossRef CAS.
- S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2002, 124, 104 CrossRef CAS PubMed.
- H. Sun, U. K. Tottempudi, J. D. Mottishaw, P. N. Basa, A. Putta and A. G. Sykes, Cryst. Growth Des., 2012, 12, 5655 CrossRef CAS.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05612a |
| This journal is © The Royal Society of Chemistry 2019 |