
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed 1,4-alkylarylation of 1,3-enynes with masked alkyl electrophiles†
Changqing
Ye
ab,
Yajun
Li
 a,
Xiaotao
Zhu
a,
Shengmin
Hu
a,
Daqiang
Yuan
a and
Hongli
Bao
*ab
a,
Xiaotao
Zhu
a,
Shengmin
Hu
a,
Daqiang
Yuan
a and
Hongli
Bao
*ab
aKey Laboratory of Coal to Ethylene Glycol and Its Related Technology, State Key Laboratory of Structural Chemistry, Center for Excellence in Molecular Synthesis, Fujian Institute of Research on the Structure of Matter, 155 Yangqiao Road West, Fuzhou, Fujian 350002, P. R. China
bUniversity of Chinese Academy of Sciences, No. 19(A) Yuquan Road, Shijingshan District, Beijing 100049, P. R. China. E-mail: hlbao@fjirsm.ac.cn
First published on 19th February 2019
Abstract
Classical 1,4-dicarbofunctionalization of 1,3-enynes employs organometallic reagents as nucleophiles to initiate the reaction. We report a copper-catalyzed 1,4-alkylarylation of 1,3-enynes with alkyl diacyl peroxides as masked alkyl electrophiles and aryl boronic acids as nucleophiles, selectively affording structurally diversified tetrasubstituted allenes under mild conditions. Mechanistic studies suggest that an allenyl radical might be involved.
Allenes are important structural motifs found in natural products and are key intermediates in the synthesis of complex and bioactive molecules.1 Useful methods1d,2 for the synthesis of allenes include molecular rearrangement,3 nucleophilic substitution,4 1,4-addition5 and other methods.6 Among these methods, 1,4-difunctionalization of 1,3-enynes is particularly appealing because it enables two functionalities to be incorporated simultaneously. Regioselective introduction of an alkyl and an aryl group into allene moieties is of great interest as it can significantly diversify the structures of the resulting allenes. There is however only one example of 1,4-alkylarylation of 1,3-enynes. In this reaction, developed by Ma et al. (Fig. 1a),7 alkyl lithium reagents were used as nucleophiles to initiate the reaction, followed by Li–Zn exchange and cross coupling with aryl halides serving as the electrophiles. Allenyl anions were proposed as the intermediates. Using alkyl-metallic reagents, groups led by Kambe,8 Yoshida,9 and Kimura5e have developed efficient 1,4-dicarbofunctionalizations of 1,3-enynes with other electrophiles.
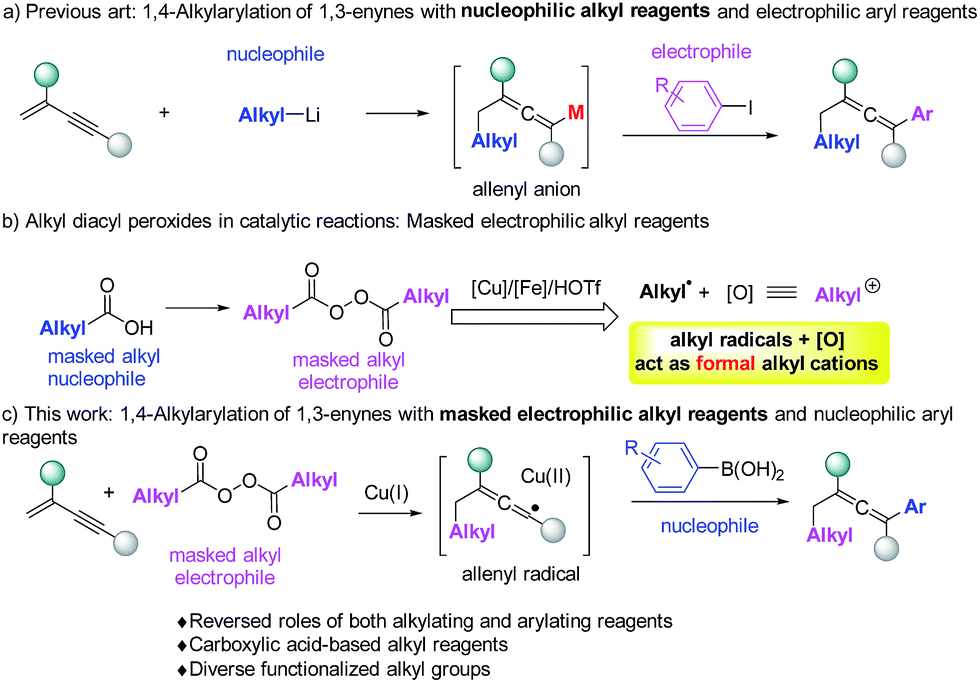 | ||
| Fig. 1 1,4-Alkylarylation of 1,3-enynes. | ||
Although alkyl-metallic reagents are highly reactive, there are some limitations associated with such compounds. For example, organometallic reagents are sensitive to moisture and reactions involving them usually have low functional group compatibilities both in the organometallic reagents and in other reaction partners. To meet the needs of alkylating reactions in organic synthesis, several non-traditional types of alkylating reagents, including alkyl boronates,10 redox-active esters,11 and alkyl peroxides12 have been developed and studied recently.
Our previous studies revealed that alkyl peroxides generated from the corresponding alkyl carboxylic acids, can in the presence of a copper,13 iron12 and HOTf14 catalyst act simultaneously as alkylating reagents and internal oxidants. Actually, the alkyl diacyl peroxides in a catalytic system can be simply regarded as masked alkyl electrophiles (Fig. 1b). Thus, we wondered whether alkyl peroxides can initiate the 1,4-alkylarylation reaction of 1,3-enynes to form allenes. This would offer an unprecedented strategy to allenes, most importantly, overcoming many of the difficulties associated with organometallic reagents.
Herein, we report that alkyl peroxides enable 1,4-alkylarylation of 1,3-enynes to form allenes, a reaction in which alkyl peroxides serve as the masked alkyl electrophiles and aryl boronic acids serve as nucleophiles (Fig. 1c). Mechanistic studies suggest that this reaction proceeds through an allenyl radical15 pathway,16 which is relatively rare in allene synthesis.
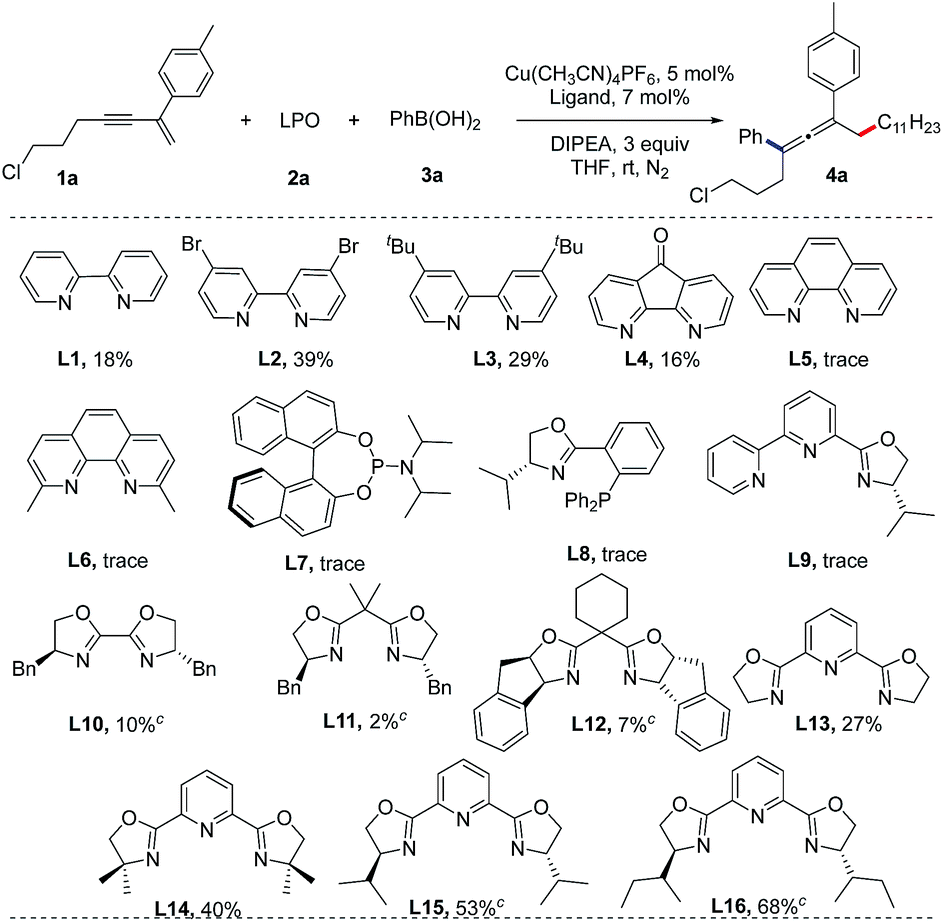
We initiated our studies by screening the reaction conditions employing a 1,3-enyne (1a), lauroyl peroxide (LPO, 2a) and phenylboronic acid (3a) as the starting materials and N,N-diisopropylethylamine (DIPEA) as a base (Table 1). The results of screening of the reaction conditions are detailed in the ESI.† Our experiments revealed that Cu(CH3CN)4BF4 is optimal and the ligand is the key element for this reaction. As shown in Table 1, bipyridine ligands (L1–L4) show low reactivities, while phenanthrolines, phosphoramidite, Phox and bipyridine oxazoline ligands (L5–L9) fail to afford the desired product. Chiral bisoxazoline ligands (L10–L12) on the other hand, proven to be excellent ligands for the copper-catalyzed asymmetric synthesis of CF3-substituted 1,1-diarylethanes, but they show poor reactivity and enantioselectivity in the present reaction.17 A dramatic increase in the yield of the reaction was observed when tridentate Py-Box ligands (L13–L16) were used. Although the enantioselectivities are still not satisfactory for the chiral ligands, the reactivity for L16 is useful. Thus, (±)-L16 was studied, providing an identical yield (70%).
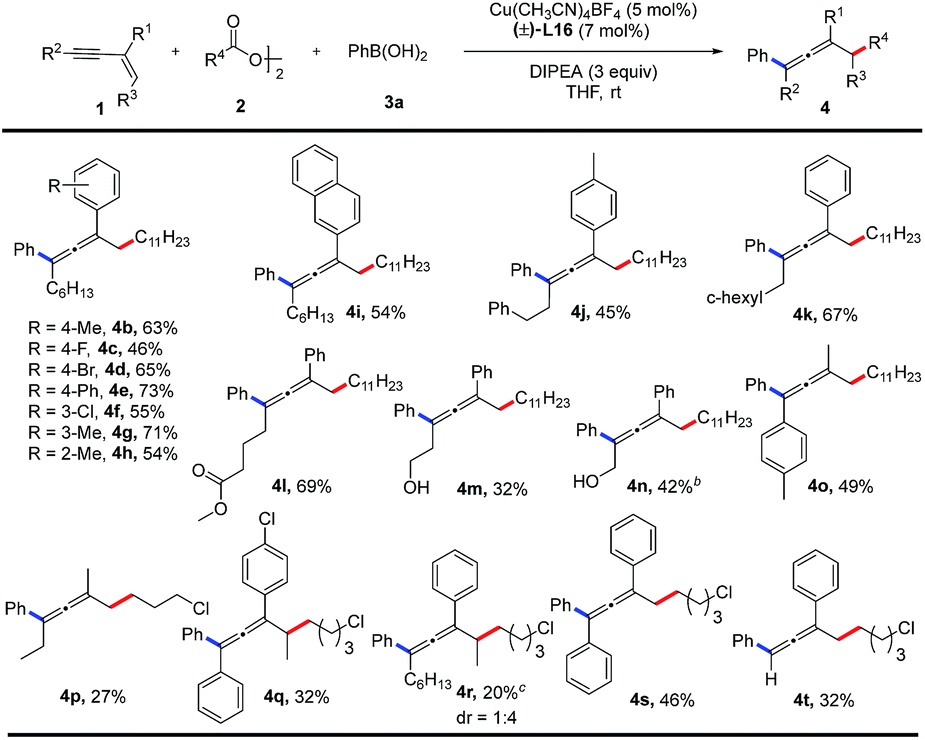
With the optimal reaction conditions in hand, the substrate scope of 1,3-enynes was explored as shown in Table 2. Both electron-donating and electron-withdrawing substituents on the aromatic ring of R1 are suitable for this coupling reaction, which affords the tetrasubstituted allene products (4b–4h) in moderate to good yields (46–73%). A naphthyl group is also compatible with this reaction, producing 4i in 54% yield. In the substituent R2 group, functional groups such as phenyl, cyclohexyl, ester, and free hydroxyl group are tolerated, giving the desired products (4j–4n) in 32–69% yields. Products 4o and 4p are obtained in 49% and 27% yields respectively, when R1 is a methyl group. Internal alkenes can also participate in the reaction, albeit with relatively lower yields of the corresponding products (4q, 4r). Diphenyl substituted 1,3-enyne affords a triphenyl allene (4s) in 46% yield. A terminal alkyne can also be employed in this reaction and provides the corresponding product (4t) in 32% yield. The accompanied side reactions toward the terminal C(sp)–H bond might be responsible for the low yield.
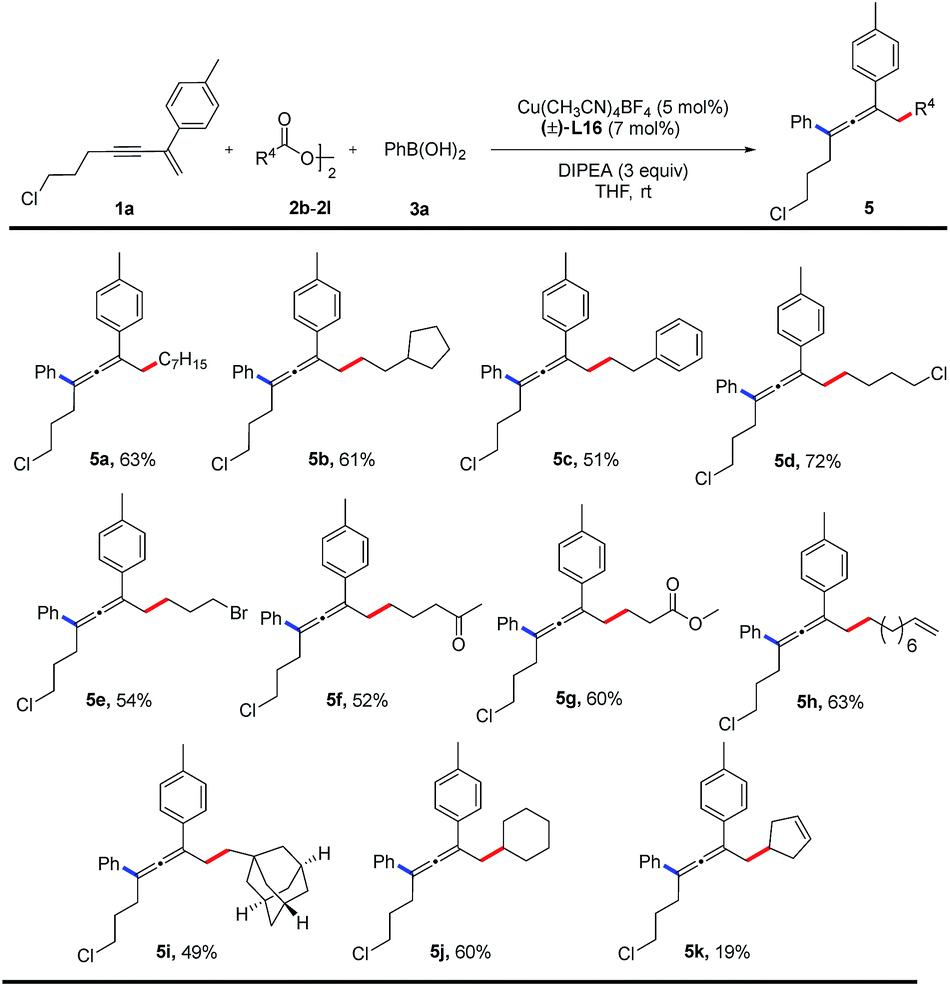
The substrate scope of alkyl diacyl peroxides was studied (Table 3). Primary alkyl diacyl peroxides functionalized with halide, carbonyl, ester, alkenyl or adamantanyl groups are converted into the desired products (5a–5i) in 49–72% yields. Furthermore, secondary alkyl diacyl peroxides can also be employed in the reaction, providing the corresponding products (5j, 5k) in moderate yields.
| a Reaction conditions: 1a (0.2 mmol, 1 equiv.), 2 (0.3 mmol, 1.5 equiv.), 3a (0.6 mmol, 3 equiv.), Cu(CH3CN)4BF4 (5 mol%), ligand (7 mol%), DIPEA (0.6 mmol, 3 equiv.), THF (1 mL), rt. |
|---|
|
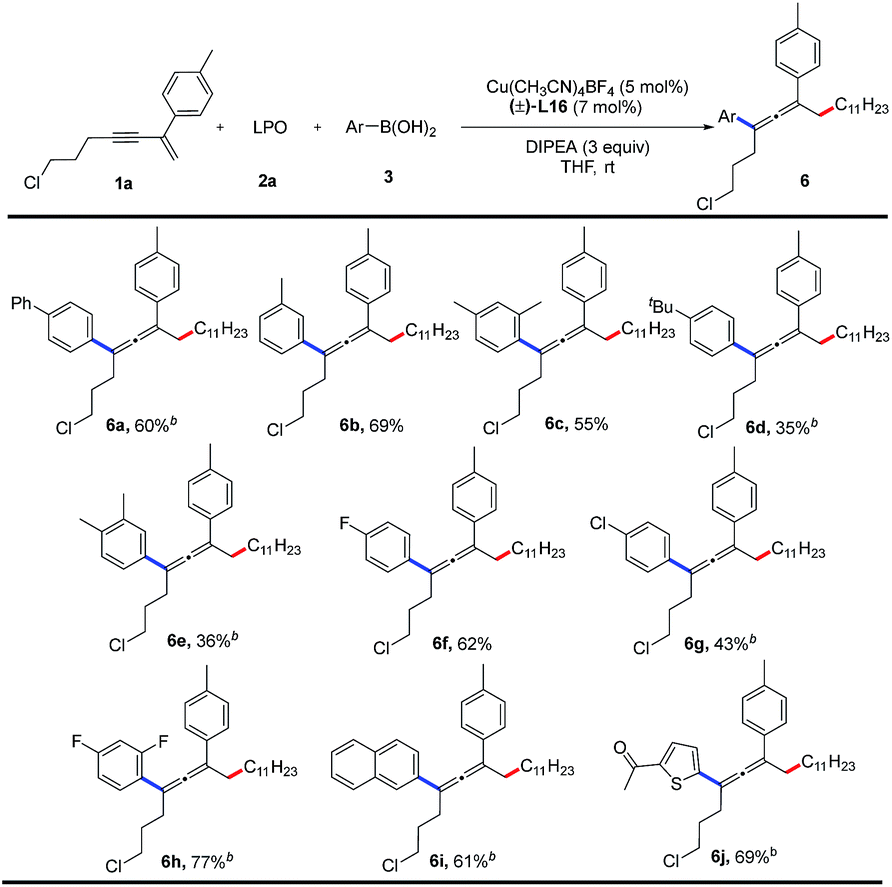
Finally, we examined the substrate scope of aryl boronic acids in this transformation (Table 4). A range of aryl boronic acids bearing substituent(s) such as phenyl group, methyl group, chloride, or fluoride, can afford the desired products (6a–6h) in 35–77% yields. Product 6i bearing a naphthyl group is formed in 61% yield. Heterocyclic aryl groups, such as thienyl, are also compatible with this reaction, and the corresponding product 6j is obtained in 69% yield. However, alkyl boronic acids are unreactive.
| a Reaction conditions: 1a (0.2 mmol, 1 equiv.), 2a (0.3 mmol, 1.5 equiv.), 3 (0.6 mmol, 3 equiv.), Cu(CH3CN)4BF4 (5 mol%), ligand (7 mol%), DIPEA (0.6 mmol, 3 equiv.), THF (1 mL), rt. b Reaction conditions: 1a (0.2 mmol, 1 equiv.), 2a (0.3 mmol, 1.5 equiv.), 3 (0.6 mmol, 3 equiv.), Cu(CH3CN)4BF4 (5 mol%), ligand (7 mol%), DMF (1 mL), THF (1 mL), rt. |
|---|
|
The synthetic applications of this method have been demonstrated (Fig. 2). The allene (4a) can react with N-iodosuccinimide (NIS) to afford an indenyl iodide in 75% yield.6l When treated under acidic reaction conditions, the allene (6i) is transformed into the 1H-indene (8) in 60% yield. Moreover, proton- or NIS-mediated cyclization of 4p afford 2H-pyrans (9, 10) in 51% and 56% yields, respectively.
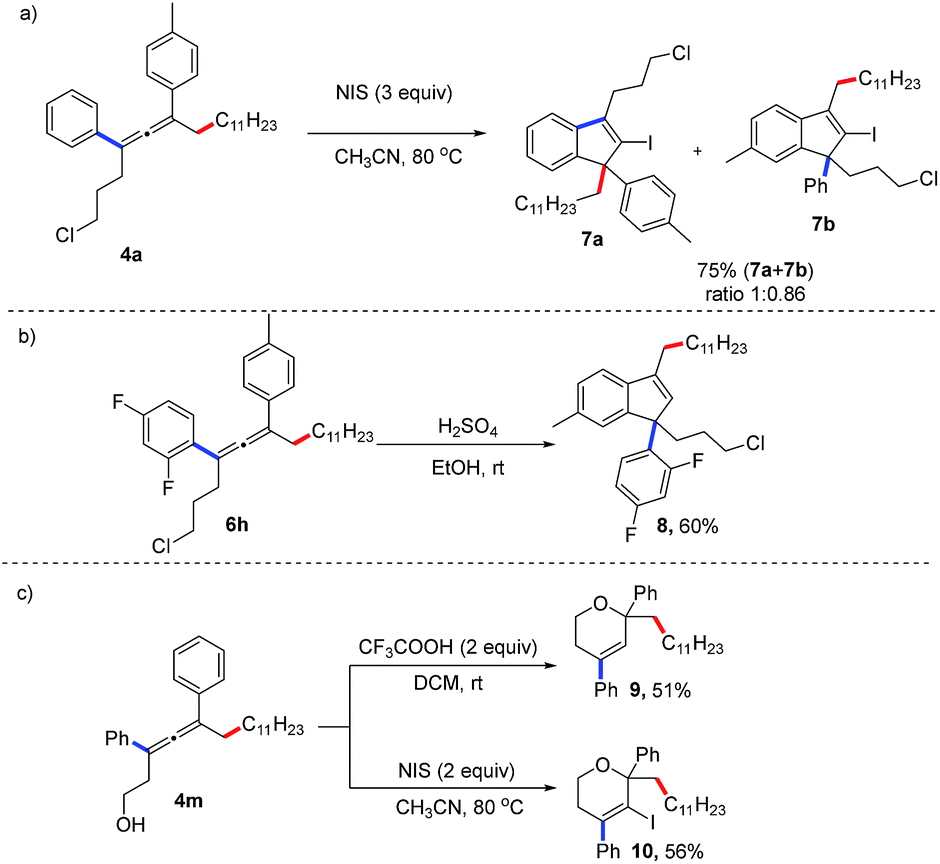 | ||
| Fig. 2 Synthetic applications. | ||
Preliminary mechanistic studies were carried out in an effort to obtain more information about the mechanism of this reaction (Fig. 3). First, an experiment with three equivalents of 2,2,6,6-tetra-methyl-1-piperidinyloxy (TEMPO), a radical-trapping reagent, was performed. The expected product (4a) was not formed, and compound 11 was identified by GC-MS analysis, indicating the formation of an alkyl radical species (Fig. 3a). Second, radical clock experiments with three different substrates were performed. Interestingly, substrates 12 and 14 were found only to produce allenyl compounds (13, 15) in moderate yields. No ring-opening product from 12 or ring-closing product from 14 was detected, suggesting that the rate of formation of the allene product is the much faster reaction (Fig. 3b). When an alkyl diacyl peroxide (16) was used as the alkylating reagent, both the vinylated allene (17a) and cyclopentylated allene (17b) were obtained. This reaction further supports the possibility of the formation of alkyl radicals. Notably, a trace amount the homocoupling product was detected in almost all of the reactions.18 More of the homocoupling product was observed with bidentate bisoxazoline ligands. When using L10 as a bidentate ligand, a mixture of homocoupling dimers was obtained in 60% yield along with 6% yield of the allene product 19 (Fig. 3c). Remarkably, the propargyl radical dimer 20 was crystallized from the mixture and the structure was confirmed by X-ray crystallography. This result suggests the existence of a propargyl radical which is a resonance structure of allenyl radical.15b
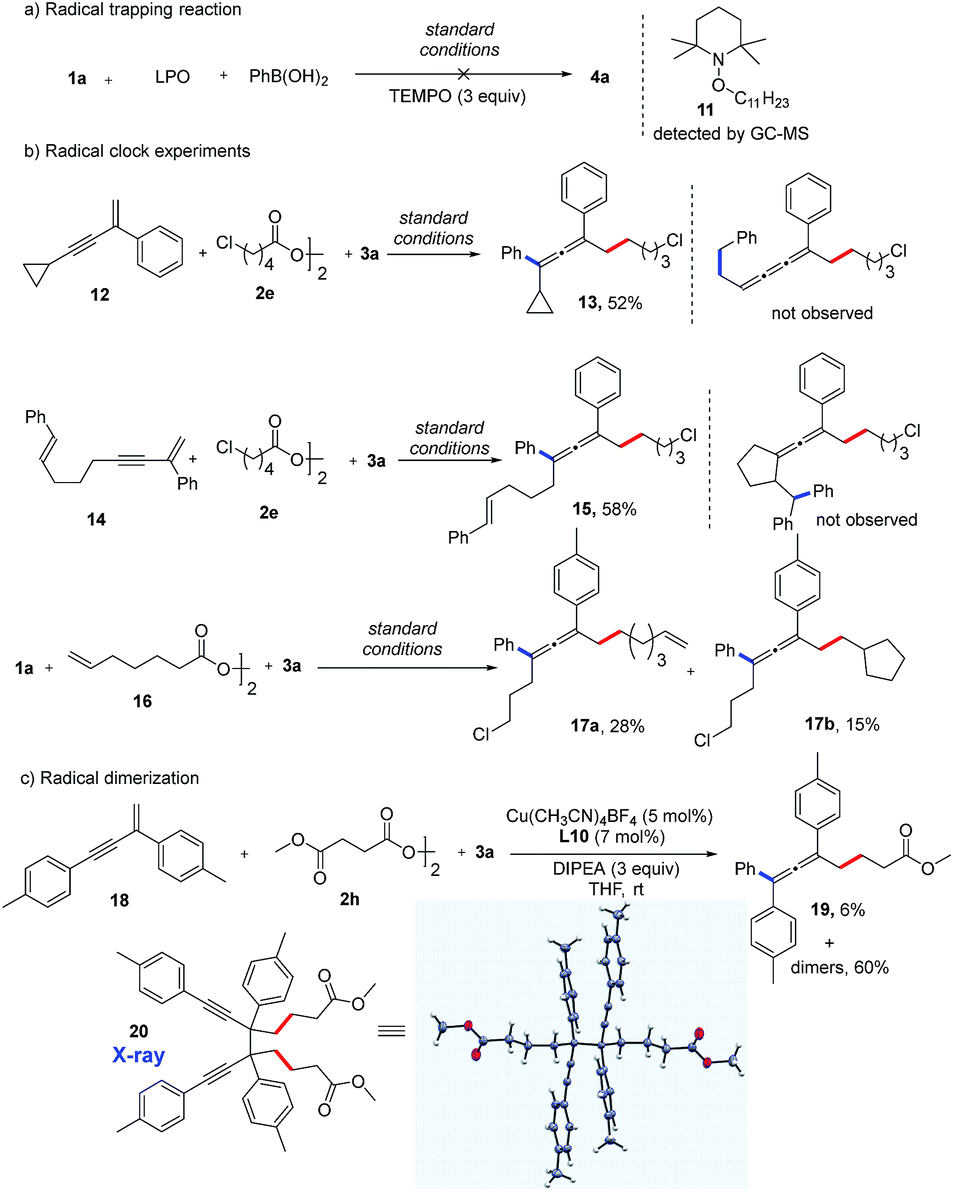 | ||
| Fig. 3 Mechanistic studies. | ||
Based on these studies, a plausible mechanism is proposed and is shown in Fig. 4. The Cu(I) species (A) reacts with an alkyl diacyl peroxide generating an alkyl radical and a Cu(II) species (B) via a single electron transfer. Then the alkyl radical attacks a 1,3-enyne to give a propargyl radical (C), which can be converted to the allenyl radical species (D). The Cu(II) species (B) can exchange its ligand with ArB(OH)2 to generate the Ar–Cu(II) species (E). Two possible pathways might be involved in the catalytic cycle: path a (outer-sphere) is the radical homolytic substitution interaction19 between the Ar–Cu(II) species (E) and the allenyl radical, leading to the product and regenerating the copper catalyst. In path b (inner-sphere), a Cu(II) species (E) combines with the allenyl radical species (D) to form a Cu(III) intermediate (F), which by reductive elimination, generates the target product and the Cu(I) species (A). As shown in the mechanistic cycle, the three-component reaction involves 1,3-enyne as a neutral substrate, aryl boronic acid as a mild nucleophile, and alkyl peroxide as an electrophile in the absence of extra oxidant or reductant. The alkyl peroxide can generate an alkyl radical and oxidize the Cu(I) to Cu(II), a subsequent oxidant to turn over the reaction. Thus, the alkyl peroxide acts as, more precisely, a masked alkyl electrophile.
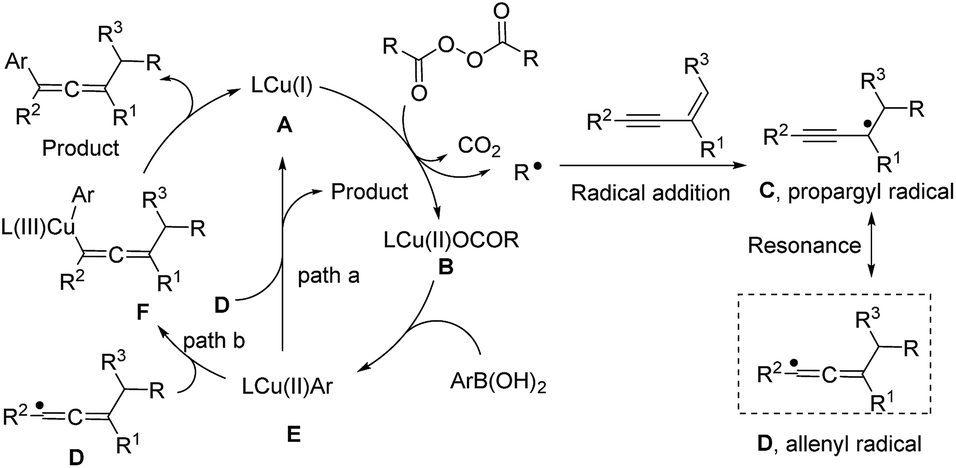 | ||
| Fig. 4 Plausible catalytic cycle. | ||
Conclusions
In conclusion, we have developed a copper-catalyzed 1,4-alkylarylation of 1,3-enynes which affords diverse tetra-substituted allenes under mild reaction conditions. Functionalized alkyl diacyl peroxides are used as masked alkyl electrophiles and aryl boronic acids as aryl nucleophiles. The tetrasubstituted allenes obtained in this way can be easily transformed into useful specific compounds such as 1H-indenes, 2H-pyrans, and their iodo-derivatives. Preliminary mechanistic studies suggest the involvement of radical species and a radical addition mechanism for this reaction is proposed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Key R&D Program of China (Grant No. 2017YFA0700103), the NSFC (Grant No. 21502191, 21672213, 21871258), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000), the Haixi Institute of CAS (Grant No. CXZX-2017-P01) for financial support. The X-ray crystallographic coordinates for structure reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 1883091.†Notes and references
- (a) N. Krause, A. Stephen and K. Hashimi, Modern Allene Chemistry, Wiley-VCH, Weinheim, 2004 CrossRef; (b) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196 CrossRef PubMed; (c) S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef PubMed; (d) K. M. Brummond and J. E. DeForrest, Synthesis, 2007, 795 CrossRef CAS; (e) S. Ma, Acc. Chem. Res., 2009, 42, 1679 CrossRef CAS PubMed; (f) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef CAS PubMed; (g) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed; (h) C.-Y. Wang, G.-H. Pan, F. Chen and J.-H. Li, Chem. Commun., 2017, 53, 4730 RSC.
- (a) S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384 RSC; (b) J. Ye and S. Ma, Org. Chem. Front., 2014, 1, 1210 RSC.
- (a) B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 15978 CrossRef CAS PubMed; (b) Z. Li, V. Boyarskikh, J. H. Hansen, J. Autschbach, D. G. Musaev and H. M. Davies, J. Am. Chem. Soc., 2012, 134, 15497 CrossRef CAS PubMed; (c) D. A. Mundal, K. E. Lutz and R. J. Thomson, J. Am. Chem. Soc., 2012, 134, 5782 CrossRef CAS PubMed; (d) Y. Jiang, A. B. Diagne, R. J. Thomson and S. E. Schaus, J. Am. Chem. Soc., 2017, 139, 1998 CrossRef CAS PubMed; (e) H. Liu, D. Leow, K. W. Huang and C. H. Tan, J. Am. Chem. Soc., 2009, 131, 7212 CrossRef CAS PubMed.
- (a) H. Ito, Y. Sasaki and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 15774 CrossRef CAS PubMed; (b) H. Ohmiya, U. Yokobori, Y. Makida and M. Sawamura, Org. Lett., 2011, 13, 6312 CrossRef CAS PubMed; (c) M. Yang, N. Yokokawa, H. Ohmiya and M. Sawamura, Org. Lett., 2012, 14, 816 CrossRef CAS PubMed; (d) S. N. Kessler and J. E. Backvall, Angew. Chem., Int. Ed., 2016, 55, 3734 CrossRef CAS PubMed.
- (a) J. W. Han, N. Tokunaga and T. Hayashi, J. Am. Chem. Soc., 2001, 123, 12915 CrossRef CAS PubMed; (b) C. Luken and C. Moberg, Org. Lett., 2008, 10, 2505 CrossRef PubMed; (c) T. Nishimura, H. Makino, M. Nagaosa and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 12865 CrossRef CAS PubMed; (d) Y. Xiao and J. Zhang, Chem. Commun., 2010, 46, 752 RSC; (e) Y. Mori, G. Onodera and M. Kimura, Chem. Lett., 2014, 43, 97 CrossRef CAS; (f) A. Nishimura, E. Tamai, M. Ohashi and S. Ogoshi, Chem.–Eur. J., 2014, 20, 6613 CrossRef CAS PubMed; (g) Y. Huang, J. Del Pozo, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2018, 140, 2643 CrossRef CAS PubMed; (h) H. L. Sang, S. Yu and S. Ge, Org. Chem. Front., 2018, 5, 1284 RSC; (i) X. Zhu, W. Deng, M. F. Chiou, C. Ye, W. Jian, Y. Zeng, Y. Jiao, L. Ge, Y. Li, X. Zhang and H. Bao, J. Am. Chem. Soc., 2019, 141, 548 CrossRef CAS PubMed; (j) D.-W. Gao, Y. Xiao, M. Liu, Z. Liu, M. K. Karunananda, J. S. Chen and K. M. Engle, ACS Catal., 2018, 8, 3650 CrossRef CAS PubMed; (k) S. Yu, H. L. Sang, S.-Q. Zhang, X. Hong and S. Ge, Commun. Chem., 2018, 1, 64 CrossRef.
- (a) E. Mainetti, L. Fensterbank and M. Malacria, Synlett, 2002, 923 CrossRef CAS; (b) Z. Yang, W. J. Hao, S. L. Wang, J. P. Zhang, B. Jiang, G. Li and S. J. Tu, J. Org. Chem., 2015, 80, 9224 CrossRef CAS PubMed; (c) S. Ma and Q. He, Angew. Chem., Int. Ed., 2004, 43, 988 CrossRef CAS PubMed; (d) M. Ogasawara, Y. Ge, K. Uetake and T. Takahashi, Org. Lett., 2005, 7, 5697 CrossRef CAS PubMed; (e) W. Zhang, H. Xu, H. Xu and W. Tang, J. Am. Chem. Soc., 2009, 131, 3832 CrossRef CAS PubMed; (f) D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298 CrossRef CAS PubMed; (g) H. Qian, X. Yu, J. Zhang and J. Sun, J. Am. Chem. Soc., 2013, 135, 18020 CrossRef CAS PubMed; (h) Z. Wang, X. Li and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 14219 CrossRef CAS PubMed; (i) M. Wang, Z. L. Liu, X. Zhang, P. P. Tian, Y. H. Xu and T. P. Loh, J. Am. Chem. Soc., 2015, 137, 14830 CrossRef CAS PubMed; (j) W. D. Chu, L. Zhang, Z. Zhang, Q. Zhou, F. Mo, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2016, 138, 14558 CrossRef CAS PubMed; (k) J. Zhao and K. J. Szabo, Angew. Chem., Int. Ed., 2016, 55, 1502 CrossRef CAS PubMed; (l) D. Qian, L. Wu, Z. Lin and J. Sun, Nat. Commun., 2017, 8, 567 CrossRef PubMed; (m) M. Guisan-Ceinos, V. Martin-Heras, R. Soler-Yanes, D. J. Cardenas and M. Tortosa, Chem. Commun., 2018, 54, 8343 RSC; (n) P. H. Poulsen, Y. Li, V. H. Lauridsen, D. K. B. Jorgensen, T. A. Palazzo, M. Meazza and K. A. Jorgensen, Angew. Chem., Int. Ed., 2018, 57, 10661 CrossRef CAS PubMed; (o) N. Liu, Y. Zhi, J. Yao, J. Xing, T. Lu and X. Dou, Adv. Synth. Catal., 2018, 360, 642 CrossRef CAS.
- (a) J. Zhao, Y. Liu, Q. He, Y. Li and S. Ma, Chem.–Eur. J., 2009, 15, 11361 CrossRef CAS PubMed; (b) S. Ma, Q. He and X. Jin, Synlett, 2005, 514 CrossRef CAS.
- H. Todo, J. Terao, H. Watanabe, H. Kuniyasu and N. Kambe, Chem. Commun., 2008, 1332 RSC.
- Y. Tomida, A. Nagaki and J.-i. Yoshida, J. Am. Chem. Soc., 2011, 133, 3744 CrossRef CAS PubMed.
- (a) Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414 CrossRef CAS; (b) H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280 CrossRef CAS PubMed; (c) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef PubMed; (d) H. Huang, K. Jia and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 1881 CrossRef CAS PubMed; (e) T. Koike and M. Akita, Org. Biomol. Chem., 2016, 14, 6886 RSC; (f) G. X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407 RSC; (g) L. Zhang and Z. Q. Liu, Org. Lett., 2017, 19, 6594 CrossRef CAS PubMed; (h) K. Duan, X. Yan, Y. Liu and Z. Li, Adv. Synth. Catal., 2018, 360, 2781 CrossRef CAS; (i) W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600 RSC.
- (a) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D. H. Bao, F. L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213 CrossRef CAS PubMed; (b) J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C. M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174 CrossRef CAS PubMed.
- (a) B. Qian, S. Chen, T. Wang, X. Zhang and H. Bao, J. Am. Chem. Soc., 2017, 139, 13076 CrossRef CAS PubMed; (b) W. Jian, L. Ge, Y. Jiao, B. Qian and H. Bao, Angew. Chem., Int. Ed., 2017, 56, 3650 CrossRef CAS PubMed.
- (a) Y. Li, Y. Han, H. Xiong, N. Zhu, B. Qian, C. Ye, E. A. Kantchev and H. Bao, Org. Lett., 2016, 18, 392 CrossRef CAS PubMed; (b) C. Ye, Y. Li and H. Bao, Adv. Synth. Catal., 2017, 359, 3720 CrossRef CAS.
- H. Zhou, L. Ge, J. Song, W. Jian, Y. Li, C. Li and H. Bao, iScience, 2018, 3, 255 CrossRef CAS PubMed.
- (a) N. J. Reilly, D. L. Kokkin, M. Nakajima, K. Nauta, S. H. Kable and T. W. Schmidt, J. Am. Chem. Soc., 2008, 130, 3137 CrossRef CAS PubMed; (b) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 15620 CrossRef CAS PubMed.
- (a) C. Alameda-Angulo, B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 2006, 47, 913 CrossRef CAS; (b) H. G. Benson, A. J. Bowles, A. Hudson and R. A. Jackson, Mol. Phys., 1971, 20, 713 CrossRef; (c) J. Terao, F. Bando and N. Kambe, Chem. Commun., 2009, 7336 RSC; (d) F. Wang, D. Wang, Y. Zhou, L. Liang, R. Lu, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2018, 57, 7140 CrossRef CAS PubMed.
- (a) D. Wang, L. Wu, F. Wang, X. Wan, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 6811 CrossRef CAS PubMed; (b) L. Wu, F. Wang, X. Wan, D. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 2904 CrossRef CAS PubMed.
- F. Wang, D. Wang, X. Wan, L. Wu, P. Chen and G. Liu, J. Am. Chem. Soc., 2016, 138, 15547 CrossRef CAS PubMed.
- (a) C. H. Schiesser and L. M. Wild, Tetrahedron, 1996, 52, 13265 CrossRef CAS; (b) S. Tang, P. Wang, H. Li and A. Lei, Nat. Commun., 2016, 7, 11676 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1883091. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05689g |
| This journal is © The Royal Society of Chemistry 2019 |