
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-catalyzed dearomative arylborylation of indoles†
Chong
Shen‡
a,
Nicolas
Zeidan‡
 b,
Quan
Wu‡
a,
Christian B. J.
Breuers
b,
Ren-Rong
Liu
a,
Yi-Xia
Jia
*ac and
Mark
Lautens
*b
b,
Quan
Wu‡
a,
Christian B. J.
Breuers
b,
Ren-Rong
Liu
a,
Yi-Xia
Jia
*ac and
Mark
Lautens
*b
aCollege of Chemical Engineering, State Key Laboratory Breeding Base of Green-Chemical Synthesis Technology, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: yxjia@zjut.edu.cn
bDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, Canada. E-mail: mark.lautens@utoronto.ca
cShanghai Key Laboratory for Molecular Engineering of Chiral Drugs, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, China
First published on 25th January 2019
Abstract
A palladium-catalyzed dearomative arylborylation of indoles is reported, which provides straightforward access to structurally diverse indolines bearing vicinal tetrasubstituted and borylated trisubstituted stereocenters in moderate to good yields with excellent diastereoselectivities. By using a BINOL-based chiral phosphoramidite ligand and an sp2–sp3 mixed-boron reagent, an enantioselective dearomative arylborylation was achieved and chiral boron-containing products were accessed in up to 94% ee. Synthetic tranformations of the resulting organoborons were conducted to afford a number of unique indoline derivatives.
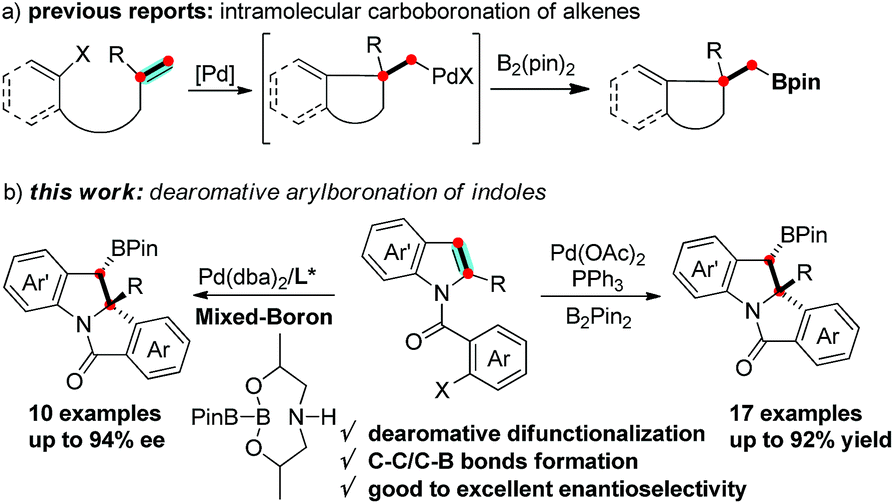
Boron containing molecules serve as important building blocks due to their capability of participating in carbon–carbon and carbon–heteroatom bond forming reactions and thus are ubiquitous intermediates in the synthesis of various natural products and bioactive compounds.1 The palladium-catalyzed 1,2-difunctionalization of olefins involving Heck/anionic-capture sequences has been intensely studied and proceeds via a carbopalladation followed by the capture of the alkyl-Pd species with a variety of nucleophiles.2 Domino Heck-borylation reactions employing boron reagents as nucleophiles have enabled an efficient method to synthesize Csp3-based organoboron compounds (Scheme 1a).3,4 It is worthwhile to extend this methodology to the synthesis of structurally diverse, chiral organoboron heterocycles.
 | ||
Scheme 1 Palladium-catalyzed arylborylation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. C bonds. | ||
The transition-metal-catalyzed dearomative functionalization of aromatics has recently emerged as a useful approach to the synthesis of unique aliphatic cyclic molecules.5 In this context, dearomative functionalization of indoles has rendered the synthesis of indolines, a frequently occurring key substructure of natural products and alkaloids, extremely straightforward and efficient.6 Documented reports include the intramolecular C3-arylation of an indolic enolate,7 the Heck arylation of N-tethered 2,3-disubstituted indoles,8 and the reductive-Heck reaction,9 which results in the dearomative mono-functionalization of indole derivatives. Furthermore, the dearomative difunctionalization of indoles was realized through the trapping of the benzyl-Pd intermediate of aryl-palladation using a series of trapping agents, such as cyanides,10 boroxines,11 terminal alkynes,12 propiolic acid,13 and heteroarenes,14 efficiently delivering 2,3-disubstituted indolines. While the formation of the C–H and C–C bonds is documented in the aforementioned reports, there are no examples reported for C–B bond formation to afford chiral organoboron compounds. We envisioned a dearomative Heck-borylation domino reaction of indole to provide benzylic-boron indolines; the protocol mainly relies on the capture of the in situ generated benzyl-Pd species with diboron compounds. Herein, we report this dearomative arylborylation reaction using bis(pinacolato)diboron (B2pin2), and its enantioselective variant with a pre-activated sp2–sp3 mixed-boron reagent and a new BINOL-based chiral phosphoramidite ligand, which leads to a series of structurally unique tetracyclic indolines in moderate to good yields, excellent diastereoselectivities, and good to excellent enantioselectivities (Scheme 1b).
The reaction of 1a with B2pin22 was chosen as the starting condition for optimization. An initial test using Pd(OAc)2 (5 mol%), PPh3 (10 mol%), and tBuOLi (2.0 equiv.) in CH2Cl2 (0.2 M) at 100 °C led to the desired arylborylation product 3a in 25% yield with >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr (Table 1, entry 1). To improve the yield, some bases were screened (entries 2–5). A poor yield was observed when using tBuOK (Table 1, entry 2), while K3PO4, K2CO3, and Na2CO3 significantly improve the yield with 3a isolated in 66% in the case of K2CO3 (Table 1, entries 3–5). Other commercially available ligands, such as P(p-tolyl)3, PtBu3, and PCy3, were then tested, none of which increased the yield (Table 1, entries 6–8). Moreover, poor yields were also observed for bidentate phosphine ligands, e.g. dppe and xantphos. A lower yield was observed when changing the catalyst from Pd(OAc)2 to Pd(dba)2 (Table 1, entry 9). Higher yields could be obtained by lowering the temperature and 3a was isolated in 83% yield when the reaction was run at 60 °C (Table 1, entries 10 and 11). Finally, the solvent effect was examined (Table 1, entries 12–15). Comparable yields were observed in toluene and CH3CN, while the best yield of 3a was achieved in DCE solvent (Table 1, entry 15).
:1 dr (Table 1, entry 1). To improve the yield, some bases were screened (entries 2–5). A poor yield was observed when using tBuOK (Table 1, entry 2), while K3PO4, K2CO3, and Na2CO3 significantly improve the yield with 3a isolated in 66% in the case of K2CO3 (Table 1, entries 3–5). Other commercially available ligands, such as P(p-tolyl)3, PtBu3, and PCy3, were then tested, none of which increased the yield (Table 1, entries 6–8). Moreover, poor yields were also observed for bidentate phosphine ligands, e.g. dppe and xantphos. A lower yield was observed when changing the catalyst from Pd(OAc)2 to Pd(dba)2 (Table 1, entry 9). Higher yields could be obtained by lowering the temperature and 3a was isolated in 83% yield when the reaction was run at 60 °C (Table 1, entries 10 and 11). Finally, the solvent effect was examined (Table 1, entries 12–15). Comparable yields were observed in toluene and CH3CN, while the best yield of 3a was achieved in DCE solvent (Table 1, entry 15).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | L | T (°C) | Solvent | Yield (%) |
|
a Reaction conditions: 1a (0.2 mmol), 2 (2 eq.), 5 mol% Pd(OAc)2, 10 mol% ligand, and 2 eq. base in solvent (2 mL) at 100 °C; isolated yield, dr > 20:1; DCE = 1,2-dichloroethane.
b 5 mol% Pd(dba)2 was used.
|
|||||
| 1 | t BuOLi | PPh3 | 100 | CH2Cl2 | 25 |
| 2 | t BuOK | PPh3 | 100 | CH2Cl2 | 27 |
| 3 | K3PO4 | PPh3 | 100 | CH2Cl2 | 57 |
| 4 | K2CO3 | PPh3 | 100 | CH2Cl2 | 66 |
| 5 | Na2CO3 | PPh3 | 100 | CH2Cl2 | 43 |
| 6 | K2CO3 | P(p-tolyl)3 | 100 | CH2Cl2 | 53 |
| 7 | K2CO3 | PtBu3·HBF4 | 100 | CH2Cl2 | 24 |
| 8 | K2CO3 | PCy3·HBF4 | 100 | CH2Cl2 | 17 |
| 9b | K2CO3 | PPh3 | 100 | CH2Cl2 | 38 |
| 10 | K2CO3 | PPh3 | 80 | CH2Cl2 | 74 |
| 11 | K2CO3 | PPh3 | 60 | CH2Cl2 | 83 |
| 12 | K2CO3 | PPh3 | 60 | THF | 68 |
| 13 | K2CO3 | PPh3 | 60 | toluene | 84 |
| 14 | K2CO3 | PPh3 | 60 | MeCN | 83 |
| 15 | K2CO3 | PPh3 | 60 | DCE | 87 |
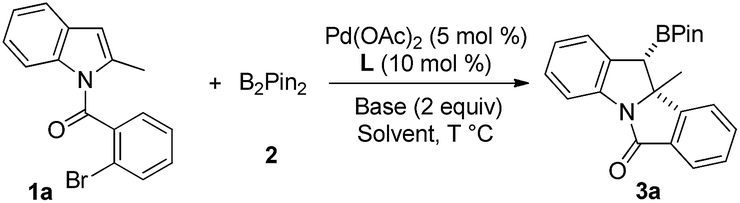
With the optimal conditions in hand, we then examined the scope of the reaction by varying the substituents on the halobenzene and indole rings. As shown in Scheme 2, substituent effect on the benzene ring of the 2-bromobenzoyl moiety was first investigated. Moderate to excellent yields of products 3a–3h were achieved for indoles bearing substituents (methyl, methoxyl, and chloride) at the C3–C5 position of the benzene ring. Higher yields were generally obtained for substrates having an electron-donating group than for those bearing an electron-withdrawing substituent (3e and 3gvs.3b–3d and 3f). Product 3h having two methoxyl groups was isolated in 92% yield. Of note, 3b, having a sterically congested methyl group at the C3 position of the bromobenzoyl moiety, was obtained in 78% yield. Next, the substituent effect on the indole ring was examined. A range of C2-alkylated and C2-arylated indoles were subjected to the reaction at 70 °C, which led to the arylborylated products 3i–3n in moderate yields. In contrast, the yields of these borylated indolines were lower than those achieved for 2-methyl products 3a–3h. It is noteworthy that the reaction of a 2-furyl indole substrate successfully delivered 3n in 53% yield. Moreover, the substituent at the C5-position of 2-substituted indoles was examined and the reactions of substrates having MeO, iPr, and Me groups afforded 3o–3q in moderate yields.
 | ||
| Scheme 2 Scope of the indole substituent. aReaction conditions: 1 (0.2 mmol), 2 (2.0 equiv.), 5 mol% Pd(OAc)2, 10 mol% PPh3, and 2 eq. K2CO3 in DCE (2.0 mL) at 60 °C for 12–72 h. bIsolated yield, dr > 20:1. cIn DCE (2 mL) at 70 °C for 18–96 h. | ||
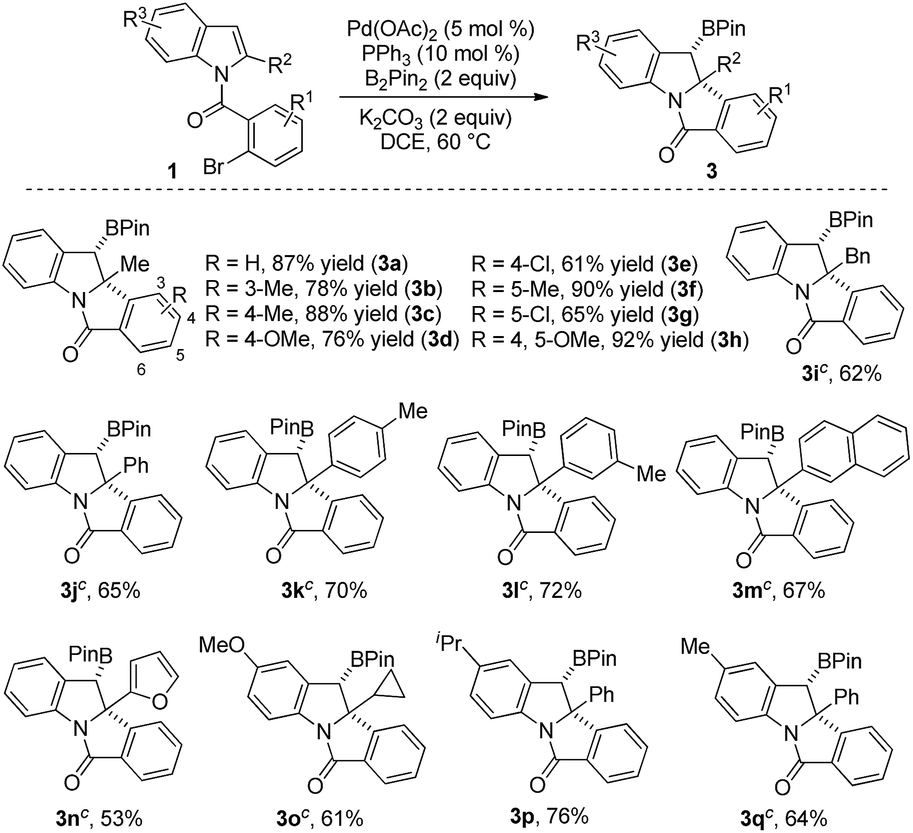
To demonstrate the synthetic utility of this reaction, a gram-scale reaction (4.0 mmol) was carried out under the optimal conditions and afforded 3a in 77% yield (Scheme 3). Synthetic transformations of 3a were then conducted. Oxidation of 3a using NaBO3·4H2O in THF/H2O led to alcohol 4 as a single isomer in almost quantitative yield.15 Compound 4 was further oxidized to ketone 5 in 85% yield with PCC as an oxidant at room temperature. Subjecting 3a to KHF2 in THF/H2O led to the corresponding potassium trifluoroborate 6 in 91% yield. Compound 6 was further converted to amide 7 in MeCN with 64% yield, as a single isomer through a Cu(OAc)2-promoted oxidative nucleophilic substitution.16 The relative configurations of alcohol 4 and amide 7 were determined from their 2D-NOESY spectra.
 | ||
| Scheme 3 Gram-scale synthesis of 3a and transformations of 3a. PCC: pyridinium chlorochromate. | ||
An enantioselective Heck/borylation reaction of 1a was then investigated using phosphoramidite L1 as the chiral ligand12b (Table 2, for more details see the ESI†). Early on, we observed that the benzylic boron was susceptible to inorganic-base promoted proto-deborylation at the high temperatures necessary for this metal–ligand system (vide infra). In order to avoid the use of the inorganic base necessary to activate B2Pin2, we utilized an sp2–sp3 mixed boron reagent first reported by Santos for copper-catalyzed hydroboration reactions.17 To the best of our knowledge, this reagent has not been used in palladium-catalyzed borylations. It was necessary to change the solvent from DCE to MTBE since the former was not efficient in the enantioselective variant (Table 2, entry 1 and 2). Although the bromo- and iodo- substrates 1a and 1a′′ delivered product in higher yields than the aryl-chloride, it was evident that the smaller halide improved the enantioselectivities (Table 2, entries 2–4). The absolute stereochemistry of 3a was assigned by single-crystal X-ray analysis.18
|
|
|||||
|---|---|---|---|---|---|
| Entry | X | Additive | Changes to condition | Yield (%) | ee (%) |
| a Standard conditions: 1 (0.2 mmol), mixed-boron reagent (2 eq.), 5 mol% Pd(dba)2, 6 mol% L1, and additive in MTBE (2 mL) at 100 °C for 18 h; isolated yield; ee was determined by chiral HPLC; TMG = tetramethyl guanidine; n.r. = no reaction. | |||||
| 1 | Br | None | None | 73 | 64 |
| 2 | Br | None | DCE as solvent | 34 | 20 |
| 3 | I | None | None | 40 | 50 |
| 4 | Cl | None | None | 15 | 88 |
| 5 | Cl | K2CO3 (2 eq.) | B2Pin2·(2 eq.) | n.r. | — |
| 6 | Cl | NEt3 (3 eq.) | None | 50 | 88 |
| 7 | Cl | NEt3 (3 eq.) | using L2 | 65 | 88 |
|
|||||
| Entries 8–11 using L2 | |||||
| 8 | Cl | i Pr2NEt (3 eq.) | None | 27 | — |
| 9 | Cl | NEt3 (3 eq.) | 80 °C | n.r. | — |
| 10 | Cl | NEt3 (3 eq.) | 10 mol% L2 | 68 | 91 |
| 11 | Cl | NEt3 (5 eq.) | 3 eq. mixed-boron reagent | 74 | 94 |
|
|||||
| Entries 12–15 using conditions in entry 11 | |||||
| 12 | Cl | NEt3 (5 eq.) | L3 | 17 | 78 |
| 13 | Cl | NEt3 (5 eq.) | L4 | 70 | 91 |
| 14 | Cl | NEt3 (5 eq.) | L5 | Trace | — |
| 15 | Cl | NEt3 (5 eq.) | L6 | Trace | — |
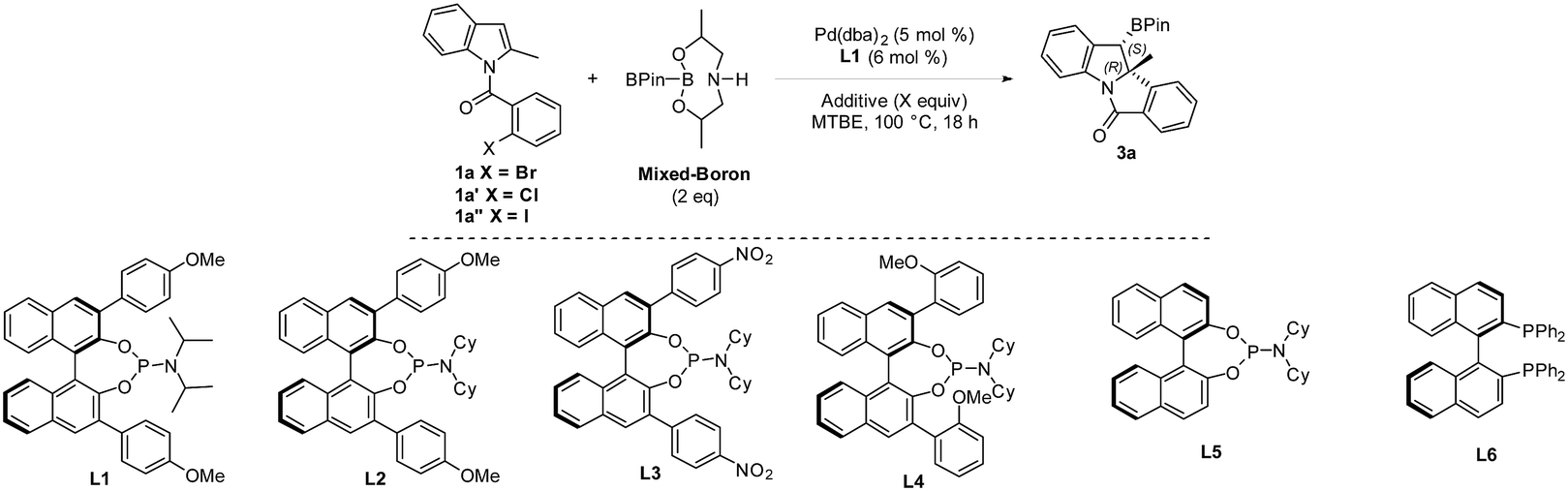
In the case of aryl-chloride, there was no reaction using B2Pin2 (Table 2, entry 5). We employed an organic base to neutralize the by-product of the boron-reagent and the conversion was improved to 50% while maintaining the ee (Table 2, entry 6). Further increasing the steric bulk of the ligand improved the yield of the reaction (Table 2, entry 7). Other amines (Table 2, entry 8) or lowering the temperature (Table 2, entry 9) were not effective. By increasing ligand and reagent loading the product was delivered in 74% yield and 94% ee (Table 2, entry 10 and 11). With respect to the ligand, the nitro variant L3 was not an effective ligand for the transformation (Table 2, entry 12). The 3,3′-orthoanisole ligand L4 also did not improve the yield (Table 2, entry 13). The importance of the 3,3′ rings was evident as the simple BINOL-derived phosphoramidite L5 did not produce the product (Table 2, entry 14). Contrary to the dearomative reductive-Heck,9a BINAP was not effective in catalyzing the reaction (Table 2, entry 15).
We then examined the scope of the Heck/borylation on various aryl-chloride substrates (Scheme 4). The steric influence at the halide (3r), as well as functionalities para to the amide tether (3d and 3s) provided products in diminished enantioselectivities. In contrast, the substitution para to chloride (3t) or on the indole moiety yielded 3t–3v in moderate yields and excellent enantioselectivities. The aryl functionality at R2 resulted in 3j and 3w in moderate and good yields with excellent enantioselectivities. Finally, heterocycle containing scaffold 3x was accessed in good yield albeit in a diminished enantioselectivity. Oxidation of compound 3a provided the chiral alcohol (+)-4 and ketone (+)-5 with no loss in enantiomeric excess.
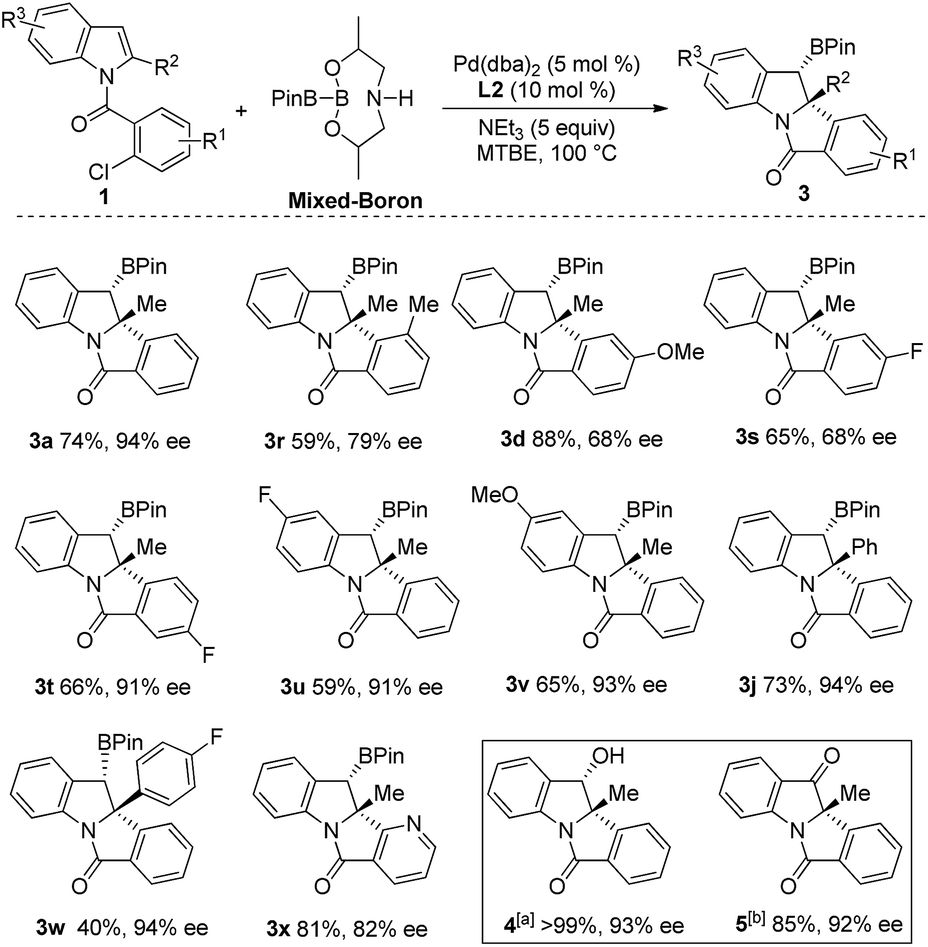 | ||
| Scheme 4 Scope of Heck/borylation of indoles. Reaction conditions: 1a′ (0.2 mmol), (mixed-boron reagent) (0.6 mmol), Pd(dba)2 (5 mol%), L2 (10 mol%), NEt3 (5 eq.), 100 °C, 18 h. [a] Reaction of 4a (0.5 mmol) with NaBO3 (5 eq.), THF:H2O (1:1), rt. [b] Reaction of 5 (0.2 mmol) with PCC (3 eq.), CHCl3, 35 °C. | ||
Conclusions
In conclusion, we have developed a dearomative difunctionalization of indoles through a palladium-catalyzed intramolecular arylborylation reaction. Indolines possessing vicinal borylated trisubstituted and tetrasubstituted stereocenters were accessed in good yields and excellent diastereoselectivities. The asymmetric variant of this reaction was explored with a new BINOL-based chiral phosphoramidite ligand and moderate to excellent enantioselectivities were obtained (up to 94% ee). Transformations of the benzylic boron to alcohol and amide were presented to show the synthetic utilities of this reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for generous support from the National Natural Science Foundation of China (nos. 21522207, 21702184, and 21772175). We thank the Natural Science and Engineering Research Council (NSERC), the University of Toronto, and Alphora Inc. for financial support. N. Z acknowledges the province of Ontario for a Graduate Scholarship (OGS). N. Z. thanks Dr Darcy Burns and the NMR staff at the University of Toronto, as well as Dr Alan Lough for the X-ray results.References
- (a) A. Pelter, K. Smith and H. C. Brown, Borane Reagents, Academic Press, London, 1988 Search PubMed; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (c) M. Davison, A. K. Hughes, T. B. Marder and K. Wade, Contemporary Boron Chemistry; RSC, Cambridge, U.K., 2000 Search PubMed; (d) Boronic Acids: Preparation and Applications in Organic Synthesis Medicine and Materials, ed. D. G. Hall, 2nd edn, Wiley-VCH, Weinheim, 2011 Search PubMed.
- For reviews, see (a) R. Grigg and V. Sridharan, J. Organomet. Chem., 1999, 576, 65 CrossRef CAS; (b) G. Poli, G. Giambastiani and A. Heumann, Tetrahedron, 2000, 56, 5959 CrossRef CAS; (c) T. Vlaar, E. Ruijter and R. V. A. Orru, Adv. Synth. Catal., 2011, 353, 809 CrossRef CAS; (d) J. Muzart, Tetrahedron, 2013, 69, 6735 CrossRef CAS; For selected recent examples, see: (e) J. Hu, H. Hirao, Y. Li and J. Zhou, Angew. Chem., Int. Ed., 2013, 52, 8676 CrossRef CAS PubMed; (f) X. Wu, H.-C. Lin, M.-L. Li, L.-L. Li, Z.-Y. Han and L.-Z. Gong, J. Am. Chem. Soc., 2015, 137, 13476 CrossRef CAS PubMed; (g) W. Kong, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2015, 137, 16028 CrossRef CAS PubMed; (h) W. Kong, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 9714 CrossRef CAS PubMed; (i) S. Tong, A. Limouni, Q. Wang, M.-X. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 14192 CrossRef CAS PubMed; (j) J. Ye, Z. Shi, T. Sperger, Y. Yasukawa, C. Kingston, F. Schoenebeck and M. Lautens, Nat. Chem., 2017, 9, 361 CrossRef CAS PubMed; (k) H. Yoon, M. Rölz, F. Landau and M. Lautens, Angew. Chem., Int. Ed., 2017, 56, 10920 CrossRef CAS PubMed; (l) Y. J. Jang, E. M. Larin and M. Lautens, Angew. Chem., Int. Ed., 2017, 56, 11927 CrossRef CAS PubMed.
- (a) H. Yoon, Y. J. Jang and M. Lautens, Synthesis, 2016, 48, 1483 CrossRef CAS; (b) D. D. Vachhani, H. H. Butani, N. Sharma, U. C. Bhoya, A. K. Shah and E. V. Van der Eycken, Chem. Commun., 2015, 51, 14862 RSCF. Wei, L. Wei, L. Zhou, C.-H. Tung, Y. Ma and Z. Xu, Asian J. Org. Chem., 2016, 5, 971 CrossRef CAS; Z. Jiang, L. Hou, C. Ni, J. Chen, D. Wang and X. Tong, Chem. Commun., 2017, 53, 4270 RSC.
- For selected other examples of transition-metal-catalyzed carboborylation of olefins, see: (a) K. B. Smith, K. M. Logan, W. You and M. K. Brown, Chem. - Eur. J., 2014, 20, 12032 CrossRef CAS PubMed; (b) K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 7567 CrossRef CAS PubMed; (c) T. Jia, P. Cao, B. Wang, Y. Lou, X. Yin, M. Wang and J. Liao, J. Am. Chem. Soc., 2015, 137, 13760 CrossRef CAS PubMed; (d) K. M. Logan and M. K. Brown, Angew. Chem., Int. Ed., 2017, 56, 851 CrossRef CAS PubMed; (e) K. B. Smith and M. K. Brown, J. Am. Chem. Soc., 2017, 139, 7721 CrossRef CAS PubMed; (f) S. R. Sardini and M. K. Brown, J. Am. Chem. Soc., 2017, 139, 9823 CrossRef CAS PubMed; (g) Y. Huang, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2017, 56, 13314 CrossRef CAS PubMed; (h) K. M. Logan, S. R. Sardini, S. D. While and M. K. Brown, J. Am. Chem. Soc., 2018, 140, 159 CrossRef CAS PubMed.
- For reviews, see: (a) A. R. Pape, K. P. Kaliappan and E. P. Kündig, Chem. Rev., 2000, 100, 2917 CrossRef CAS PubMed; (b) D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557 CrossRef CAS PubMed; (c) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed; (d) C.-X. Zhuo, C. Zheng and S.-L. You, Acc. Chem. Res., 2014, 47, 2558 CrossRef CAS PubMed.
- For reviews, see: (a) S. P. Roche, J.-J. Y. Tendoung and B. Treguier, Tetrahedron, 2015, 71, 3549 CrossRef CAS; (b) J.-B. Chen and Y.-X. Jia, Org. Biomol. Chem., 2017, 15, 3550 RSC.
- K.-J. Wu, L.-X. Dai and S.-L. You, Org. Lett., 2012, 14, 3772 CrossRef CAS PubMed.
- (a) L. Zhao, Z.-Y. Li, L. Chang, J.-Y. Xu, H.-Q. Yao and X.-M. Wu, Org. Lett., 2012, 14, 2066 CrossRef CAS PubMed; (b) K. Douki, H. Ono, T. Taniquchi, J. Shimokawa, M. Kitamura and T. Fukuyama, J. Am. Chem. Soc., 2016, 138, 14578 CrossRef CAS PubMed; (c) X. Li, B. Zhou, R.-Z. Yang, F.-M. Yang, R.-X. Liang, R.-R. Liu and Y.-X. Jia, J. Am. Chem. Soc., 2018, 140, 13945 CrossRef CAS PubMed.
- (a) C. Shen, R.-R. Liu, R.-J. Fan, Y.-L. Li, T.-F. Xu, J.-R. Gao and Y.-X. Jia, J. Am. Chem. Soc., 2015, 137, 4936 CrossRef CAS PubMed; (b) R.-R. Liu, Y. Xu, R.-X. Liang, B. Xiang and Y.-X. Jia, Org. Biomol. Chem., 2017, 15, 2711 RSC; (c) R.-X. Liang, R.-Z. Yang, R.-R. Liu and Y.-X. Jia, Org. Chem. Front., 2018, 5, 1840 RSC.
- D. A. Petrone, A. Yen, N. Zeidan and M. Lautens, Org. Lett., 2015, 17, 4838 CrossRef CAS PubMed.
- D. A. Petrone, M. Kondo, N. Zeidan and M. Lautens, Chem. - Eur. J., 2016, 22, 5684 CrossRef CAS PubMed.
- (a) R.-R. Liu, T.-F. Xu, Y.-G. Wang, B. Xiang, J.-R. Gao and Y.-X. Jia, Chem. Commun., 2016, 52, 13664 RSC; (b) R.-R. Liu, Y.-G. Wang, Y.-L. Li, B.-B. Huang, R.-X. Liang and Y.-X. Jia, Angew. Chem., Int. Ed., 2017, 56, 7475 CrossRef CAS PubMed.
- (a) S. Chen, X.-X. Wu, J. Wang, X.-H. Hao, Y. Xia, Y. Shen, H. Jing and Y.-M. Liang, Org. Lett., 2016, 18, 4016 CrossRef CAS PubMed; (b) Y. Wang, R. Liu, J. Gao and Y. Jia, Chin. J. Org. Chem., 2017, 37, 691 CrossRef CAS.
- N. Zeidan, T. Beisel, R. Ross and M. Lautens, Org. Lett., 2018, 20, 7322 CrossRef PubMed.
- K. Kubota, K. Hayama, H. Iwamoto and H. Ito, Angew. Chem., Int. Ed., 2015, 54, 8809 CrossRef CAS PubMed.
- C. Cazorla, E. Métay and M. Lemaire, Tetrahedron, 2011, 67, 8615 CrossRef CAS.
- (a) M. Gao, S. B. Thorpe and W. L. Santos, Org. Lett., 2009, 11, 3478 CrossRef CAS PubMed; (b) S. B. Thorpe, X. Guo and W. L. Santos, Chem. Commun., 2011, 47, 424 RSC; (c) M. Gao, S. B. Thorpe, C. Kleeberg, C. Slebodnick, T. B. Marder and W. L. Santos, J. Org. Chem., 2011, 76, 3997 CrossRef CAS PubMed.
- CCDC 1855293 ((+)-3a).
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1855293. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05737k |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |