DOI:
10.1039/C9SC00086K
(Edge Article)
Chem. Sci., 2019,
10, 4667-4672
Biomimetic synthesis of the natural product salviadione and its hybrids: discovery of tissue-specific anti-inflammatory agents for acute lung injury†
Received
7th January 2019
, Accepted 20th March 2019
First published on 21st March 2019
Abstract
Acute lung injury (ALI) is an inflammatory disease with no effective pharmacological treatment. The therapeutic potential of the anti-inflammatory natural product tanshinone IIA (2) for ALI is seriously impaired by its poor pharmacokinetic (PK) properties. Inspired by the unique benzo[def]carbazole-3,5-dione (BCD) core of the natural product salviadione (5), a series of furan-fused BCD hybrids of 5 with 2 was rationally designed with the aim to improve both PK properties and the anti-inflammatory activity. A biomimetic synthetic approach featuring one-pot tandem N-heterocyclization was first developed for convenient assembly of salviadione (56% overall yield over 2 steps) and the designed hybrids (35–85% yields in one step). Compared to 2, most of the resulting compounds exhibited a markedly enhanced inhibitory effect against LPS-induced release of pro-inflammatory cytokines in macrophages. Particularly, compound 15a not only possessed the most potent activity in vitro, but also exhibited significantly improved metabolic stability (4- to 7-fold enhancement), pharmacokinetic properties (T1/2 = 4.05 h; F = 30.2%), and preferable lung tissue distribution (11- to 300-fold selectivity). An in vivo study in mice showed that pretreatment with 15a at 5 mg kg−1 distinctly attenuated LPS-induced ALI via lung tissue-specific anti-inflammatory actions, indicating that the furan-fused BCD core presents a unique chemotype with promising therapeutic potential for ALI.
Introduction
Acute lung injury (ALI) is an unmet medical need and currently has no effective therapies.1 It is believed that the inflammatory storm is a critical contributor to the occurrence of ALI,2 and many released pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and IL-1β, are key players in the progression of ALI.3 As such, blocking the excessive production of these cytokines by anti-inflammatory agents including corticosteroids represents a promising strategy to prevent and treat this disease.4 However, most of these agents have no favorable benefits to ALI patients due to low efficacy and severe side effects.5 Therefore, novel anti-inflammatory agents with high efficacy and better safety are highly needed for ALI treatment.
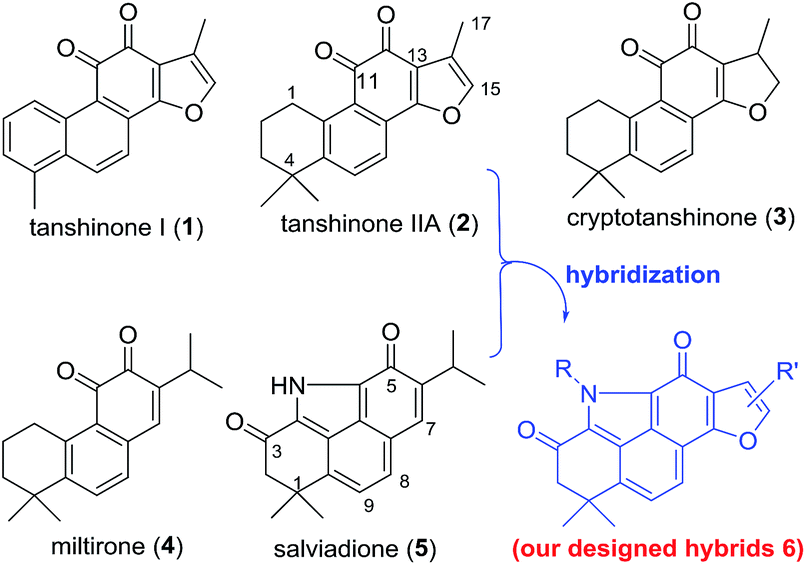
Natural products are generally valuable starting points for drug discovery. Tanshinones, such as tanshinone I (1), tanshinone IIA (2), cryptotanshinone (3), and miltirone (4), represent a group of lipophilic quinoidal diterpenes from the traditional Chinese anti-inflammatory medicine Salvia miltiorrhiza (“Danshen”) (Fig. 1).6 The anti-inflammatory effect of 2 with therapeutic potential for ALI has been well documented.7 Nevertheless, further clinical investigation of 2 to treat ALI is seriously impaired by its modest potency and poor pharmacokinetic (PK) properties likely arising from the ortho-quinone moiety.8 Since ortho-quinone is a major structural determinant for the bioactivity of tanshinones, attempts to replace or mask this moiety often counteract its bioactivity.9 Therefore, development of novel chemical approaches to modify the ortho-quinone of 2 both to improve PK properties and to retain or improve the anti-inflammatory activity still remains an unsolved challenge.
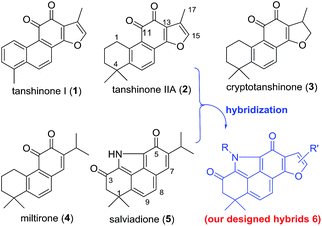 |
| | Fig. 1 Tanshinones (1–4), salviadione (5), and our designed hybrids 6. | |
Salviadione (5) is another lipophilic ingredient also isolated from “Danshen” but lacking the ortho-quinone moiety (Fig. 1).10 Though 5 was barely studied due to its extremely low natural abundance, its unique structure bearing a benzo[def]carbazole-3,5-dione (BCD) core attracted our attention. Considering the same origin as that of 2, we propose that hybridization of 5 with 2 would deliver a new compound series 6 bearing the furan-fused BCD core (Fig. 1) that may retain or improve the anti-inflammatory activity of 2. Although the tetracyclic alkaloid 5 was first synthesized in 2011 by Söderberg and co-workers with a palladium-catalyzed CO-mediated reductive N-heterocyclization as the key step (Fig. 2a),11 long reaction steps and harsh reaction conditions make this approach unfeasible for the construction of 5 on a large scale. Since both compounds 5 and 4 were isolated from the same plant, it is likely that 5 is biogenetically derived from 4 under an ammonia atmosphere (Fig. 2b). As such, hybrid 6 might be accessed from 2 through a similar reaction pathway (Fig. 2c). In this report, we first describe our efforts to establish a biomimetic synthesis of 5 from 4, and then apply this strategy to synthesize 6 from 2 (or its analogues). Subsequently, the anti-inflammatory activity of these compounds potentially for treating ALI is investigated as well.
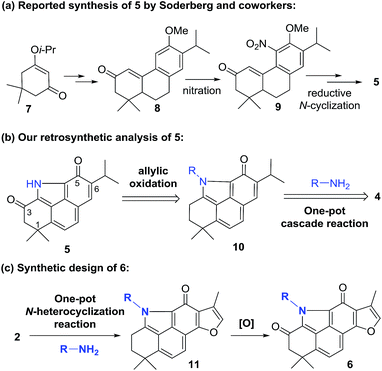 |
| | Fig. 2 Reported synthesis of 5 and our synthetic strategy for 5 and 6. | |
Results and discussion
Biomimetic synthesis of natural product 5
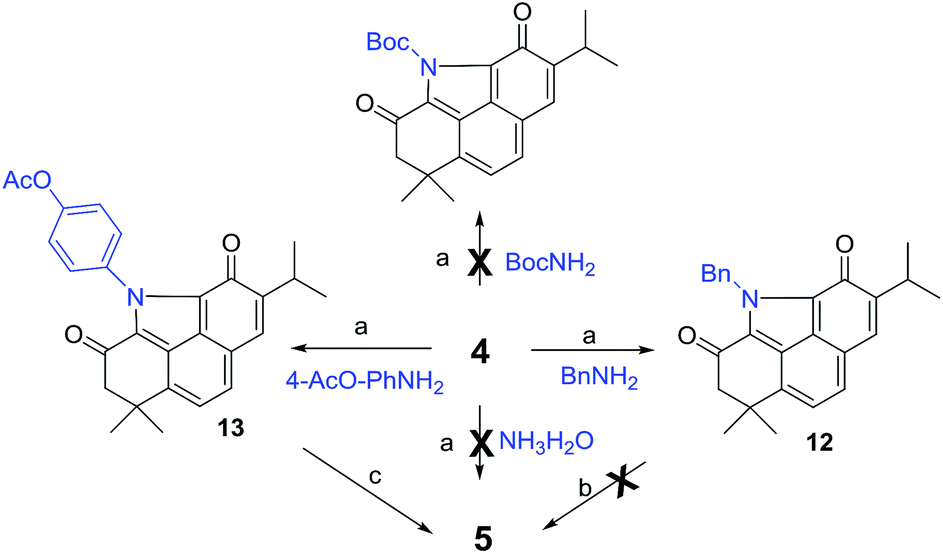
Inspired by our recent success in the Csp3–H acyloxylation or aryloxylation of 2,12 we attempted to convert 4 to 5 through amination with various amines by using TEMPO as the oxidant (Scheme 1). Initially, with ammonia (7.0 M solution in MeOH) or BocNH2 as the amino source, the reaction failed to give the desired BCD products. Gratifyingly, when benzyl amine was used, the desired N-benzyl product 12 was obtained in 60% yield. After optimization of the reaction conditions (Table S1†), treating the mixture of 4, benzyl amine, and TEMPO in toluene at 120 °C was identified as the optimal condition. Unfortunately, subsequent N-deprotection of the benzyl group catalyzed by Pd/C (10%) under a H2 atmosphere resulted in a complex mixture rather than the expected 5. Alternatively, 4-aminophenyl acetate was selected as the amination reagent. Pleasingly, the corresponding benzo[def]carbazole 13 was achieved in 62% yield under the optimal conditions. Subsequent hydrolysis with K2CO3 followed by treatment with PhI(OAc)2 successfully delivered the natural product 5 in 90% yield. The spectroscopic data of the synthetic 5 were in complete agreement with those of the isolated alkaloid10,11 (Table S2†). Thus, by means of a one-pot N-heterocyclization reaction, the biomimetic synthesis of 5 was accomplished from 4 in 56% overall yield over 2 steps.
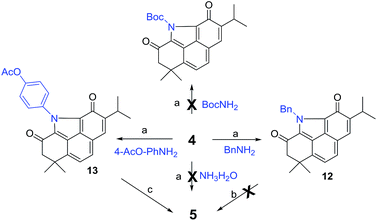 |
| | Scheme 1 Biomimetic synthesis of 5. Reagents and conditions: (a) TEMPO, toluene, 120 °C, 48 h, and 60–62%; (b) 10% Pd/C, H2, and EtOH; (c) (i) K2CO3, MeOH, and rt; (ii) PhI(OAc)2, CH3CN/H2O, 0 °C, and 90% (2 steps). | |
Synthesis of hybrid derivatives of the natural product 2 with 5
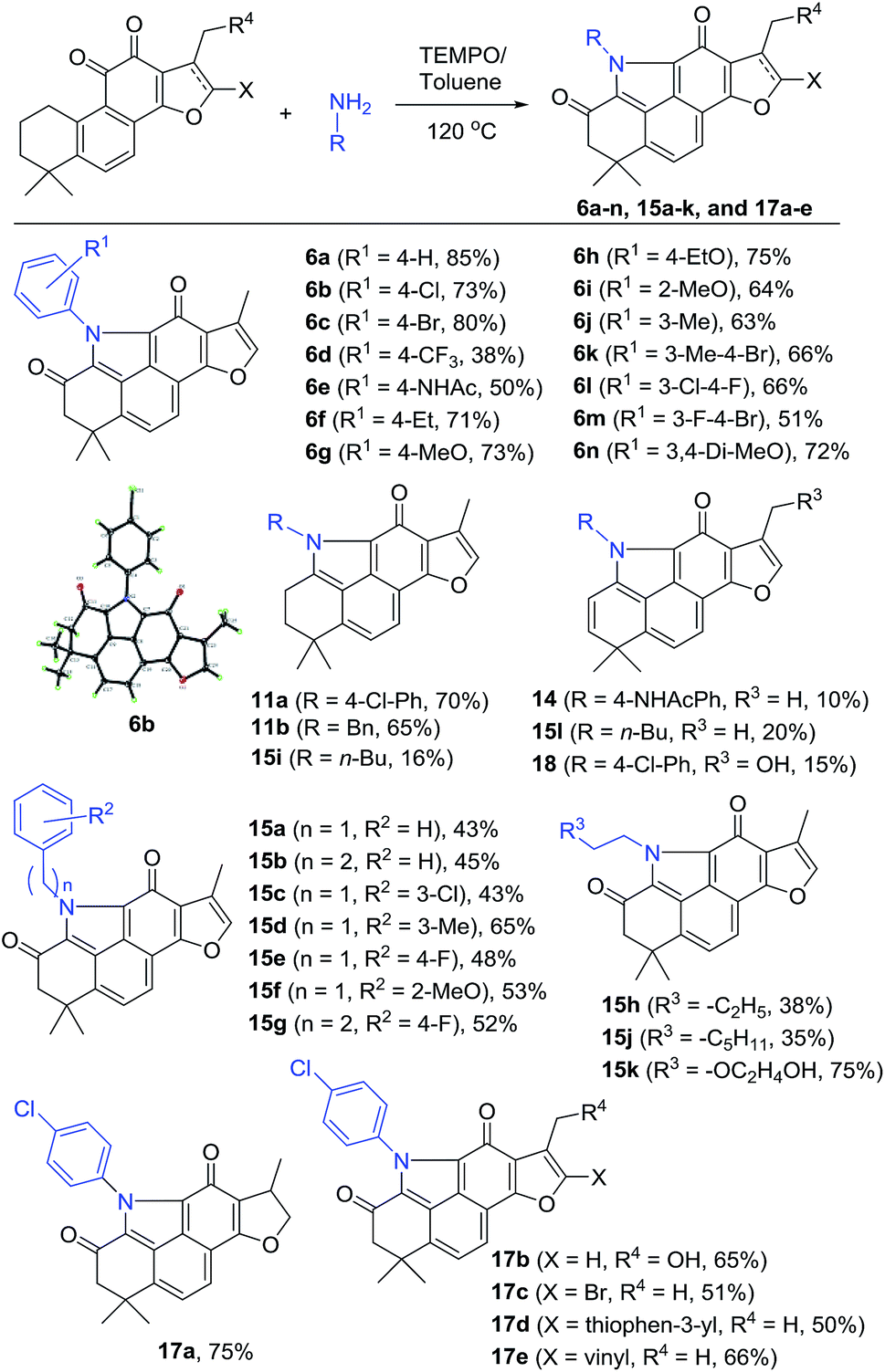
To incorporate the unique structural feature of alkaloid 5 into 2, we designed compound series 6 bearing the benzo[def]carbazole core. As shown in Scheme 2, treating 2 with various anilines under the same reaction conditions as those for 12 provided the N-aryl products 6a–n with furan-fused BCDs in 38–85% yields (Scheme 2). Among them, aniline exhibited the highest yield (85% for 6a). para-Halogenated anilines were found to give high yields (73% for 6b and 80% for 6c). The structure of 6b was unambiguously confirmed by single X-ray crystallographic analysis (Fig. S1†). Nevertheless, the aniline substrate with the para-trifluoromethyl group led to a dramatically decreased yield (38% for 6d). Anilines with electron-donating groups at the ortho-, meta-, and para-positions were well tolerated to provide 6e–j in 50–75% yields. In the case of N-(4-aminophenyl)acetamide, in addition to the major product 6e (50%), a side product 14 with the 2-ene functionality was obtained in 10% yield. 3,4-Di-substituted anilines were also compatible with the reaction and the corresponding products 6k–n were obtained in 51–72% yields.
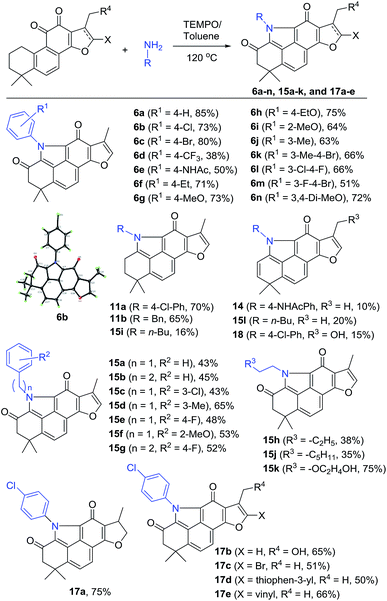 |
| | Scheme 2 Synthesis of compounds 6a–n, 15a–k, and 17a–e. | |
In addition to anilines, various alkyl amines were found to take part in the reaction as well providing the N-alkyl products 15a–h and 15j–k in relatively lower yields (mostly 35–53% except 15d and 15k), likely due to the high nucleophilic properties of alkyl amines that might undergo other side reactions (Scheme 2). For example, in the case of butan-1-amine, byproducts 15i and 15l were isolated in 16% and 20% yields, respectively, together with the major product 15h (38%). Other tanshinone analogues including 3 were also suitable as the substrate to generate the corresponding products 17a–e in 50–75% yields under the standard reaction conditions. In the case of the 17-hydroxyl substrate, the expected product 17b was obtained in 65% yield, together with the 2-ene byproduct 18 in 15% yield.
The isolated byproducts 11a–b, 14, 15i, 15l, and 18 indicated that the reaction may occur through a unique tandem N-heterocyclization process including condensation of 11-ketone of 2 with amines followed by successive imine–enamine tautomerism, intramolecular 1,6-aza-conjugate addition, and dehydrogenative aromatization to generate a key intermediate 11, such as 11a–b and 15i. Further allylic oxidation of 11 with TEMPO provides the final products. Alternatively, allylic hydroxylation of 11 followed by elimination led to side products 2-enes, such as 14, 15l and 18 (Fig. S2 in the ESI†). Indeed, in our control experiments, treatment of 2 with 4-chloroaniline or benzylamine in an argon atmosphere without oxidants yielded 11a–b in 70% and 65% yields, respectively, which were further transferred to corresponding 6b and 15a by TEMPO, respectively (Scheme S1†).
In vitro bioactivity of selected hybrid derivatives
Since TNF-α and IL-6 are two well-known pro-inflammatory cytokines that mainly contribute to inflammation-induced ALI, we first tested the inhibitory effects of compounds 2, 5, 6a–n, 15a–k and 17a–e at 10 μM against their release induced by LPS in mouse primary peritoneal macrophages. L6H21 (19)4b was used as the positive control. Most of the synthesized hybrids show markedly improved inhibitory effects (inhibition rates >50%) than the parent compounds 2 and 5 (Fig. S3†), while lacking significant cytotoxic effects on the cell viability at 10 μM (Fig. S4†), indicating that their anti-inflammatory activity was not ascribed to the cell death or injury. Compounds 6b–c, 15a–b, 15f, and 15h suppressed LPS-induced production of IL-6 and TNF-α in a dose-dependent manner (Fig. S5†), further validating their significant anti-inflammatory effects. Particularly, 15a exhibited the most potent activity against the release of IL-6 and TNF-α with IC50 values of 1.62 μM and 6.37 μM, respectively. Compound 15a was further found exhibiting negligible hERG inhibition with an IC50 value greater than 40 μM, indicating its low risk of cardiac toxicity (Table S3†).
Metabolic and pharmacokinetic properties of 15a
Compare to 2, compound 15a possessed about 4- to 7-fold enhanced metabolic stability in both human and rat microsomes (Table 1). Also, it exhibited markedly improved overall pharmacokinetic (PK) properties with a half-life (T1/2) of 4.05 h and an oral bioavailability (F) of 30.2% (Table S4†) when compared to those of 2 (T1/2 = 0.34 h; F < 3.5% (ref. 8a)). A tissue distribution study showed that 15a was mainly distributed in the lung with time extending. At 12 h, the lung drug concentration is about 11 to 300 times higher than that in other tissues (Table 2), indicating the selective lung accumulation of 15a suitably for treating ALI.
Table 1
In vitro metabolic stability in liver microsomes
| Compound |
Species |
T
1/2 (min) |
Clint in vitro (mL min−1 g−1 protein) |
|
15a
|
Human |
34.09 |
61.62 |
| Rat |
23.48 |
89.44 |
|
2
|
Human |
7.58 |
277.17 |
| Rat |
3.15 |
666.40 |
Table 2 Tissue distribution of 15a following a single intravenous administration (2 mg kg−1)a
| Tissue |
Concentration of 15a |
| 0.25 h |
2 h |
12 h |
|
Six rats were assigned to each group.
|
| Heart plasma (ng mL−1) |
144 |
31.2 |
1.90 |
| Portal vein plasma (ng mL−1) |
142 |
33.1 |
2.04 |
| Heart (ng g−1) |
992 |
223 |
19.8 |
| Liver (ng g−1) |
2557 |
518 |
57.3 |
| Lung (ng g−1) |
3320 |
847 |
633 |
| Kidney (ng g−1) |
806 |
187 |
33.0 |
| Brain (ng g−1) |
224 |
93.9 |
3.11 |
In vivo anti-ALI effect of 15a
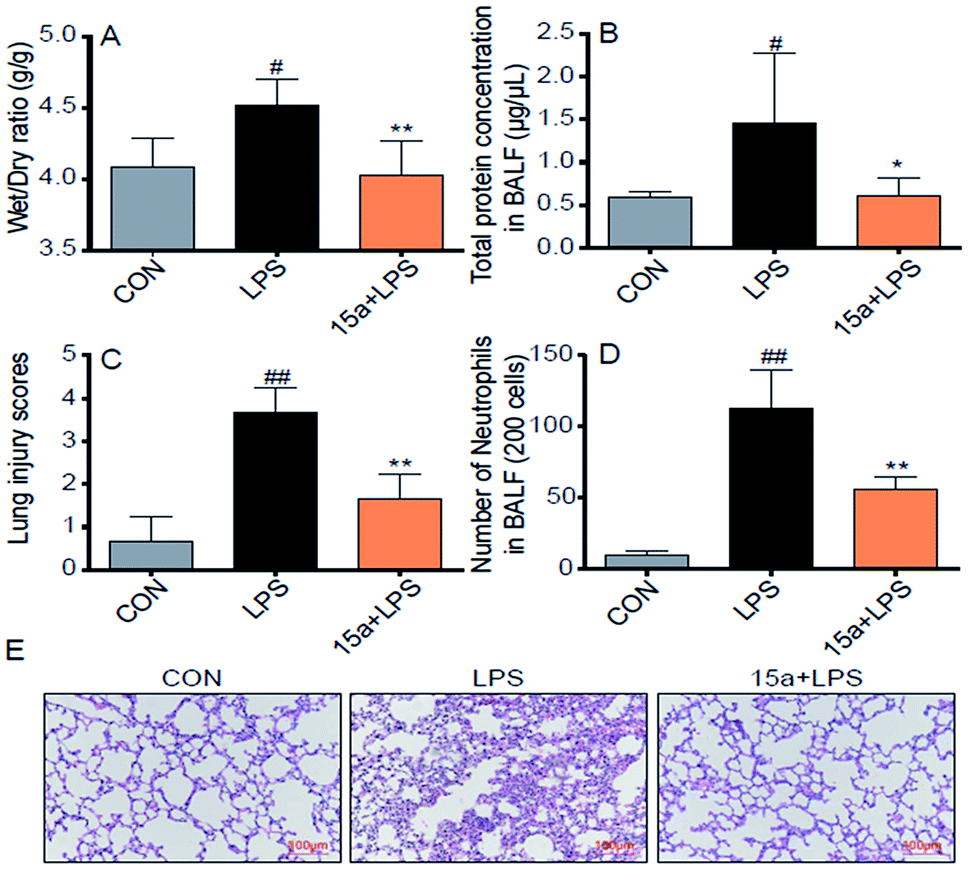
Encouraged by the potent in vitro anti-inflammatory activity as well as the promising drug-like properties, the in vivo anti-ALI effect of 15a was further evaluated. As shown in Fig. 3A, the lung wet/dry weight ratio, an index of lung edema, was significantly increased after LPS-stimulation, compared with the control group. Nevertheless, pretreatment with 15a at 5 mg kg−1 effectively reduced the lung wet/dry ratio, indicating that 15a suppressed LPS-induced lung edema. The protein concentration in mice bronchial alveolar lavage fluid (BALF) is an important indicator for the structural integrity of the alveolar wall. It was found that the protein concentration of mice BALF was markedly increased after LPS instillation, whereas the increase was significantly inhibited by pretreatment with 15a (Fig. 3B). Recruitment of neutrophils into the pulmonary compartment is also an important characteristic of ALI. The LPS challenge led to a strong increase in the number of neutrophils in the BALF (Fig. 3D), which was also significantly decreased by pretreatment with 15a. In addition, the effects of 15a on the histopathological features of ALI mice lungs were also tested (Fig. 3C and E). Compared to the normal structure of mice lung tissues, the LPS treatment remarkably increased the alveolar wall thickness, hemorrhage, alveolar collapse, and inflammatory infiltration in the lungs (Fig. 3E, middle panel). As a comparison, the group pretreated with 5 mg kg−1 of 15a displayed very little histopathological changes, when compared to the normal lung structure. In Fig. 3C, the injury score of lung tissues is well correlated with the histopathological changes as shown in Fig. 3E. All the results indicate that 15a effectively attenuates LPS-induced ALI in mice with optimal treatment potential for ALI.
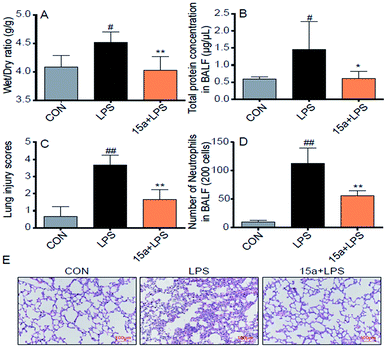 |
| | Fig. 3 Compound 15a attenuated the LPS-induced ALI in mice. (A) Wet/dry ratio. (B) The protein concentration in BALF. (C) Number of neutrophils in BALF. (D) The histogram of lung injury scores. (E) Compound 15a attenuated the LPS-induced histopathological change in lung tissue (H&E staining). Data were presented as mean ± S.E.M. *p < 0.05, **p < 0.01 vs. LPS group, #p < 0.05, ##p < 0.01 vs. CON group, and n = 7 per group. | |
Considering that pro-inflammatory cytokines play critical roles in the progression of ALI, the production levels of TNF-α and IL-6 in the BALF and serum from ALI mice were then examined as biomarkers of the in vivo anti-ALI effects of 15a. As shown in Fig. 4A and B, the levels of TNF-α and IL-6 in the BALF of LPS-induced ALI mice were distinctly suppressed by pretreatment with 15a at 5 mg kg−1; in contrast, no significant inhibition was observed in the serum (Fig. 4C and D) probably due to the low distribution of 15a in the plasma, demonstrating that 15a possesses a unique anti-inflammatory activity selectively in lung tissue, thus probably avoiding potential side effects arising from systematic inhibition of pro-inflammatory cytokines.
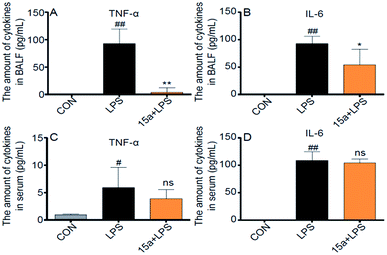 |
| | Fig. 4 Effects of compound 15a on LPS-induced acute lung inflammation in mice. (A) The amount of TNF-α in BALF. (B) The amount of IL-6 in BALF. (C) The amount of TNF-α in the serum. (D) The amount of IL-6 in the serum. Data were presented as mean ± S.E.M. *p < 0.05, **p < 0.01 vs. LPS group, #p < 0.05, ##p < 0.01, ns = not significant, vs. CON group, and n = 7 per group. | |
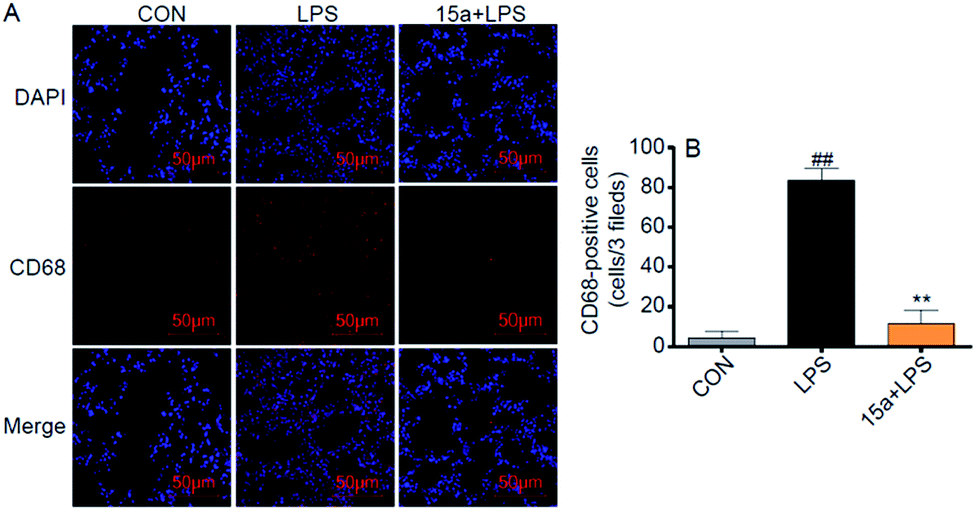
Besides, 15a markedly suppressed the mRNA expression of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-1β, and the adhesion molecule intercellular cell adhesion molecule-1 (ICAM-1), in the LPS-treated lung tissues (Fig. S6†). To validate the inhibitory effect of 15a against macrophage infiltration into the lung, immunohistochemistry analysis of CD68, a macrophage marker, was performed in the lung tissue. Pretreatment with 15a at 5 mg kg−1 significantly attenuated LPS-induced lung macrophage infiltration into the lung, as evidenced by CD68-immunostaining in Fig. 5. These data indicate that 15a improves ALI therapy via its anti-inflammatory actions including decreasing inflammatory gene expression and inhibiting macrophage infiltration.
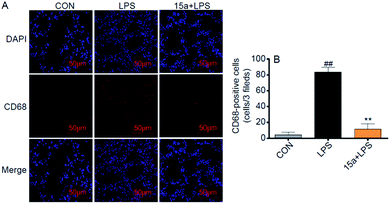 |
| | Fig. 5 Compound 15a exhibited the inhibitory effect against macrophage infiltration. (A) Representative images of CD68/DAPI immunohistochemical staining in the mice lung tissues (CD68 = red; nuclei = blue); scale bars = 50 μm. (B) Quantification of CD68 positive cells from 7 mice. **p < 0.01 vs. LPS group and ##p < 0.01 vs. CON group. | |
Conclusions
In summary, we have developed a biomimetic synthetic approach featuring one-pot tandem N-heterocyclization that allows for convenient assembly of the natural product salviadione in 56% overall yield over 2 steps as well as a series of unique hybrids of 5 with 2 bearing a furan-fused BCD core. Most of the resulting compounds exhibited markedly improved in vitro anti-inflammatory effects. Particularly, 15a was identified as the most potent with an IC50 value of 1.62 μM against the release of IL-6 induced by LPS in macrophages. In addition to its negligible cellular and cardiovascular toxicity, when compared to the natural product 2, the derivative 15a exhibited significantly improved drug-like properties including metabolic stability (4- to 7-fold enhancement), pharmacokinetic properties (T1/2 = 4.05 h; F = 30.2%) and preferable tissue distribution in the lung (11- to 300-fold selectivity). More importantly, 15a effectively attenuates LPS-induced ALI in mice via lung tissue-specific anti-inflammatory actions, thus confirming the furan-fused BCD core as a unique chemotype with ALI therapeutic potential.
Ethical statements
Protocols involving the use of animals were approved by the Wenzhou Medical University Animal Policy and Welfare Committee, and all experiments were performed in compliance with the Animal Ethics Procedures and Guidelines of the People's Republic of China.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We gratefully acknowledge financial support for this work through grants from the National Natural Science Foundation of China (21877120 and 81430080) and the National Program on Key Basic Research Project of China (2015CB910603). Support from the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program“, China (Number: 2018ZX09711002-010-001), as well as a grant from the ‘‘Personalized Medicines—Molecular Signature-based Drug Discovery and Development”, Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA12020374) are also highly appreciated.
Notes and references
-
(a) A. P. Wheeler and G. R. Bernard, Lancet, 2007, 369, 1553–1564 CrossRef;
(b) J. E. Levitt and M. A. Matthay, Crit. Care, 2012, 16, 223 CrossRef PubMed;
(c) M. Zambon and J. L. Vincent, Chest, 2008, 133, 1120–1127 CrossRef PubMed.
- J. Grommes and O. Soehnlein, Mol. Med., 2011, 17, 293–307 CAS.
-
(a) Y. C. Lu, W. C. Yeh and P. S. Ohashi, Cytokine, 2008, 42, 145–151 CrossRef CAS PubMed;
(b) M. Rincon, Trends Immunol., 2012, 33, 571–577 CrossRef CAS.
-
(a) C. S. Calfee and M. A. Matthay, Chest, 2007, 131, 913–920 CrossRef;
(b) J. Wu, J. Li, Y. Cai, Y. Pan, F. Ye, Y. Zhang, Y. Zhao, S. Yang, X. Li and G. Liang, J. Med. Chem., 2011, 54, 8110–8123 CrossRef CAS PubMed;
(c) T. Ozawa, K. Mihara and N. Yasuno, Journal of Pharmaceutical Health Care and Sciences, 2016, 2, 19 CrossRef PubMed.
- F. Gao Smith, G. D. Perkins, S. Gates, D. Young, D. F. McAuley, W. Tunnicliffe, Z. Khan and S. E. Lamb, Lancet, 2012, 379, 229–235 CrossRef.
- X. H. Wang, S. L. Morris-Natschke and K. H. Lee, Med. Res. Rev., 2007, 27, 133–148 CrossRef CAS PubMed.
-
(a) S. I. Jang, S. I. Jeong, K. J. Kim, H. J. Kim, H. H. Yu, R. Park, H. M. Kim and Y. O. You, Planta Med., 2003, 69, 1057–1059 CrossRef CAS PubMed;
(b) M. Xu, M. Q. Dong, F. L. Cao, M. L. Liu, Y. X. Wang, H. Y. Dong, Y. F. Huang, Y. Liu, X. B. Wang, B. Zhang, P. T. Zhao, Y. Luo, W. Niu, Y. Cui and Z. C. Li, Eur. J. Pharmacol., 2009, 607, 194–200 CrossRef CAS PubMed.
-
(a) H. Hao, G. Wang, N. Cui, J. Li, L. Xie and Z. Ding, Planta Med., 2006, 72, 1311–1317 CrossRef CAS PubMed;
(b) X. Y. Yu, S. G. Lin, Z. W. Zhou, X. Chen, J. Liang, P. Q. Liu, W. Duan, B. Chowbay, J. Y. Wen, C. G. Li and S. F. Zhou, Curr. Drug Metab., 2007, 8, 325–340 CrossRef CAS PubMed.
-
(a) H. Luo, J. Gao and J. Zheng, J. China Pharm. Univ., 1988, 19, 258–262 CAS;
(b) S. Xu and P. Liu, Expert Opin. Ther. Pat., 2013, 23, 149–153 CrossRef CAS PubMed.
- M. J. Don, C. C. Shen, Y. L. Lin, W. J. Syu, Y. H. Ding and C. M. Sun, J. Nat. Prod., 2005, 68, 1066–1070 CrossRef CAS PubMed.
- M. M. Cummings, R. W. Clawson Jr, S. B. Sharma, R. A. Byerly, N. G. Akhmedov and B. C. G. Söderberg, Tetrahedron, 2011, 67, 4753–4757 CrossRef CAS.
-
(a) C. Ding, J. Li, M. Jiao and A. Zhang, J. Nat. Prod., 2016, 79, 2514–2520 CrossRef CAS PubMed;
(b) B. Liang, S. Yu, J. Li, F. Wang, G. Liang, A. Zhang and C. Ding, Tetrahedron Lett., 2017, 58, 1822–1825 CrossRef CAS;
(c) J. Li, Y. Xue, Z. Fan, C. Ding and A. Zhang, J. Org. Chem., 2017, 82, 7388–7393 CrossRef CAS PubMed;
(d) C. Ding, Q. Tian, J. Li, M. Jiao, S. Song, Y. Wang, Z.-H. Miao and A. Zhang, J. Med. Chem., 2018, 61, 760–776 CrossRef CAS PubMed;
(e) F. Wang, H. Yang, S. Yu, Y. Xue, Z. Fan, G. Liang, M. Geng, A. Zhang and C. Ding, Org. Biomol. Chem., 2018, 16, 3376–3381 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of spectroscopic data and 1H and 13C NMR spectral copies of the products. CCDC 1520901. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00086k |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ac,
Hongjin
Chen‡
b,
Bin
Liang
a,
Mingkun
Jiao
a,
Guang
Liang
*b and
Ao
Zhang
ac,
Hongjin
Chen‡
b,
Bin
Liang
a,
Mingkun
Jiao
a,
Guang
Liang
*b and
Ao
Zhang