
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstructing new metal–organic frameworks with complicated ligands from “One-Pot” in situ reactions†
Xiang-Jing
Kong
,
Tao
He
 ,
Yong-Zheng
Zhang
,
Xue-Qian
Wu
,
Si-Nan
Wang
,
Ming-Ming
Xu
,
Guang-Rui
Si
and
Jian-Rong
Li
*
,
Yong-Zheng
Zhang
,
Xue-Qian
Wu
,
Si-Nan
Wang
,
Ming-Ming
Xu
,
Guang-Rui
Si
and
Jian-Rong
Li
*
Beijing Key Laboratory for Green Catalysis and Separation, Department of Chemistry and Chemical Engineering, College of Environmental and Energy Engineering, Beijing University of Technology, Beijing 100124, P. R. China. E-mail: jrli@bjut.edu.cn
First published on 28th February 2019
Abstract
Metal–organic frameworks (MOFs) have emerged as one of the most fascinating libraries of porous materials. In spite of their myriad merits, practical application of most MOFs is restricted due to their high preparation cost because of the complicated organic ligands involved. To address this limitation, we propose to use simple and cheap organic precursors to synthesize MOFs with complicated ligands via “one-pot” in situ reactions of these precursors along with the formation of new MOFs. In this work, we have carefully screened several organic reactions, through which target ligands were generated in situ from easily available reactants during the MOF construction. With this “one-pot” approach, the fabrication of a series of novel MOFs by integrating the organic covalent bond and the coordinate bond has thus been realized through the judicious selection of organic reactions, which effectively simplifies the MOF synthesis process and thus reduces the cost.
Introduction
Considerable efforts have been devoted to the rapid development of metal–organic frameworks (MOFs).1 Up to now, abundant MOFs with intriguing structures and unique properties have been constructed under the guidance of classical/efficient synthetic approaches of coordination networks, such as reticular chemistry, topology-guided design and synthesis, ligand exchange/insertion, and post-synthesis modification.2 In the course of expanding the MOF family, the structure of involved organic ligands is becoming more and more complicated, accordingly demanding researchers to have higher organic synthetic skills and inevitably raising the production expenditure of MOFs.3 Due to the use of complicated ligands, the desired MOFs are obtained not only at the expense of long reaction time and high cost but also with great restriction on their further development from academic exploration to practical industrial application. Therefore, new strategies have been expected to extend the methodology of MOF synthesis to effectively simplify the synthetic procedure and reduce the cost of exploiting new MOFs.A combination of in situ ligand generation and hydro- or solvo-thermal coordination reaction has already been adopted for the discovery of new organic ligands and MOFs.4 This eco-friendly strategy evades the necessity of ligand synthesis prior to building MOFs, in particular that of complicated ones with arduous preparation. Furthermore, it also provides significant insight into the self-assembly of MOFs driven by the co-occurrence of organic covalent bonds and coordinate bonds, for further optimizing the crystallinity of the target compounds. However, previous reports on in situ ligand synthesis mostly involve simple reactions, in which products are usually unpredictable.5 Herein, we intend to develop an avenue to target new MOFs with intricate ligands, which can be generated from the in situ reaction of simple starting materials or readily available precursors, through deft crystal engineering. These ligands are always difficult or even impossible to obtain by using common organic synthetic methods. Albeit being easy to speculate, such exquisite integration between organic reaction and MOF synthesis is quite difficult as it requires precise matching of MOF precursor reactivity and solvo-thermal coordination conditions. To realize this speculation, it is a requisite to establish the subtle balance of several competing forces, which could result in a range of kinetic or thermodynamic favored products. Hence, the desired compounds can only be formed under a narrow set of reaction conditions.
Through a careful comparison between the conditions adopted in organic ligand and MOF synthesis, several evident similarities have been found, including a polar solvent, acidic environment or protonic modulator, and heating in a sealed system.6 Such similarities imply the possibility to merge the in situ generation of eligible ligands with the fabrication of MOFs. With plenty of alternative reactions and resulting ligands, it is reasonably believed that the synergistic evolution from simple compounds to complex ones and from discrete fragments to unified frameworks, largely depending on the suitable reaction conditions, is feasible.
Among various organic reactions, here four benign reactions were tentatively selected to examine the viability of the in situ ligand transformation strategy, including (a) reduction and (b) diazo coupling of nitro-compounds, (c) condensation of boronic acids, and (d) imidization between anhydride and amine, as depicted in Scheme 1. As a proof-of-concept, this approach has indeed worked well, and a series of ligands, 4′-amino-[1,1′-biphenyl]-3,5-dicarboxylate (L62−), (E)-5′,5′′′′-(diazene-1,2-diyl)bis(([1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylate)) (L74−), 4,4′,4′′-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)-tribenzoate (L83−), and 4,4′-(1,3,6,8-tetraoxobenzo[lmn] [3,8]phenanthroline-2,7(1H,3H,6H,8H)-diyl)dibenzoate (L92−), were afforded with carefully chosen reactants in model reactions. Meanwhile, corresponding MOFs of BUT-101–109 (BUT = Beijing University of Technology) were constructed through the self-assembly of these newly formed ligands with various metal species in a “one-pot” reaction.
 | ||
| Scheme 1 In situ ligand transformation for the (a) reduction and (b) diazo coupling of nitro-compounds, (c) condensation of boronic acids, and (d) imidization between anhydride and amine. | ||
Results and discussion
The reduction of nitro-compounds is a significant reaction for the production of various amine compounds. The common approaches always focus on catalytic hydrogenation using metal catalysts or stoichiometric reducing agents, being not environmentally sustainable or accompanied by safety hazards.7 Therefore, facile reduction under mild conditions has attracted great interest. Taking the similarity of reaction conditions between the reduction of nitro- to amino-compounds and solvo-thermal MOF synthesis into consideration, such as the reductive environment and a polar solvent, an idea to use nitro-precursors directly for constructing MOFs based on amine ligands was proposed. To test our hypothesis, a series of nitro-containing compounds (HL1′–H2L6′) (Fig. S1†) varying in type, size, symmetry, and connectivity were designed and tested in the reaction. Among them, 4′-nitro-[1,1′-biphenyl]-3,5-dicarboxylic acid (H2L6′) presented the best adaptation to this system, offering a new amine compound and contributing to four new MOFs with different metal clusters (Fig. 1).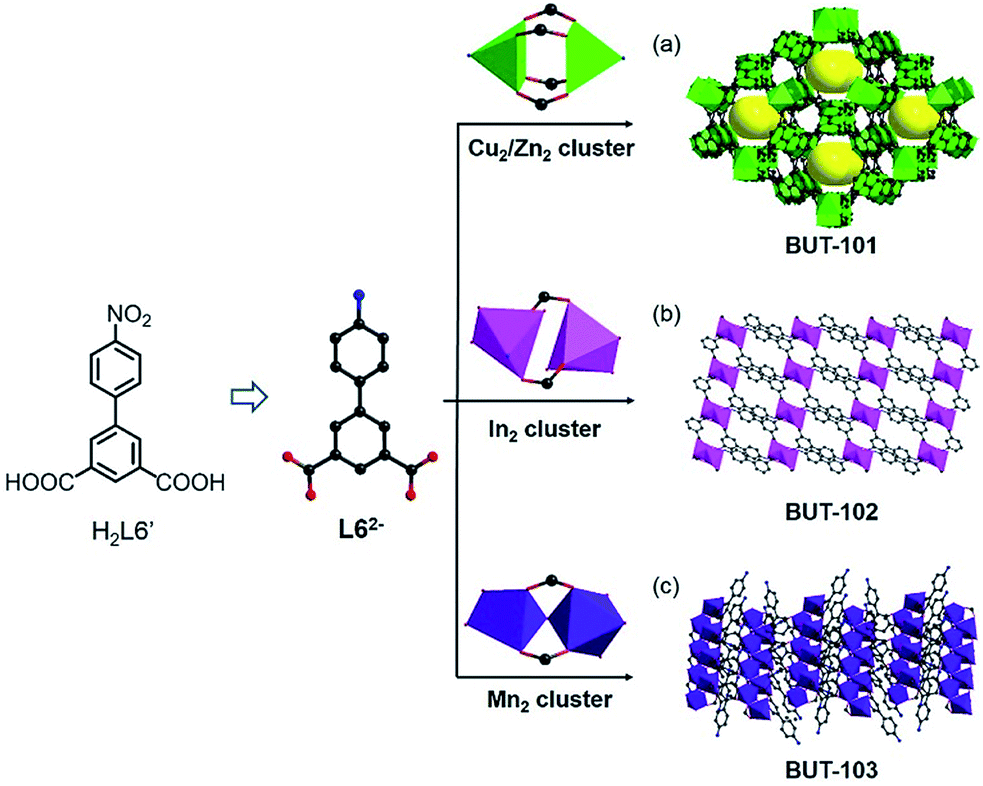 | ||
| Fig. 1 Construction and structure of (a) BUT-101, (b) BUT-102, and (c) BUT-103 with the L62− ligand generated in situ from H2L6′. Hydrogen atoms are omitted for clarity except those in the ligand precursor. | ||
The reaction of H2L6′ with Cu(NO3)2·3H2O in a DMA/H2O mixture gave crystals of BUT-101(Cu), [Cu(L6)(H2O)2], and the reaction with Zn(NO3)2·6H2O afforded BUT-101(Zn), [Zn(L6)(H2O)2]. Single-crystal X-ray diffraction (SXRD) analysis shows that BUT-101(Cu) and BUT-101(Zn) consisting of the classical M2(COO)4 paddle-wheel SBUs (SBUs = Secondary Building Units) have isostructural framework structures, resembling that of JUC-141 (ref. 8) with the eea topology (Fig. 1a and S2†). BUT-102, [Cd(L6)(H2O)], was obtained as crystals via the reaction between H2L6′ and Cd(NO3)2·4H2O in a DMF/MeOH/H2O mixture. In the structure of BUT-102, Cd2 SBUs link with L62− ligands to form a 2D layer (Fig. 1b and S3†). Similarly, the reaction between H2L6′ and MnCl2·4H2O yielded crystals of BUT-103, [Mn4(L6)4(DMF)6]·(DMF)3. It has a 2D structure in which L62− ligands are connected by Mn2 clusters (Fig. 1c and S4†). As expected, the H2L6′ in all four cases has been reduced to 4′-amino-[1,1′-biphenyl]-3,5-dicarboxylate (L62−) without purposive addition of any reductant. For BUT-101 and 102, the amino groups on the di-carboxylate ligand L62− also participate in the coordination with metal ions, generally serving as a 3-connected linker, while in BUT-103, they are free in voids and the L62− only acts as a 2-connected linker. The unique geometry and electric effect of H2L6′, probably, enable it to match well with the reductivity of these reaction systems in the presence of metal ions. These features provide it the chance to give L62−in situ, thus avoiding the pre-synthesis of the latter with environmental contamination and dangerous operation.
Inspired by the successful trial of the in situ reduction of nitro-compounds during MOF synthesis, the diazo coupling, as a tandem reaction of partial nitro compound reduction and coupling of resulting amines with intact nitro compounds in acid media,9 has sparked our next interest. It would be more challenging and interesting to obtain the azoic ligand due to the more complicated conversion pattern and sensitive reaction conditions. The above-mentioned ligands designed for nitro reduction were tested again in this reaction by varying factors such as the metal salt, dosage ratio, modulator, solvent, and temperature, but to no avail, only with the reduced compounds as before or amorphous materials being obtained. Given the size and geometry of the target ligand, directed by the concept of reticular chemistry and topology-guided synthesis, a new nitro-containing compound, 5′-nitro-[1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic acid (H2L7′) (Fig. S1†), was selected to verify this speculation. As expected, the azoic ligand (E)-5′,5′′′′-(diazene-1,2-diyl)bis(([1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylate)) (L7-a4− and -d4−) and its azoxy analogue (L7-b4− and -c4−) have been generated in situ under specific conditions and simultaneously resulted in four other new MOFs with different metal nodes (Fig. 2).
 | ||
| Fig. 2 Construction and structure of (a) BUT-104, (b) BUT-105, (c) BUT-106, and (d) BUT-107 based on L74− ligands generated in situ from H2L7′. Hydrogen atoms are omitted for clarity except those in the ligand precursor. | ||
The reaction between H2L7′ and ZrOCl2·8H2O in DMF at 120 °C afforded crystals of BUT-104, [Zr6O4(OH)8(L7-a)2(H2O)4], using formic acid as the modulator. SXRD analysis reveals that BUT-104 is constituted by the diazo-bond-coupled tetra-topic carboxylate ligand L7-a4− and typical 8-connected Zr6O8 cluster, and the entire network is similar to that of NU-901,10 NU-902,11 and NPF-300 (ref. 12) with an scu topology (Fig. 2a and S5†). In this structure, the L7-a4− ligand exhibits a planar rhomboid geometry. In contrast, the TBAPy4− in NU-901, the TCPP4− in NU-902, and 5′,5′′′′-(buta-1,3-diyne-1,4-diyl)bis(([1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylate)) in NPF-300 can all be regarded as rectangular linkers. The reaction of H2L7′ with Zn(NO3)2·6H2O in a mixed solvent of DMF/MeOH yielded crystals of [Zn5(OH)2(L7-b)2(H2O)4] (BUT-105). In addition, [Cd7(L7-c)4(H2O)12] (BUT-106) was obtained as crystals through the reaction of H2L7′ and CdCl2·2.5H2O in the presence of trace 4 M HCl aqueous solution. Interestingly, different from the azoic ligand L7-a4− in BUT-104, both the L7-b4− ligand in BUT-105 (Fig. 2b) and the L7-c4− ligand in BUT-106 (Fig. 2c) were produced in an azoxy form, probably due to the lower reaction temperature and thereby an incomplete conversion during reaction. However, the azoxy L7-c4− displays two different configurations in BUT-106 to adjust to the complex coordination of mixed Cd3 clusters and Cd(II) ions, with varying dihedral angles between the central plane and four peripheral benzene rings, while for L7-b4− in BUT-105, there is only one configuration observed (Fig. S6 and S7†). The slight structural imparity between these two azoxy ligands has rendered them suitable for different requirements of Zn(II) and Cd(II) coordination (Fig. 2b and c). Crystals of BUT-107, [In(L7-d)2], were harvested after the reaction of H2L7′ with In(NO3)3·8H2O using 4 M HCl aqueous solution as the modulator. Generally, azoic compounds are more stable in a planar conjugated structure and thus hard to distort due to the strong torsion.13 The ligands L7-a4−, L7-b4−, and L7-c4− in BUT-104, BUT-105 and BUT-106 are all nearly coplanar with respect to the connection of the respective inorganic node. It is noteworthy that, in BUT-107, the two terphenyl moieties of the azoic ligand L7-d4− are arranged in an almost perpendicular mode, which is scarce in isolated organic compounds (Fig. S8b†). This self-adapting configuration of L7-d4− makes it match well the coordination geometry of the low-symmetric single In(III) node to afford BUT-107 (Fig. 2d and S8†), representing another advantage of the in situ ligand synthesis.
It should be pointed out that the conventional synthesis of MOFs from the exact ligand is usually practicable in most cases, while for some complex ligands, especially for those inaccessible by the expertise of synthetic chemistry, this route can hardly take effect. Actually, the L74− linker is difficult to acquire under common conditions available for simple azoic ligands such as 4,4′-azobenzene-dicarboxylate and 3,3′,5,5′-azobenzene-tetracarboxylate.14 Herein, weakly reductive formic acid as well as hydrogen-donating reagents, namely methanol and water, serves as the reductant, slowly giving rise to the target linker in situ. This approach not only carves out a new way to synthesize the L74− ligand under mild conditions but also provides a series of novel MOFs, hence proven to have obvious strengths over the traditional means.
Besides these covalent bonds formed at specific reactive sites with irreversible transformation, there are many reversible reactions contributing to the diversity of covalent bonds for the assembly of covalent organic frameworks (COFs).15 Dynamic reversible reactions based on the breaking and re-formation of covalent bonds are essential for the construction of COFs, whose reversibility nature also endows them with good applicability for the assembly of MOFs based on reversible coordination bonds. This compatibility between the organic covalent bond and coordinate bond formation is amenable to the concurrent generation of target organic linkers and related coordination frameworks. Given the fact that connection of discrete arylboronic acid molecules via covalent bonds could afford larger building moieties16 and that the coordination of carboxylate ligands to Zr(IV) or Hf(IV) ions under acidic conditions usually leads to various appealing structures, the strategy for binding a new covalent bond and coordinate bond into one compound by integrating the COF bricks into a MOF using simple precursors has been audaciously proposed. Thus, we designed two model reactions: (1) the self-condensation of boronic acids by using 4-boronobenzoic acid (HL8′) as the precursor and (2) the cross-condensation between boronic acids and catechols, using HL8′ and 2,3,6,7,10,11-hexahydroxytriphenylene (L8′′) as starting materials. Through a “one-pot” approach, ZrOCl2·8H2O and HfCl4 were used in these reactions for building MOFs comprising cyclotrimerized boronic acid and dioxaborole ligands.
The reaction between HL8′ and ZrOCl2·8H2O in DMF with formic acid as the modulator offered crystals of BUT-108(Zr), [Zr6O4(OH)4(L8)2(HCOO)6(H2O)2], and similar reaction with HfCl4 gave crystals of BUT-108(Hf), [Hf6O4(OH)4(L8)2(HCOO)6(H2O)2]. SXRD analysis shows that BUT-108(Zr) and BUT-108(Hf) are isostructural and both have a two-fold interpenetrated framework. Their single framework structure, composed of distorted tetrahedral cages and a large mesoporous cage, is analogous to that of PCN-777 (ref. 17) (Fig. 3). In BUT-108, the boroxine-centred tri-carboxylate ligand L83− (4,4′,4′′-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tribenzoate) is generated from the in situ condensation of HL8′. L83− serves as a 3-connected linker, coordinating with the 6-connected Zr6 or Hf6 clusters to form the overall 3D structures of BUT-108(Zr) and BUT-108(Hf), respectively. In PCN-777, the TATB ligand displays a trigonal-planar geometry, enclosing tetrahedral cages with Zr6 clusters, while in BUT-108, three peripheral benzoic acids of the boroxine ligand are in a flat tripod shape, causing each face of the tetrahedral cage to bulge slightly (Fig. S9†). This difference can be attributed to the weak interaction between the boron atom and the O atom of HCOO− which is monodentately coordinated to the Zr6 or Hf6 cluster in another framework with high disorder, leading to the bulge of the central boroxine ring. In contrast, a lot of attempts on the reaction between HL8′ and L8′′ with Zr(IV) or Hf(IV) salts failed, with just floccule being obtained, possibly due to the bad match between cyclization and coordination conditions.
 | ||
| Fig. 3 (a) Formation of the L83− ligand from HL8′ in situ. (b) The 6-connected Zr6/Hf6 cluster in BUT-108. (c) The framework structure of BUT-108 with distorted tetrahedral cages (pink) and a large mesoporous cage (yellow). Hydrogen atoms are omitted for clarity except those in the ligand precursor. | ||
To investigate the universality of this strategy, several other reversible reactions such as the Schiff base synthesis and imidization were next examined. A Schiff base-type linker with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N linkage, 4-carboxybenzylidene-4-aminobenzoate, was then selected as the target ligand, which could be produced in situ from 4-amino benzoic acid (HL9′) and 4-formylbenzoic acid. However, it is very labile to easily decompose into aldehyde and amine compounds through hydrolysis.18 Compared with the imine connection with extreme fragility, imide-based linkers with the C–N linkage can be formed via a reversible condensation of aromatic amines and anhydrides catalyzed by aqueous acetic acid and/or at high temperature,19 exactly conforming to the conditions required for the synthesis of Zr(IV)/Hf(IV)-MOFs. Because of the higher reactivity of isochromeno[6,5,4-def]isochromene-1,3,6,8-tetraone (L9′′) in imidization compared to that of pyromellitic dianhydride (PMDA), the temperature required in the following solvothermal reaction of L9′′ could be lower, which will be more energy-effective. Therefore, in the model condensation reaction, the organic precursors HL9′ and L9′′ were employed to fabricate MOFs based on the newly generated linear imide ligand.
N linkage, 4-carboxybenzylidene-4-aminobenzoate, was then selected as the target ligand, which could be produced in situ from 4-amino benzoic acid (HL9′) and 4-formylbenzoic acid. However, it is very labile to easily decompose into aldehyde and amine compounds through hydrolysis.18 Compared with the imine connection with extreme fragility, imide-based linkers with the C–N linkage can be formed via a reversible condensation of aromatic amines and anhydrides catalyzed by aqueous acetic acid and/or at high temperature,19 exactly conforming to the conditions required for the synthesis of Zr(IV)/Hf(IV)-MOFs. Because of the higher reactivity of isochromeno[6,5,4-def]isochromene-1,3,6,8-tetraone (L9′′) in imidization compared to that of pyromellitic dianhydride (PMDA), the temperature required in the following solvothermal reaction of L9′′ could be lower, which will be more energy-effective. Therefore, in the model condensation reaction, the organic precursors HL9′ and L9′′ were employed to fabricate MOFs based on the newly generated linear imide ligand.
The reaction between HL9′, L9′′, and ZrOCl2·8H2O in DMF using acetic acid as the modulator yielded crystals of BUT-109(Zr). SXRD analysis shows that BUT-109(Zr) has a 3D framework structure constructed from Zr6 clusters and L92− ligands, seemingly adopting an ftw topology with two-fold interpenetration (Fig. 4).20 Due to the crystallographically imposed symmetry, the L92− ligand in the structure model is in disorder (Fig. S10†). The structural refinement of BUT-109(Zr) indicates that the occupancy of L92− ligands is close to 0.5. The linear di-carboxylate ligand, 4,4′-(1,3,6,8-tetraoxobenzo[lmn][3,8]-phenanthroline-2,7(1H,3H,6H,8H)-diyl)dibenzoate (L92−), was produced from the in situ condensation of HL9′ and L9′′, acting as a 2-connected linker actually. Statistically, the Zr6 clusters are linked by six L92− ligands to form the final 3D framework. Missing linkers thus lead to defects in this pseudo 4,12-connected framework and OH/OH2 entities complete the remaining coordination of the Zr6O4(OH)4 cluster. Overall, BUT-109(Zr) might have a 6-connected network with full occupancy of L92− ligands, or other cases, and the formula could be [Zr6O4(OH)10(L9)3(H2O)6]. Similarly, we performed the reaction between HL9′, L9′′, and HfCl4. Only microcrystalline materials were harvested even after numerous trials, unable to be characterized by SXRD. Nevertheless, the PXRD pattern of this MOF (BUT-109(Hf)), [Hf6O4(OH)10(L9)3(H2O)6], reveals an agreement with that of BUT-109(Zr), confirming their homogeneous structure (Fig. S13c†).
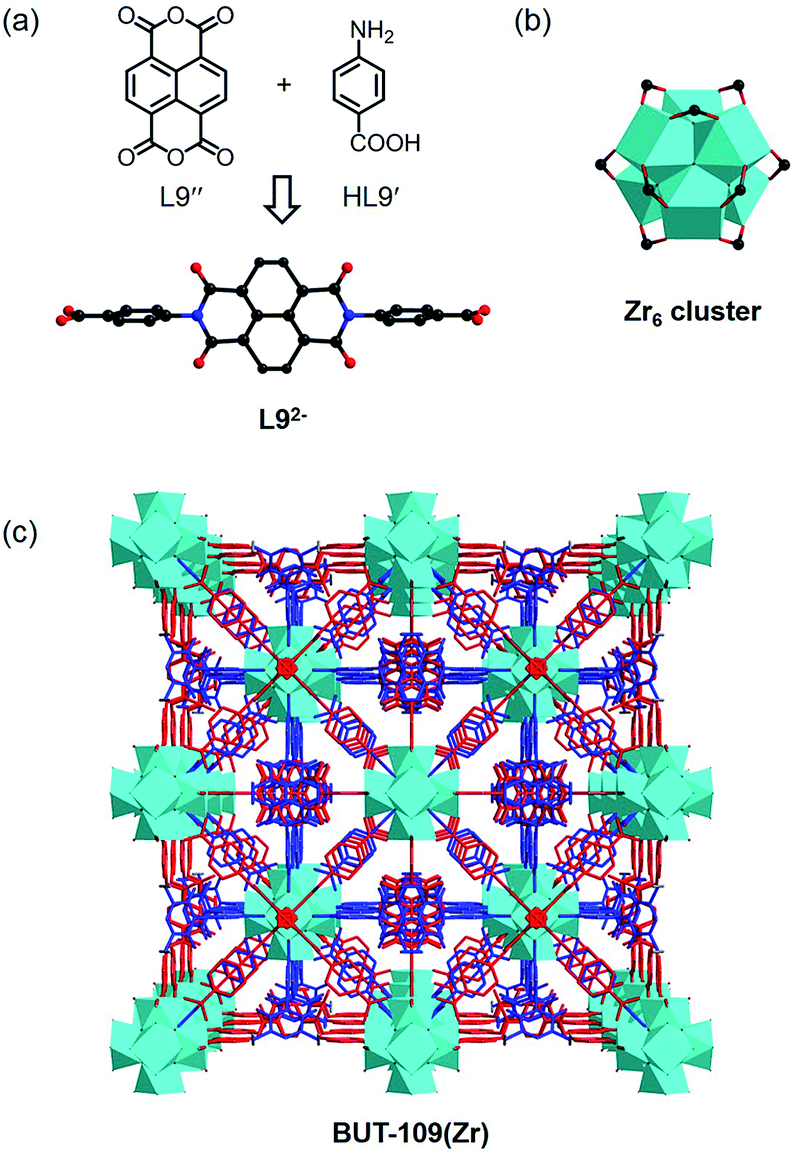 | ||
| Fig. 4 (a) Formation of the L92− ligand from HL9′ and L9′′ in situ. (b) The Zr6 cluster in BUT-109(Zr). (c) The pseudo framework structure of BUT-109(Zr) adopting an ftw net with two-fold interpenetration. Hydrogen atoms are omitted for clarity except those in the ligand precursor. | ||
In these two reversible reactions, on the subtle synergetic effect of acid modulators and water molecules from the aggregation of ligand precursors, namely the covalent bond formation, good crystallization has been achieved in the final MOFs. In the case of BUT-108, the boroxine-based ligand L83− can hardly remain intact upon heating or exposure to moisture as a pre-synthesized ligand because it is vulnerable to hydrolysis,21 hindering its direct usage in MOF assembly. For L92−, although the condensation between two precursors is possible, the “one-pot” strategy can effectively facilitate the MOF synthesis and reduce the preparation time. As a result, these two preliminary explorations contribute a new approach to the cohesion of COF and MOF construction in one framework by taking advantage of the cooperativity of covalent and coordinate bonding, thus opening a new door for synthesizing MOFs with complex organic linkers just from readily available starting materials. It should be pointed out that the existence of defects in BUT-109 due to missing linkers indeed reduce the framework stability, thus limiting the measurement of its gas adsorption. However, such a strategy also offers a new way to create hierarchical pores in target MOFs.18,22
Then, all the newly obtained MOFs (BUT-101–109) were subjected to basic characterization, including powder X-ray diffraction (PXRD), N2 adsorption/desorption, Fourier transform infrared (FT-IR), thermogravimetric analysis (TGA), and elemental analysis. To test the phase purity, samples of BUT-101–109 were checked by PXRD. As shown in Fig. S11–S13,† peaks of the simulated patterns generated from SXRD structural data and those of experimental ones are in good agreement with each other, demonstrating the phase purity of these MOFs. The elemental analysis results of twelve newly synthesized MOFs are also basically consistent with the calculations (see S9 in ESI†). To evaluate their porosity, N2 sorption measurements of activated samples were conducted at 77 K. The adsorption/desorption isotherms are shown in Fig. 5, which show N2 uptakes of 377, 363, 757, 803, 177, 573, 355 and 270 cm3 g−1 for BUT-101(Cu), BUT-101(Zn), BUT-104, BUT-105, BUT-106, BUT-107, BUT-108(Zr), and BUT-108(Hf), and their Brunauer–Emmett–Teller (BET) surface areas evaluated from the N2 adsorption data are 1150, 1071, 2346, 2410, 513, 1788, 1118, and 865 m2 g−1, respectively. No obvious uptakes were observed for BUT-102, BUT-103 and BUT-109, which may be due to their unique structural features and/or framework collapse. The FT-IR spectra of BUT-101–109 are displayed in Fig. S14–S17.† As can be found from the spectra, the stretching vibration absorption of the carboxyl groups in acid ligand precursors of H2L6′–HL9′ is in the range of 1691–1733 cm−1. After being involved in coordination with metal nodes, their characteristic peaks appear in the range of 1683–1724 cm−1 with a clear red shift. TGA curves show that the activated samples can be stable up to the temperature range of 256–452 °C, indicating moderate or good thermal stabilities of these new MOFs (Fig. S18†).
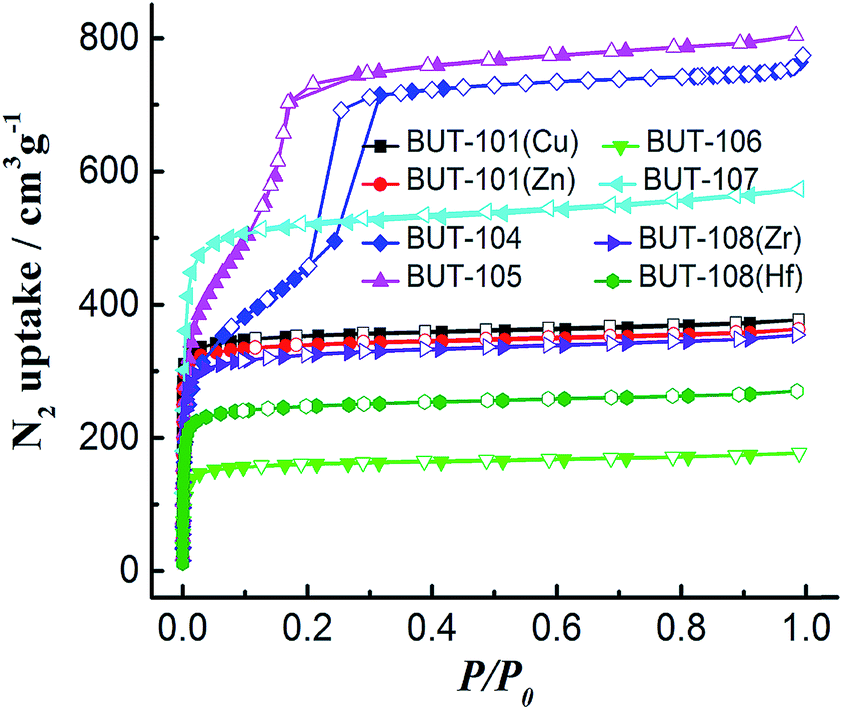 | ||
| Fig. 5 N2 adsorption/desorption isotherms of BUT-101 and BUT-104–108 at 77 K. | ||
Conclusions
In conclusion, by screening and inspecting four model organic reactions, a general strategy has been established for constructing novel MOFs via integrating the in situ generation of complicated ligands with their coordination to metal species under suitable reaction conditions. We have experimentally verified its viability through building a series of new MOFs BUT-101–109. It was demonstrated that MOFs can serve as flexible platforms to incorporate discrete organic fragments and metal ions into a highly ordered arrangement, allowing their multiple connections to provide attractive architectures. The direct use of simple or even commercially available materials for the fabrication of MOFs involving intricate linkers presents the possibility of accessing unattainable ligands without pre-synthesis, thereby saving a lot of money and time. Furthermore, the bright outlook is also forecasted to prepare MOFs with target structures and properties only starting from simple and cheap materials based on precise design at the molecular level, thus unveiling extraordinary perspectives towards their scale-up preparation and practical application in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (No. 21576006, 51621003, and 21771012).Notes and references
- (a) S. Kaskel, The Chemistry of Metal–Organic Frameworks: Synthesis, Characterization, and Applications, Wiley-VCH, Weinheim, 1st edn, 2016 Search PubMed; (b) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (c) Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef CAS PubMed; (d) C. H. Hendon, A. J. Rieth, M. D. Korzyński and M. Dincă, ACS Cent. Sci., 2017, 3, 554–563 CrossRef CAS PubMed.
- (a) N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS PubMed; (b) B. Wang, X.-L. Lv, D. Feng, L.-H. Xie, J. Zhang, M. Li, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef CAS PubMed; (c) H. Wang, X. Dong, J. Lin, S. J. Teat, S. Jensen, J. Cure, E. V. Alexandrov, Q. Xia, K. Tan, Q. Wang, D. H. Olson, D. M. Proserpio, Y. J. Chabal, T. Thonhauser, J. Sun, Y. Han and J. Li, Nat. Commun., 2018, 9, 1745 CrossRef PubMed; (d) T. Islamoglu, S. Goswami, Z. Li, A. J. Howarth, O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2017, 50, 805–813 CrossRef CAS PubMed; (e) V. Valtchev, G. Majano, S. Mintova and J. Pérez-Ramírez, Chem. Soc. Rev., 2013, 42, 263–290 RSC.
- (a) W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC; (b) F. A. Almeida Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC; (c) Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- (a) X.-M. Zhang, Coord. Chem. Rev., 2005, 249, 1201–1219 CrossRef CAS; (b) X.-M. Chen and M.-L. Tong, Acc. Chem. Res., 2007, 40, 162–170 CrossRef CAS PubMed; (c) H. Zhao, Z.-R. Qu, H.-Y. Ye and R.-G. Xiong, Chem. Soc. Rev., 2008, 37, 84–100 RSC; (d) H.-B. Zhu and S.-H. Gou, Coord. Chem. Rev., 2011, 255, 318–338 CrossRef CAS.
- (a) A. J. Blake, N. R. Champness, S. S. M. Chung, W.-S. Li and M. Schröder, Chem. Commun., 1997, 1675–1676 RSC; (b) S. Hu, J.-C. Chen, M.-L. Tong, B. Wang, Y.-X. Yan and S. R. Batten, Angew. Chem., Int. Ed., 2005, 44, 5471–5475 CrossRef CAS PubMed; (c) F. Debatin, A. Thomas, A. Kelling, N. Hedin, Z. Bacsik, I. Senkovska, S. Kaskel, M. Junginger, H. Müller, U. Schilde, C. Jäger, A. Friedrich and H.-J. Holdt, Angew. Chem., Int. Ed., 2010, 49, 1258–1262 CrossRef CAS PubMed; (d) Y. Han, H. Zheng, K. Liu, H. Wang, H. Huang, L.-H. Xie, L. Wang and J.-R. Li, ACS Appl. Mater. Interfaces, 2016, 8, 23331–23337 CrossRef CAS PubMed; (e) X.-L. Wang, R. Zhang, X. Wang, H.-Y. Lin and G.-C. Liu, Inorg. Chem., 2016, 55, 6384–6393 CrossRef CAS PubMed; (f) Y. Xu, W. Liu, D. Li, H. Chen and M. Lu, Dalton Trans., 2017, 46, 11046–11052 RSC; (g) X. Zhang, Z.-J. Wang, S.-G. Chen, Z.-Z. Shi, J.-X. Chen and H.-G. Zheng, Dalton Trans., 2017, 46, 2332–2338 RSC; (h) T. A. Grigolo, S. D. de Campos, F. Manarin, G. V. Botteselle, P. Brandão, A. A. Amaral and E. A. de Campos, Dalton Trans., 2017, 46, 15698–15703 RSC; (i) L.-I. Hung, P.-L. Chen, J.-H. Yang, C.-H. Peng and S.-L. Wang, Chem.–Eur. J., 2017, 23, 13583–13586 CrossRef CAS PubMed; (j) F. Rouhani and A. Morsali, Chem.–Eur. J., 2018, 24, 5529–5537 CrossRef CAS PubMed.
- (a) N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed; (b) F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry, Part B: Reactions and Synthesis, Springer, 5th edn, 2007 Search PubMed.
- (a) A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed; (b) N. R. Lee, A. A. Bikovtseva, M. Cortes-Clerget, F. Gallou and B. H. Lipshutz, Org. Lett., 2017, 19, 6518–6521 CrossRef CAS PubMed.
- N. Zhao, F. Sun, P. Li, X. Mu and G. Zhu, Inorg. Chem., 2017, 56, 6938–6942 CrossRef CAS PubMed.
- L. Schweighauser, M. A. Strauss, S. Bellotto and H. A. Wegner, Angew. Chem., Int. Ed., 2015, 54, 13436–13439 CrossRef CAS PubMed.
- C.-W. Kung, T. C. Wang, J. E. Mondloch, D. Fairen-Jimenez, D. M. Gardner, W. Bury, J. M. Klingsporn, J. C. Barnes, R. V. Duyne, J. F. Stoddart, M. R. Wasielewski, O. K. Farha and J. T. Hupp, Chem. Mater., 2013, 25, 5012–5017 CrossRef CAS.
- P. Deria, D. A. Gómez-Gualdrón, I. Hod, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 14449–14457 CrossRef CAS PubMed.
- X. Zhang, B. L. Frey, Y.-S. Chen and J. Zhang, J. Am. Chem. Soc., 2018, 140, 7710–7715 CrossRef CAS PubMed.
- (a) N. B. Shustova, B. D. McCarthy and M. Dincă, J. Am. Chem. Soc., 2011, 133, 20126–20129 CrossRef CAS PubMed; (b) N. B. Shustova, A. F. Cozzolino and M. Dincă, J. Am. Chem. Soc., 2012, 134, 19596–19599 CrossRef CAS PubMed.
- X.-S. Wang, S. Ma, K. Rauch, J. M. Simmons, D. Yuan, X. Wang, T. Yildirim, W. C. Cole, J. J. López, A. de Meijere and H.-C. Zhou, Chem. Mater., 2008, 20, 3145–3152 CrossRef CAS.
- L. Zhu and Y.-B. Zhang, Molecules, 2017, 22, 1149 CrossRef PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- D. Feng, K. Wang, J. Su, T.-F. Liu, J. Park, Z. Wei, M. Bosch, A. Yakovenko, X. Zou and H.-C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 149–154 CrossRef CAS PubMed.
- S. Yuan, L. Zou, J.-S. Qin, J. Li, L. Huang, L. Feng, X. Wang, M. Bosch, A. Alsalme, T. Cagin and H.-C. Zhou, Nat. Commun., 2017, 8, 15356 CrossRef CAS PubMed.
- (a) Q. Fang, Z. Zhuang, S. Gu, R. B. Kaspar, J. Zheng, J. Wang, S. Qiu and Y. Yan, Nat. Commun., 2014, 5, 4503 CrossRef PubMed; (b) S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- (a) D. Feng, H.-L. Jiang, Y.-P. Chen, Z.-Y. Gu, Z. Wei and H.-C. Zhou, Inorg. Chem., 2013, 52, 12661–12667 CrossRef CAS PubMed; (b) O. V. Gutov, W. Bury, D. A. Gomez-Gualdron, V. Krungleviciute, D. Fairen-Jimenez, J. E. Mondloch, A. A. Sarjeant, S. S. Al-Juaid, R. Q. Snurr, J. T. Hupp, T. Yildirim and O. K. Farha, Chem.–Eur. J., 2014, 20, 12389–12393 CrossRef CAS PubMed; (c) Q. Lin, X. Bu, A. Kong, C. Mao, X. Zhao, F. Bu and P. Feng, J. Am. Chem. Soc., 2015, 137, 2235–2238 CrossRef CAS PubMed; (d) S. B. Kalidindi, S. Nayak, M. E. Briggs, S. Jansat, A. P. Katsoulidis, G. J. Miller, J. E. Warren, D. Antypov, F. Corà, B. Slater, M. R. Prestly, C. Martí-Gastaldo and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2015, 54, 221–226 CrossRef CAS PubMed.
- (a) K. Ono, S. Shimo, K. Takahashi, N. Yasuda, H. Uekusa and N. Iwasawa, Angew. Chem., Int. Ed., 2018, 130, 3113–3117 CrossRef PubMed; (b) A. L. Korich and P. M. Iovine, Dalton Trans., 2010, 39, 1423–1431 RSC; (c) W. A. Marinaro, L. J. Schieber, E. J. Munson, V. W. Day and V. J. Stella, J. Pharm. Sci., 2012, 101, 3190–3198 CrossRef CAS PubMed; (d) K. L. Bhat, G. D. Markham, J. D. Larkin and C. W. Bock, J. Phys. Chem. A, 2011, 115, 7785–7793 CrossRef CAS PubMed.
- (a) H. Huang, J.-R. Li, K. Wang, T. Han, M. Tong, L. Li, Y. Xie, Q. Yang, D. Liu and C. Zhong, Nat. Commun., 2015, 6, 8847 CrossRef CAS PubMed; (b) B. Bueken, N. V. Velthoven, A. Krajnc, S. Smolders, F. Taulelle, C. Mellot-Draznieks, G. Mali, T. D. Bennett and D. De Vos, Chem. Mater., 2017, 29, 10478–10486 CrossRef CAS; (c) L. Feng, S. Yuan, L.-L. Zhang, K. Tan, J.-L. Li, A. Kirchon, L.-M. Liu, P. Zhang, Y. Han, Y. J. Chabal and H.-C. Zhou, J. Am. Chem. Soc., 2018, 140, 2363–2372 CrossRef CAS PubMed; (d) N. A. Khan, Z. Hasan and S. H. Jhung, Coord. Chem. Rev., 2018, 376, 20–45 CrossRef CAS; (e) H. N. Abdelhamid and X. Zou, Green Chem., 2018, 20, 1074–1084 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of ligands and MOFs, PXRD, FT-IR, TGA, 1H NMR, and crystal data. CCDC 1888829–1888839. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00178f |
| This journal is © The Royal Society of Chemistry 2019 |