
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe antioxidant activity of polysulfides: it's radical!†
Jean-Philippe R.
Chauvin
 ,
Markus
Griesser
and
Derek A.
Pratt
*
,
Markus
Griesser
and
Derek A.
Pratt
*
Department of Chemistry and Biomolecular Sciences, University of Ottawa, Ottawa, Ontario, Canada K1N 6N5. E-mail: dpratt@uottawa.ca
First published on 25th April 2019
Abstract
Olefin sulfurization, wherein alkenes and sulfur are heated together at high temperatures, produces branched polysulfides. Due to their anti-wear properties, they are indispensible additives to lubricants, but are also added to other petroleum-derived products as oxidation inhibitors. Polysulfides also figure prominently in the chemistry and biology of garlic and other plants of the Allium species. We previously reported that trisulfides, upon oxidation to their corresponding 1-oxides, are surprisingly effective radical-trapping antioxidants (RTAs) at ambient temperatures. Herein, we show that the homolytic substitution mechanism responsible also operates for tetrasulfides, but not trisulfides, disulfides or sulfides. Moreover, we show that this reactivity persists at elevated temperature (160 °C), enabling tetrasulfides to not only eclipse their 1-oxides as RTAs, but also hindered phenols and alkylated diphenylamines – the most common industrial antioxidant additives. The reactivity is unique to higher polysulfides (n ≥ 4), since homolytic substitution upon them at S2 yields stabilized perthiyl radicals. The persistence of perthiyl radicals also underlies the greater reactivity of polysulfides at elevated temperatures relative to their 1-oxides, since homolytic S–S bond cleavage is reversible in the former, but not in the latter. These results suggest that olefin sulfurization processes optimized for tetrasulfide production will afford materials that impart significantly better oxidation stability to hydrocarbon-based products to which polysulfides are added. Moreover, it suggests that RTA activity may contribute to the biological activity of plant-derived polysulfides.
Introduction
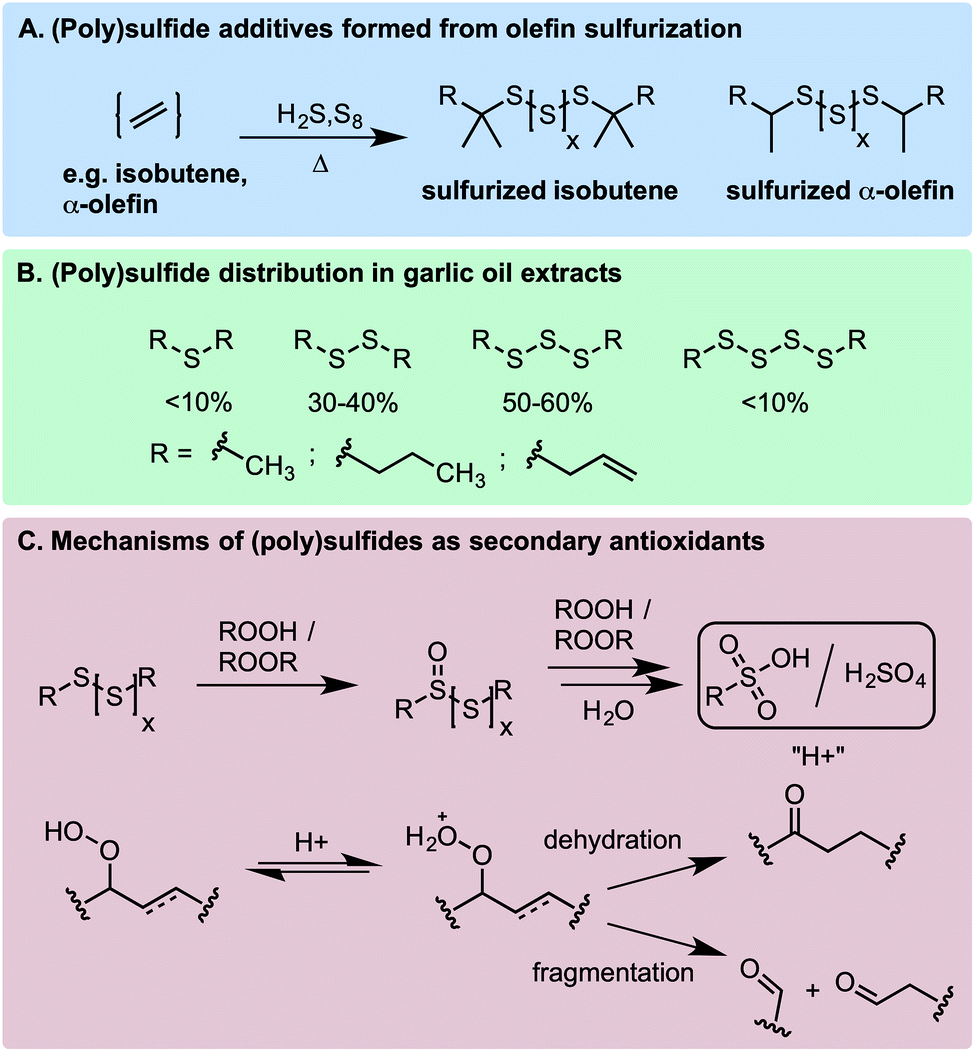
It was well recognized before the mid-20th century that the oxidative stability of normally refined petroleum used for the production of lubricants and other products was largely the result of the small quantities of organosulfur compounds naturally present and not the inherent stability of the hydrocarbon fraction itself.1,2 However, great efforts were made to remove the organosulfur compounds during the refining process due to their propensity to form corrosion-inducing acids. Over time, synthetic organosulfur compounds were introduced into lubricants (and some other petroleum-derived products) because of their excellent anti-wear properties, particularly at extreme pressures,3–5 and those compounds with the best balance of maximum anti-wear properties and minimum corrosive effects were identified, developed, and incorporated into commercial products.Sulfurized olefins (polysulfides), obtained by the treatment of alkenes with elemental sulfur at elevated temperatures (Scheme 1A), are among the most common organosulfur additives to petroleum-derived products.6,7 Polysulfides also figure prominently in the chemistry and biology of Allium species, which include garlic and petiveria (Scheme 1B).8,9 Their medicinal properties are well recognized, but the mechanism(s) responsible – often linked to their ability to inhibit oxidative stress – are poorly understood.10,11 In an industrial context, polysulfides are described as ‘secondary antioxidants’ since they are known to react with hydroperoxides to produce alcohols, inhibiting the auto-initiation of autoxidation chain reactions that degrade hydrocarbons (Scheme 1C).2,12,13 Moreover, the oxidation of polysulfides eventually leads to the formation of sulfur oxyacids, which catalyze the decomposition of hydroperoxides by either dehydration or Hock fragmentation to carbonyl compounds (Scheme 1C).14,15 This is in contrast with ‘primary’ antioxidants, such as hindered phenols (e.g. BHT) and alkylated diphenylamines, which react with chain-carrying peroxyl radicals to interrupt chain propagation (leading to their classification as radical-trapping antioxidants – RTAs).4,5
 | ||
| Scheme 1 | ||
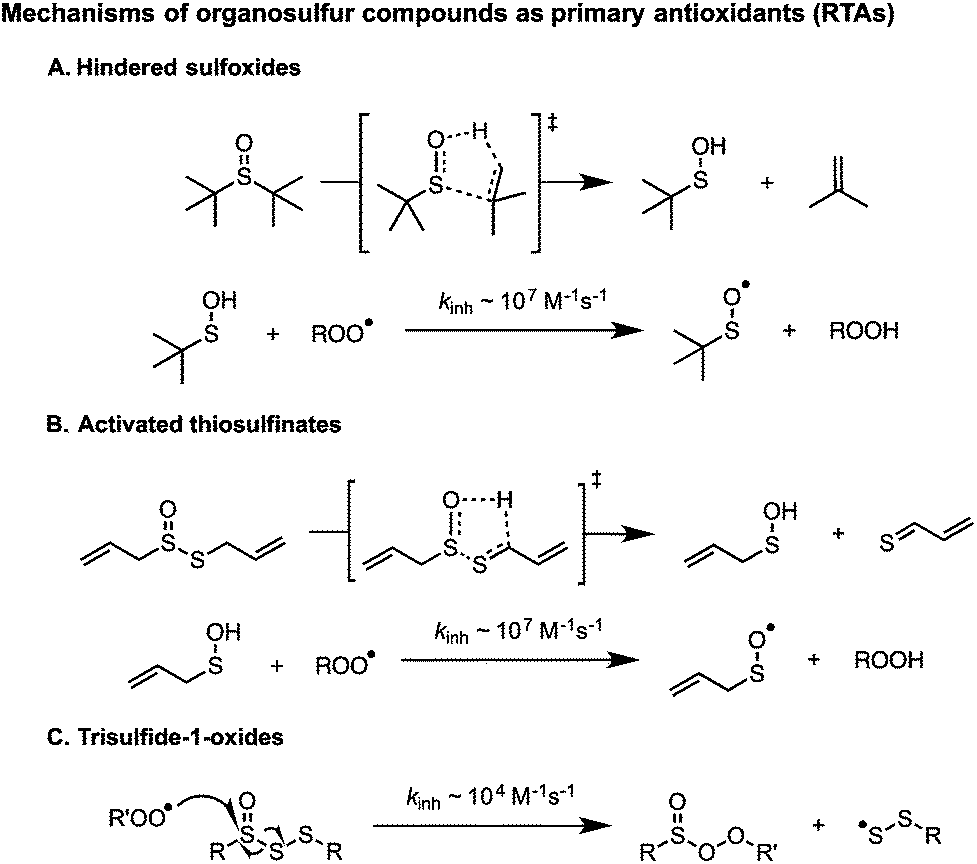
In 1974, Koelewijn and Berger16 provided evidence that organosulfur compounds can act as RTAs. They showed that di-tert-butylsulfide, upon oxidation to the corresponding sulfoxide, is a good RTA. They proposed that the sulfoxide undergoes a Cope-type elimination to yield a sulfenic acid (Scheme 2A), which they surmised would undergo fast reactions with peroxyl radicals. Since sulfenic acids are transient species that rapidly undergo self-condensation to yield thiosulfinates, they were unable to directly determine the kinetics of the reaction, but estimated a rate constant of ∼107 M−1 s−1 based on the rates of inhibited autoxidations of tetralin and sulfoxide thermolysis (to produce t-BuSOH) at 60 °C. Several years later, we showed that this sequence of reactions is also responsible for the RTA activity of allicin and petivericin, the odorous thiosulfinates derived from garlic and petiveria, respectively (Scheme 2B).17–19 Unactivated thiosulfinates were unreactive as RTAs under the same conditions (37 °C).
 | ||
| Scheme 2 | ||
We recently reported that trisulfide-1-oxides react with peroxyl radicals with kinh = 1–2 × 104 M−1 s−1 at 37 °C (Scheme 2C).20 As such, they are similarly reactive to the most common industrial RTAs (hindered phenols) and suggests that the antioxidant activity of sulfurized olefins may not stem solely from secondary antioxidant reactions, but also from their reactivity as RTAs. Experimental and computational investigations suggested that the reaction proceeds via a concerted bimolecular homolytic substitution by the peroxyl radical at the oxidized sulfur center, liberating a highly-stabilized perthiyl radical. The corresponding trisulfides were unreactive under the same conditions. We have since wondered if higher polysulfides would be amenable to direct substitution by peroxyl radicals. The rationale is straightforward; on going from the trisulfide to tetrasulfide (and higher), the S–S bond strength weakens considerably (53.4 to 36.3 kcal mol−1, respectively),21 such that it approaches the RS(O)–SSR bond strength (29.8 kcal mol−1).20 Moreover, we wondered how this reactivity would translate from the biologically-relevant temperatures at which we have studied the reactions of the activated thiosulfinates and trisulfide-1-oxides to more industrially-relevant (elevated) temperatures. Would the polysulfides and/or their 1-oxides be too labile to be effective in this capacity? Herein we address these questions as part of a comprehensive study of the RTA activity of polysulfides (n = 1–4) and their corresponding 1-oxides at 37, 100, and 160 °C.
 | ||
| Chart 1 | ||
Results
tert-Butylated (poly)sulfides were chosen as model substrates because of their structural similarity to the common sulfurized olefin, sulfurized di(isobutylene) (see Chart 1). These compounds were either obtained from commercial sources (1 and 2) or prepared in one step from tert-butylthiol and SCl2 (3) or S2Cl2 (4). The corresponding 1-oxides 5–8 were obtained by treating the parent (poly)sulfide with one equivalent of m-CPBA. Complete details are available in the ESI.†Inhibited autoxidation of 1-hexadecene
The reactivity of the tert-butyl polysulfides (1–4) and their corresponding 1-oxides (5–8) as inhibitors of hydrocarbon autoxidation was first evaluated in 1-hexadecene at 100 °C using the PBD-BODIPY co-autoxidation approach.22,23 This method, which is illustrated in Fig. 1A and B, utilizes the highly-absorbing and highly-autoxidizable PBD-BODIPY to enable monitoring of the autoxidation by conventional spectrophotometry. The reactivity of the polysulfides and their 1-oxides to peroxyl radicals can be derived from their ability to suppress the initial rate of PBD-BODIPY consumption using eqn (1) and the stoichiometry of the reaction determined from the length of the inhibited period of the autoxidation using eqn (2). Representative data for the polysulfides and their corresponding 1-oxides are given in Fig. 1C and D, respectively. Corresponding data obtained at 37 °C are shown in Fig. 1E and F, respectively.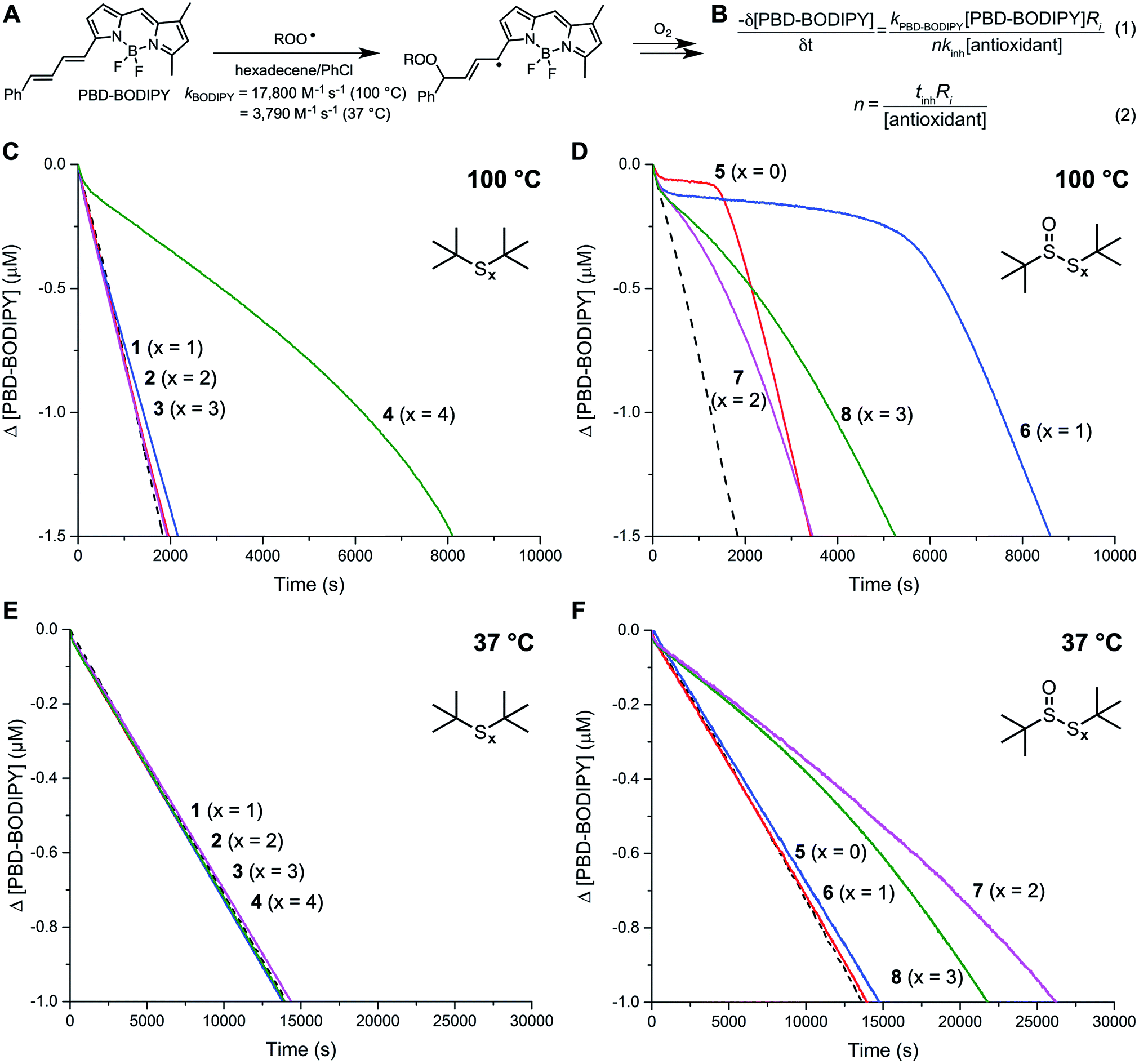 | ||
Fig. 1 PBD-BODIPY is the signal carrier in 1-hexadecene autoxidations (A), enabling determination of rate constants (kinh) and stoichiometries (n) for reactions of inhibitors with chain-carrying peroxyl radicals (B). Co-autoxidations of 1-hexadecene (2.8 M) and PBD-BODIPY (10 μM) initiated with dicumylperoxide (1 mM) in chlorobenzene at 100 °C (dashed black trace) and inhibited by 50 μM of organosulfur compounds 1–4 (C) and 5–8 (D). Reaction progress was monitored by absorbance at 587 nm (ε = 119![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 166 M−1 cm−1). Co-autoxidations of 1-hexadecene (2.8 M) and PBD-BODIPY (10 μM) initiated with AIBN (6 mM) in chlorobenzene at 37 °C (dashed black trace) and inhibited by 50 μM of organosulfur compounds 1–4 (E) and 5–8 (F). Reaction progress was monitored by absorbance at 588 nm (ε = 148700 M−1 cm−1). 166 M−1 cm−1). Co-autoxidations of 1-hexadecene (2.8 M) and PBD-BODIPY (10 μM) initiated with AIBN (6 mM) in chlorobenzene at 37 °C (dashed black trace) and inhibited by 50 μM of organosulfur compounds 1–4 (E) and 5–8 (F). Reaction progress was monitored by absorbance at 588 nm (ε = 148700 M−1 cm−1). | ||
Of the polysulfides, only the tetrasulfide (4) inhibited PBD-BODIPY oxidation, with the initial rate giving rise to kinh = (2.3 ± 0.4) × 105 M−1 s−1 at 100 °C. The stoichiometry of peroxyl radical trapping was determined to be n = 1.4 ± 0.2, indicating that each molecule of tetrasulfide halts between one and two chain reactions. The sulfide (1), disulfide (2), and trisulfide (3) were unable to suppress the rate of autoxidation. In contrast, each of the polysulfide-1-oxides were inhibitors, albeit with differing activity. The sulfoxide (5) and thiosulfinate (6) were the most reactive compounds, with kinh = (1.4 ± 0.6) × 106 and (2.8 ± 0.6) × 106 M−1 s−1, respectively, but they had significantly different stoichiometries of n = 0.4 ± 0.1 and 1.0 ± 0.1, respectively. The trisulfide-1-oxide (7) and tetrasulfide-1-oxide (8) were significantly slower to react with peroxyl radicals, yielding kinh = (1.3 ± 0.4) × 105 and (1.6 ± 0.5) × 105 M−1 s−1, while their stoichiometries were similar to that of the sulfoxide, with n = 0.4 ± 0.1 and 0.5 ± 0.1, respectively. None of the polysulfides were able to inhibit the autoxidations carried out at 37 °C, whereas the trisulfide-1-oxide and tetrasulfide-1-oxide were effective, yielding kinh = (5.6 ± 0.9) × 104 and (3.1 ± 0.8) × 104 M−1 s−1 with n = 0.7 ± 0.1 and 0.6 ± 0.1, respectively – fully consistent with previous observations.20 For comparison, under the same conditions, BHT and an alkylated diphenylamine have kinh = 3.0 × 104 and 1.7 × 105 M−1 s−1 at 37 °C and 2.0 × 105 and 9.9 × 105 M−1 s−1 at 100 °C, respectively (see ESI† for the data).
Mechanism of polysulfide reactivity
Since the tetrasulfide lacks labile H-atoms and there is no obvious reaction by which it could form a good H-atom donor in situ, there are limited mechanisms that may account for their good RTA activity. Each possibility must be consistent with the observation that the tetrasulfide is a good RTA under conditions where the trisulfide is not. Three possibilities were identified: (1) the tetrasulfide reacts with the dicumyl peroxide initiator (as a secondary antioxidant), and therefore only appears to be an RTA, (2) the tetrasulfide undergoes homolysis to yield two perthiyl radicals, which can combine with chain-carrying peroxyl radicals to yield stable products, and (3) the tetrasulfide undergoes homolytic substitution by a peroxyl radical similarly to the reaction of trisulfide-1-oxides and tetrasulfide-1-oxides at lower temperatures (i.e.Scheme 2C).20It has been reported that the rate of reaction of polysulfides with peroxides is proportional to the number of sulfur atoms in the polysulfide.24 On this basis, it is expected that the tetrasulfide would react with either the initiating peroxide – or product hydroperoxides – faster than the trisulfide. To probe the intervention of any secondary antioxidant behaviour by the polysulfides under the reaction conditions, we heated compounds 3 and 4 (1 equiv.) with either dicumyl peroxide or a model hydroperoxide (tetralin hydroperoxide) (5 equiv.) at 100 °C (see ESI†). No significant change was observed in the rate of peroxide decomposition over three hours in each case (as determined by HPLC; data not shown), excluding a role for peroxide decomposition in the antioxidant activity of the tetrasulfide under these conditions.
As mentioned in the Introduction, the central S–S bond in a tetrasulfide is 17 kcal mol−1 weaker than the S–S bond in a trisulfide due to the greater stability of perthiyl radicals over thiyl radicals. To establish whether S–S homolysis is efficient under the conditions of the autoxidation, an equimolar solution of di-iso-propyltetrasulfide (9) and di-tert-butyltetrasulfide (8) was heated at 100 °C under air and monitored by GC. After ca. 2 hours, roughly equimolar amounts of di-tert-butyltetrasulfide, di-iso-propyltetrasulfide and tert-butyl-iso-propyltetrasulfide were observed, supporting the equilibrium in Fig. 2A. It is important to note that the reaction progress was essentially identical in the presence/absence of air, consistent with our previous observations that perthiyl radicals do not react with O2.20,23 (When analogous experiments were carried out with the corresponding trisulfide, no scrambling was observed – consistent with its significantly stronger S–S bond.) The data were fit to a kinetic model depicting the reactions shown in Fig. 2A, where input of the concentrations of the starting symmetrical tetrasulfides and the known recombination rate constant (krec = 6 × 109 M−1 s−1),20 yielded a homolysis rate constant for the tetrasulfide of 2.9 × 10−4 s−1 at 100 °C, in reasonably good agreement with an estimated rate constant of 4.5 × 10−5 s−1 derived from the calculated S–S BDE (at 25 °C). Thus, although homolysis of the tetrasulfide may be competitive with chain propagation, the highly-reversible nature of the reaction suggests that insufficient perthiyl radical will accumulate to intercept a chain-carrying peroxyl radical.
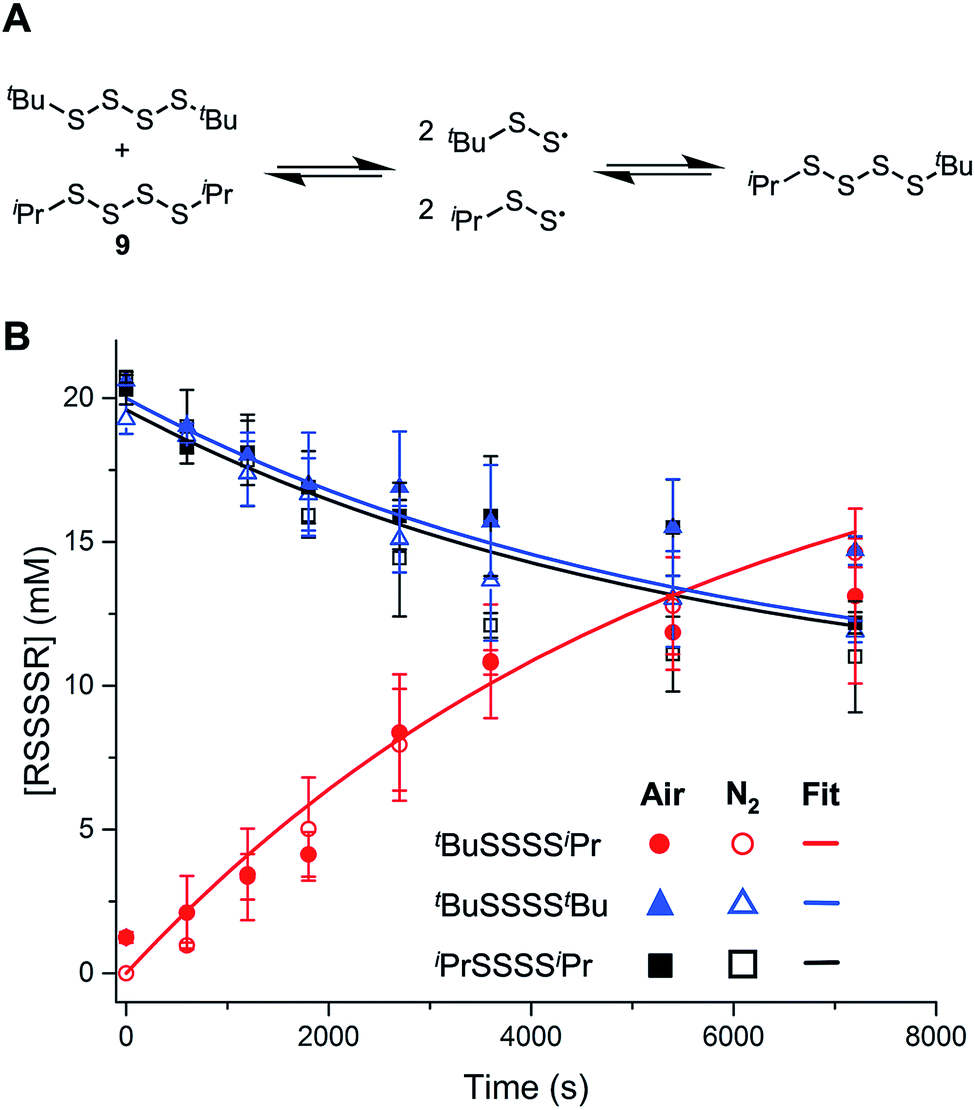 | ||
| Fig. 2 Thermal equilibration of tetrasulfides. (A) Scrambling experiment of 4 (20 mM) and 9 (20 mM) in PhCl at 100 °C under an atmosphere of air (solid symbols) or nitrogen (empty symbols). Solid lines are from numerical fitting of the data obtained under nitrogen to the reaction mechanism shown above given the known rate constant of perthiyl radical recombination of krec = 6 × 109 M−1 s−1 (B). | ||
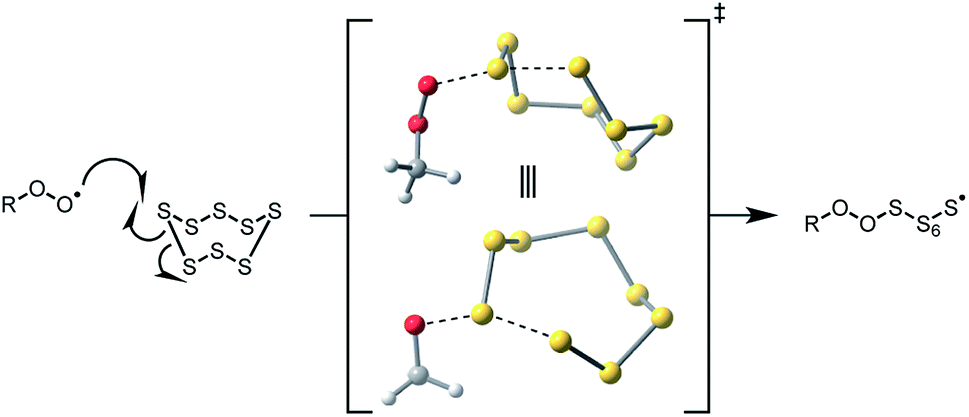
The remaining possibility – homolytic substitution – can occur at either S1 or S2 of the tetrasulfide, which yield similarly stabilized perthiyl radicals (RSSS˙ and RSS˙, respectively).20 As such, the reaction at S2 may be expected on steric grounds. In contrast, reactions of the trisulfide at S1 and S2 yield perthiyl and thiyl radicals, respectively. Given the far greater stability of the former than the latter (∼17 kcal mol−1),23 substitution at S1 is expected to be the preferred path on thermodynamic grounds, but because of steric hinderance of the adjacent t-butyl group, chemistry may be directed to S2. Indeed, in our previous work, we found substitution at the sulfinyl (S1) sulfur atom of the trisulfide-1-oxides can be subject to profound steric effects.20 This may explain why the tetrasulfide displays good RTA activity in the foregoing experiments while the trisulfide does not: the substitution on the most accessible sulfur atom yields radicals of significantly different stability.
To probe the regioselectivity of substitution, we examined the reactivity of a less hindered tetrasulfide (di-iso-propyl tetrasulfide 9) and found it to be a slightly poorer inhibitor than di-tert-butyl tetrasulfide (compare kinh = 1.3 ± 0.2 to 2.3 ± 0.4 × 105 M−1 s−1, respectively – see ESI† for the data). If substitution at S1 were the mechanism, it is expected that the di-iso-propyl tetrasulfide would be far more reactive.25 To provide some corroboration for homolytic substitution at S2 being operative in the reaction of tetrasulfides with peroxyl radicals, we turned to computations – the results of which are summarized in Fig. 3. Fully consistent with the above considerations, peroxyl radical attack is predicted by CBS-QB3 calculations to be most favourable at S2 of the tetrasulfide (compare ΔG‡ = 19.9 kcal mol−1 to 22.4 kcal mol−1 for substitution at S1 at 25 °C) and S1 of the trisulfide (compare ΔG‡ = 21.2 kcal mol−1 to 26.4 kcal mol−1 for substitution at S2 at 25 °C).26
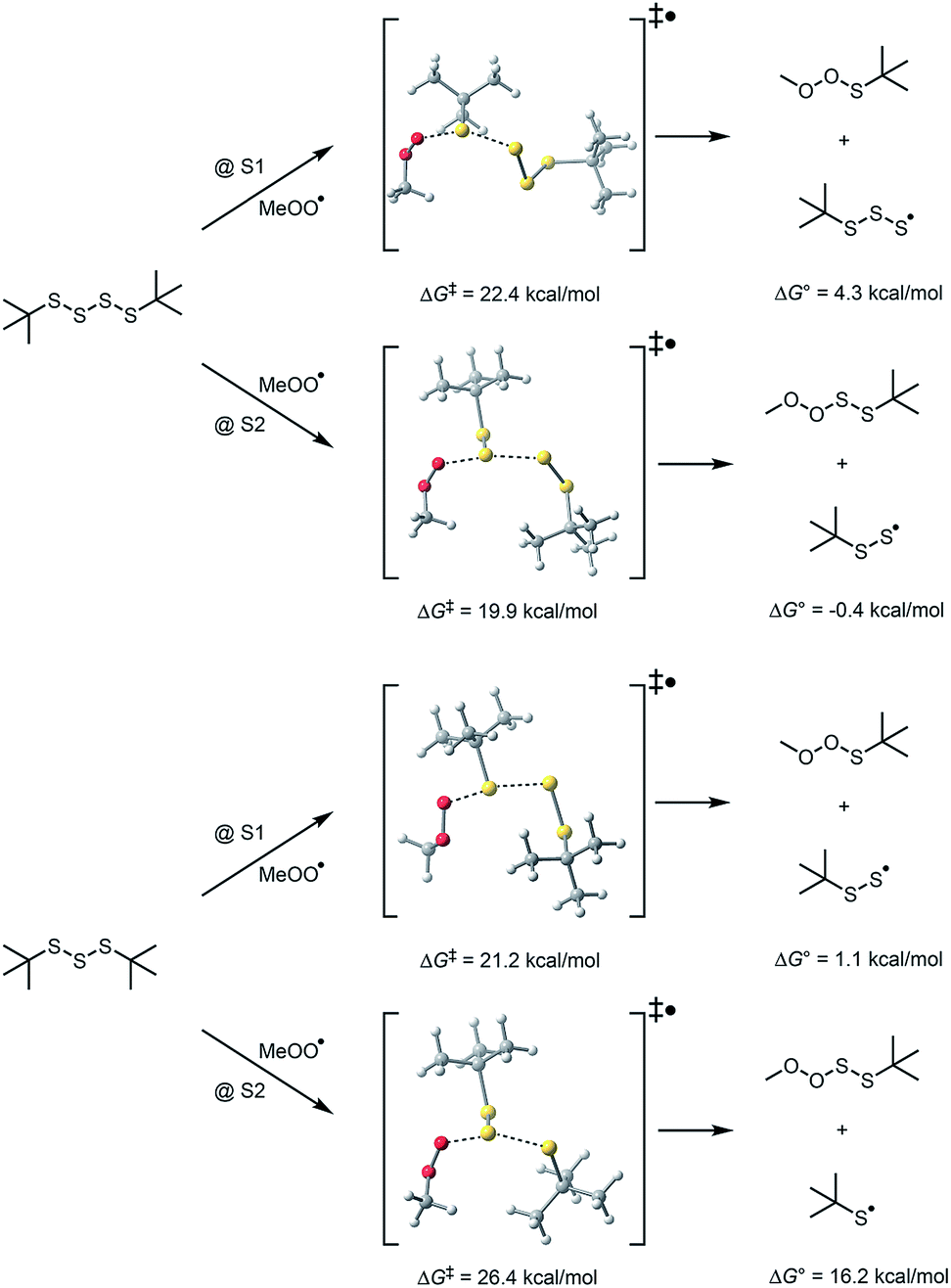 | ||
| Fig. 3 Computational insight on the regioselectivity of homolytic substitution of peroxyl radicals on trisulfides and tetrasulfides at 25 °C. | ||
The difference in calculated free energy barriers to substitution at S2 of the tetrasulfide and S1 of the trisulfide (1.3 kcal mol−1) corresponds to a difference in rate constant of a factor of 7 at 100 °C, implying that retardation of the autoxidations inhibited by the trisulfide should be observed. In fact, retardation was observed, but only at much higher concentrations of trisulfide (∼1 mM), yielding kinh ∼ 2 × 103 M−1 s−1 (see ESI†). The larger difference in reactivity between trisulfides and tetrasulfides determined experimentally (∼100-fold) may result from greater steric hinderance in the experimental system (wherein hexadecenylperoxyl radicals carry the autoxidation chain) as compared to the computational model (which employs methylperoxyl radicals for computational expediency).
Mechanisms of polysulfide-1-oxide reactivity
Although the RTA mechanisms of the hindered sulfoxides and thiosulfinates (Cope elimination followed by H-atom transfer to peroxyl radicals) and the trisulfide-1-oxides and tetrasulfide-1-oxides (homolytic substitution by peroxyl radicals) are established, the data in Fig. 1D and F underscore that their relevance is highly temperature-dependent. At 37 °C, the sulfoxide and thiosulfinate show no activity, while the trisulfide-1-oxide and tetrasulfide-1-oxide are effective inhibitors. At 100 °C, the opposite is observed. Since Cope elimination from the sulfoxide (k = 6.8 × 10−6 s−1 at 96 °C) is faster than from the thiosulfinate (k = 1.9 × 10−6 s−1 at 96 °C),27 sulfenic acid may build up that does not trap peroxyls, but instead undergoes other reactions, such as self-condensation or oxidation. When the autoxidation is inhibited by the persistent 9-triptycenesulfenic acid,28,29 similar kinh = (2.0 ± 0.6) × 106 M−1 s−1 and n = 0.5 ± 0.1 are observed (see ESI†), suggesting that oxidation depletes the sulfenic acid, since 9-triptycenesulfenic acid cannot undergo self-condensation. The importance of the competition between radical-trapping and oxidation is highlighted when autoxidations are carried out at varying rates of initiation. At a higher rate of initiation (Ri) (dicumyl peroxide concentration was doubled) the stoichiometry of the sulfoxide increased slightly from n = 0.4 to n = 0.5. Conversely, at a lower Ri (dicumyl peroxide concentration was halved) the stoichiometry decreased slightly from n = 0.4 to n = 0.3. The thiosulfinate presumably has a larger n because it produces sulfenic acid more slowly. Accordingly, its kinh increased from 1.5 to 3.5 × 106 M−1 s−1 when Ri was halved (n = 1 in both cases), but eroded when Ri was doubled. In contrast, the RTA activity of tetrasulfide 4 was independent of Ri. Please consult the ESI for all of the raw data for the results given above.In principle, at elevated temperatures, the trisulfide-1-oxide and tetrasulfide-1-oxide could undergo β-elimination in a manner similar to the thiosulfinate and sulfoxide (cf.Scheme 2). Since the propensity of polysulfide-1-oxides to undergo β-elimination has not been reported beyond thiosulfinates,27 we calculated the barriers for this process for the trisulfide-1-oxide and tetrasulfide-1-oxide using CBS-QB3 and compared them to the sulfoxide and thiosulfinate. The results are summarized in Fig. 4.
 | ||
| Fig. 4 Free energy barriers for β-elimination of excellent H-atom donors from sulfoxides, thiosulfinates and polysulfide-1-oxides at 25 °C. | ||
The calculations predict ΔG‡ = 31.2 and 31.1 kcal mol−1 for the elimination of (perthio)sulfenic acid from the trisulfide- and tetrasulfide-1-oxides, respectively – scarcely different from the thiosulfinate (ΔG‡ = 30.0 kcal mol−1). Confidence in these data can be gleaned from the good agreement between rate constants derived from the computed barriers for elimination from the thiosulfinate (1.9 × 10−5 s−1) at 100 °C with experimental data determined by Block at 96 °C (1.9 × 10−6 s−1).27 However, the S–S bond dissociation free energies of the trisulfide- and tetrasulfide-1-oxide are only 12.9 and 16.1 kcal mol−1, respectively, at 100 °C, suggesting that they will homolyze faster than they undergo Cope elimination to produce a good H-atom donor. Indeed, when we attempted to fit the experimental data from the polysulfide-1-oxide-inhibited autoxidations to a kinetic model involving initial elimination of (perthio)sulfenic acid followed by H-atom transfer, only the sulfoxide- and thiosulfinate-inhibited autoxidation data could be fit – yielding inhibition rate constants of 2.0 × 107 and 1.2 × 107 M−1 s−1, in excellent agreement with previous estimates of the reactivity of unhindered sulfenic acids to peroxyl radicals.19
The foregoing suggests that the facile bond homolysis of the higher polysulfide-1-oxides may explain their lower reactivity at elevated temperatures. In lieu of simply returning the starting compound (as is the case of the tetrasulfide, see Fig. 2), homolysis of the weak S–S bond in the polysulfide-1-oxides is expected to yield redox inactive thiosulfonates from dimerization of two sulfinyl radicals.18a Fortunately, the other product, a polysulfide, is still reactive via homolytic substitution. To support these expectations, the trisulfide-1-oxide was injected onto a gas chromatograph (inlet temperature set to 100 °C), which revealed only the tetrasulfide (tBuSSSStBu) and thiosulfonate (tBuSO2StBu) in the chromatogram. When the decomposition products of tBuS(O)SStBu were monitored over time (Fig. 5), the concentration of tetrasulfide was steady at about half of the initial concentration of trisulfide-1-oxide whereas the thiosulfonate concentration dropped to zero within 5 minutes under an air atmosphere or within 15 minutes under a nitrogen atmosphere. These observations suggest that the sulfinyl radical formed from the homolysis of the trisulfide-1-oxide reacts with O2, preventing the formation of thiosulfonate. Initially, most of the mass balance is thiosulfinate, which corresponds to the loss in thiosulfonate, and which arises from an unknown mechanism that is accelerated in air. Although we cannot exclude a role for the sulfenic acids formed by elimination from the trisulfide- and tetrasulfide-1-oxides based on the calculated energy barriers in Fig. 4, overall, these data suggest that the RTA activity of the trisulfide-1-oxide and tetrasulfide-1-oxide at 100 °C results primarily from homolytic substitution on the polysulfide derived therefrom and not the 1-oxides themselves, which are unstable.
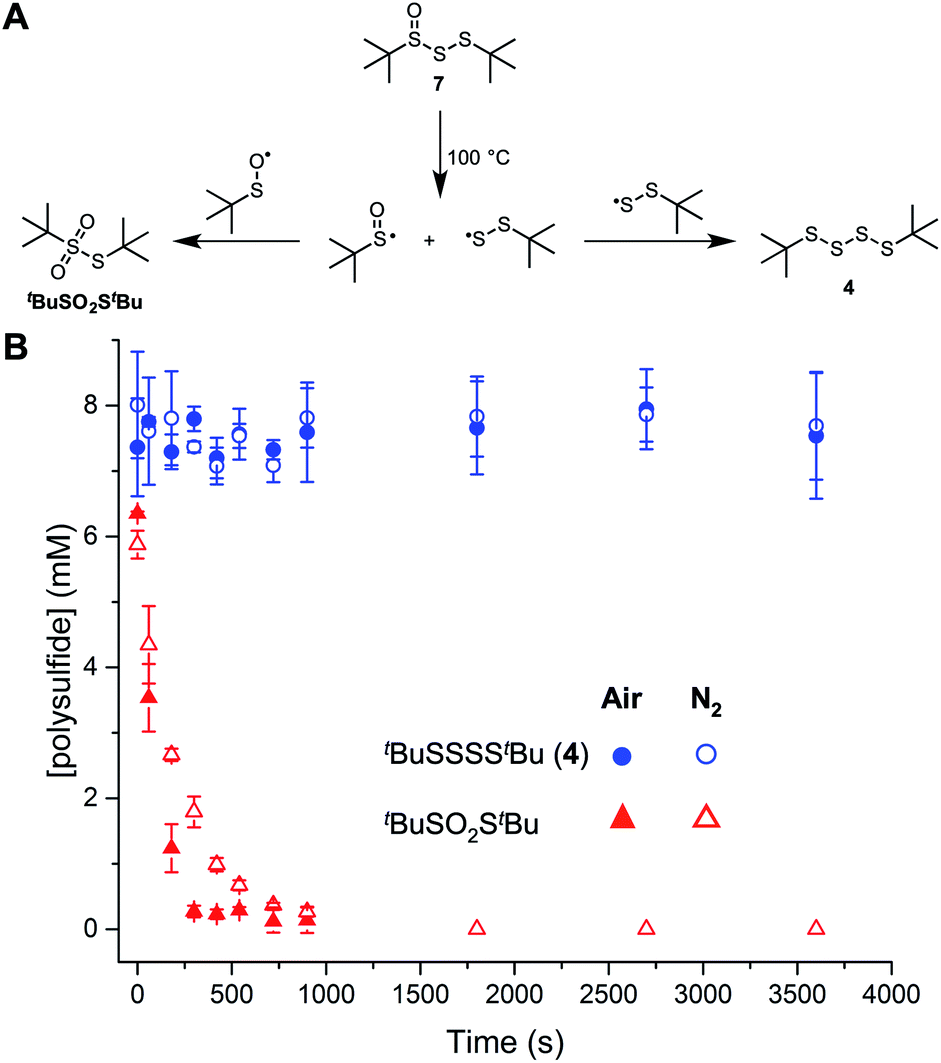 | ||
| Fig. 5 Decomposition of di-tert-butyl trisulfide-1-oxide 7 (20 mM) at 100 °C in chlorobenzene yields di-tert-butyl thiosulfonate and 4 (A). Product formation from the decomposition of 7 under air or nitrogen atmosphere as a function of time (B). | ||
Inhibited autoxidation of hexadecane at 160 °C
Given the substantial temperature dependence of the reactivity of both the tetrasulfides and the polysulfide-1-oxides, and the fact that sulfurized olefins are often used in applications at elevated temperatures, we also carried out inhibited autoxidations at 160 °C. n-Hexadecane was selected as the autoxidizable hydrocarbon for these experiments, which were carried out under a continuous flow of O2 to prevent mass transfer from limiting reaction progress.30,31 Product hydroperoxides were determined over the course of the autoxidation using a pro-fluorescent coumarin probe, which exhibits substantial fluorescence enhancement upon oxidation (Fig. 6A).15,38 Representative data sets are shown in Fig. 6B and C. | ||
| Fig. 6 A phosphine–coumarin conjugate (A) enables the quantification of hydroperoxides formed during inhibited uninitiated n-hexadecane autoxidation at 160 °C (black squares) inhibited by 500 μM organosulfur compound (B and C). Hydroperoxide production over the course of a n-hexadecane autoxidation at 160 °C initiated with 5 mM tetralin hydroperoxide (black square) and inhibited by 250, 500 and 1000 μM BHT (blue circle) or 4 (green diamond) (D). Hydroperoxide production over the course of a n-hexadecane autoxidation at 160 °C initiated with 5 mM tetralin hydroperoxide (black square) inhibited by 250, 500 and 1000 μM tBu2DPA (purple triangle) and 4 (green diamond) (E). | ||
Again, of the polysulfides, only the tetrasulfide was an efficient inhibitor, producing an inhibited period lasting for tinh ∼ 6000 s. It is important to note that autoxidations carried out at these temperatures are autocatalytic, precluding accurate determination of the RTA stoichiometry from tinh as in eqn (2) since Ri is not constant, but increasing with the formation of product peroxides. Interestingly, and in contrast with the results at 100 °C, the trisulfide-1-oxide and tetrasulfide-1-oxide were most effective of the polysulfide-1-oxides (tinh ∼ 3600 s), followed by the thiosulfinate (tinh ∼ 2700 s). The sulfoxide was completely ineffective at this temperature, suggesting that Cope elimination to sulfenic acid is so rapid that it is consumed immediately, precluding efficient RTA activity. Evidently, the slower Cope elimination from the thiosulfinate is slightly better matched to the rate of the autoxidation, enabling it to inhibit. However, it is clear that the higher polysulfide-1-oxides are best. Nevertheless, they remain less reactive than the tetrasulfide, presumably since half of the 1-oxides are used to produce polysulfides that trap peroxyl radicals – analogous to what was observed at 100 °C. Similar results were obtained with analogous higher molecular weight polysulfides and their 1-oxides, confirming that the lack of activity for some of the lower polysulfides analogues is not due to their volatility at 160 °C (see ESI† for further details).
Lastly, we sought to compare the reactivity of the tetrasulfides to that of the common primary antioxidants: a hindered phenol (BHT) and an alkylated diphenylamine (4,4′-di-tert-butyldiphenylamine, tBu2DPA). Representative results are presented in Fig. 6D and E, respectively. Interestingly, we found that the tetrasulfide was a better inhibitor of n-hexadecane autoxidation at 160 °C than either BHT or tBu2DPA. This contrasts the order of reactivity seen at 37 °C, where tBu2DPA > BHT ≫ tetrasulfide and 100 °C, where tBu2DPA > BHT ∼ tetrasulfide (see the ESI† for the data).
Discussion
The antioxidant mechanisms of organosulfur compounds are generally believed to be ionic ‘secondary antioxidant’ processes, where they react with (hydro)peroxides directly or via the eventual formation of sulfur dioxide or sulfur oxyacids.3 Moreover, it is generally accepted that all polysulfides are equally competent at inhibiting the oxidation of hydrocarbons when normalized to the number of sulfur atoms in the chain (i.e. a tetrasulfide reacts as quickly as 2 eq. of disulfide). This dogma appears to stem from the work of Bridgewater and Sexton, who studied the rates of reaction between polysulfides derived from para-sulfurization of a hindered phenol and cumene hydroperoxide.24 Thus, our observations that the tetrasulfide is a much better inhibitor of autoxidation than the corresponding trisulfide, disulfide and sulfide initially provided a very strong indication that a different mechanism was operative. In fact, tetrasulfides are shown to be potent radical-trapping antioxidants (k = 2.3 × 105 M−1 s−1 at 100 °C) under conditions where they are unreactive toward either hydroperoxides or initiating peroxides, and our experimental and computational results suggest that a homolytic substitution mechanism, which yields a stable and persistent perthiyl radical, is responsible.Similarly to the lower polysulfides, the lower 1-oxides (i.e. sulfoxide and thiosulfinate) are not activated to substitution due to their stronger S–S bonds. Nonetheless, they can be effective RTAs under certain conditions. Since their reactivity relies on the irreversible elimination of a sulfenic acid, and the sulfenic acid is not persistent under most conditions, the rate of the elimination must be similar to the rate of radical generation. With careful manipulation of laboratory conditions this is easily achieved – particularly for the sulfoxide at 100 °C, as the free energy barrier for elimination of the sulfenic acid therefrom (30.0 kcal mol−1) is almost indistinguishable from that of O–O bond homolysis of an initiating species (e.g. 30.9 kcal mol−1 for iPrO-OiPr). However, this similarity is unlikely to consistently manifest itself in real-world contexts, which is likely why sulfides and their sulfoxides are not commonly employed as RTAs.
The higher polysulfide-1-oxides are, like the tetrasulfide, activated to substitution by peroxyl radicals. This mechanism is clearly favoured over the elimination of sulfenic acid since they are effective throughout the temperature range that was investigated. If their RTA activity relied on sulfenic acid formation – which is predicted to take place at a similar rate to the thiosulfinate – we would expect it to be effective only at 100 °C under these conditions. However, for the same reason they are activated to substitution (weak S1–S2 bonds), the higher polysulfide 1-oxides decompose readily. Although the tetrasulfide has a similarly weak bond,36 its homolysis is reversible. When the polysulfide-1-oxides homolyze, the reaction is irreversible. Instead, two sulfinyl radicals combine to initially afford an unreactive thiosulfonate and the perthiyls combine to give a polysulfide (exemplified above in the decomposition of the trisulfide-1-oxide).‡18a Thus, they are only roughly 50% as effective as an authentic polysulfide (Fig. 6).
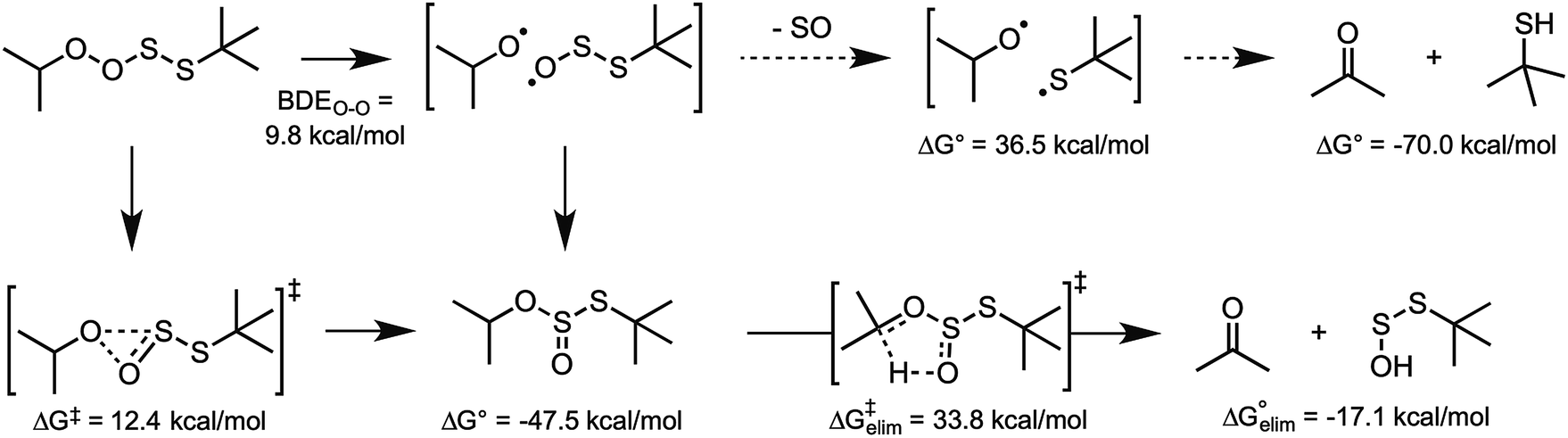 | ||
| Fig. 7 Possible fates of the peroxyl–perthiyl adduct formed from homolytic substitution of a (model) peroxyl radical on di-tert-butyltetrasulfide. | ||
Homolytic substitution of a peroxyl radical on a tetrasulfide produces a peroxyl–perthiyl adduct. The fact that this reaction is predicted to be essentially thermoneutral implies that the adduct must undergo a rapid subsequent reaction with some driving force. Moreover, given that the observed stoichiometry of radical-trapping reactivity of the tetrasulfide is close to the stoichiometry of 1.5 associated with the mechanism, the product must be relatively innocuous (i.e. it does not propagate the autoxidation). Given the structural similarity of the adduct to the tetroxide intermediate invoked when two peroxyl radicals combine to terminate an autoxidation chain, we considered an analogous fate for the peroxyl–perthiyl adduct: cleavage of the O–O bond, subsequent loss of sulfur monoxide and then in-cage comproportionation of the thiyl and alkoxyl radicals to yield a thiol and carbonyl (Fig. 7). CBS-QB3 calculations indicate that although the O–O bond is very weak, and the overall reaction is 70 kcal mol−1 exergonic, the fragmentation of sulfur monoxide from the alkylthiosulfinyl radical is not a particularly good reaction (requiring ca. 37 kcal mol−1). This suggests that the alkoxyl radical would have plenty of time to escape the solvent (substrate) cage and abstract an H-atom to initiate a new chain reaction. However, explorations of the potential energy surface near the O–O cleavage coordinate generally converged on combination of the alkoxyl and alkylthiosulfinyl radical fragments to yield a thiosulfurous acid diester, which was 47.5 kcal mol−1 exergonic relative to the perthiyl–peroxyl adduct. Moreover, we were able to identify a transition state structure for the concerted rearrangement, which has a predicted barrier of ΔG‡ = 12.4 kcal mol−1.37 Thus, it seems reasonable to suggest that the perthiyl–peroxyl adduct rearranges to a thiosulfurous acid diester – by either step-wise bond homolysis and in-cage radical recombination or concerted (1,2-)alkoxyl migration – nicely accounting for the observed stoichiometry of radical-trapping.
 | ||
| Fig. 8 Computed transition state for homolytic substitution of a (model) peroxyl radical on the most common allotrope of elemental sulfur (S8). | ||
Interestingly, we found that elemental sulfur (S8) did not inhibit the autoxidations we carried out (see ESI†) even though an analogous substitution reaction between it and a peroxyl radical could be envisioned. Although a transition state for peroxyl radical substitution on octahedral sulfur was readily identified using the same CBS-QB3 calculations that were carried out on the polysulfides (Fig. 8), the barrier for the reaction was predicted to be higher than that for the tetrasulfide (ΔG‡ = 22.9 kcal mol−1versus 19.9 kcal mol−1).35 In contrast with the thermoneutral reaction of the peroxyl radical with the tetrasulfide (ΔG° = −0.4 kcal mol−1), the substitution on S8 is endergonic (ΔG° = 8.5 kcal mol−1) due to the increased entropic cost of the reaction (essentially an addition rather than substitution). Moreover, while the facile rearrangement of the peroxyl–perthiyl adduct to the thiosulfurous acid diester appears to drive the reaction of the peroxyl radical and tetrasulfide forward, there is no obvious corresponding follow-up reaction of the peroxyl-S8 adduct to compete with the reverse reaction. Thus, it would appear that olefin sulfurization is not only a means to better solubilize sulfur for use in hydrocarbon-based products, but also to activate it to interception of peroxyl radicals.
The impressive RTA activity of the tetrasulfide is maintained at elevated temperatures (160 °C), where it is a more effective inhibitor of n-hexadecane autoxidation than either the quintessential hindered phenolic antioxidant (BHT) or a representative alkylated diphenylamine (tBu2DPA). At first glance, this result is perplexing given that the tetrasulfide is significantly less reactive than both BHT and tBu2DPA at 37 °C. However, the reactivity gap narrows upon increasing the temperature to 100 °C – fully consistent with the trends in Ea, viz. 0.8 kcal mol−1 (BHT),32 2.5 kcal mol−1 (diarylamine),33 and 9.9 kcal mol−1 (tetrasulfide, computed).34 Of course, the length of the inhibited period is dictated by the radical-trapping stoichiometry and not simply the kinetics. BHT and related hindered phenols are generally considered to trap a single radical at elevated temperatures, whereas the stoichiometry of alkylated diphenylamines vary wildly depending on reaction conditions. The data in Fig. 6D suggest that the tetrasulfide has an elevated stoichiometry relative to the hindered phenol – and more so than the 1.5 quantified at 100 °C. An explanation for this enhanced stoichiometry arises upon consideration of the fate of the initially formed peroxyl–perthiyl adduct discussed above. The product thiosulfurous acid diester can be expected to undergo a 5-centre Cope-type elimination to produce the thiosulfenic acid which is responsible for the RTA activity of the corresponding thiosulfinate. We readily identified a transition state for this reaction using CBS-QB3 which suggests that it has a higher barrier (ΔG‡ = 33.8 kcal mol−1) than the elimination from the corresponding thiosulfinate (ΔG‡ = 30.0 kcal mol−1), but still easily accessible at 160 °C. This also accounts for why the tetrasulfide has a higher stoichiometry than the thiosulfinate at 160 °C, despite a similar stoichiometry at 100 °C.
The foregoing results suggest that preparations of polysulfides enriched in tetrasulfide will impart greater oxidative stability to lubricants and other hydrocarbon-based products to which sulfurized olefins are added. Of course, this must be balanced against any negative impact this would have on the extreme pressure anti-wear properties of the material, which tend to be best exhibited by low order polysulfides (n ≤ 4) due to their increased stability. It would appear that 4 is the ‘magic’ number.
Conclusions
Inhibited autoxidations have revealed that the radical-trapping antioxidant activity of (poly)sulfides varies significantly with chain length. Of the first four members of the series of di-tert-butyl polysulfide, only the tetrasulfide is sufficiently reactive towards peroxyl radicals to inhibit the autoxidation of 1-hexadecene and n-hexadecane at 100 and 160 °C, respectively. The rate constant for the reaction of the tetrasulfide with peroxyl radicals was measured to be 2 × 105 M−1 s−1 at 100 °C; similar to hindered phenols, the most common industrial antioxidant. Moreover, the tetrasulfide bested both hindered phenols and alkylated diphenylamines at 160 °C. Computation and experiment point to a homolytic substitution of peroxyl radicals on the central sulfur atom(s) of the tetrasulfide, which liberates a highly stabilized and persistent perthiyl radical which simply combines with another to reform a tetrasulfide. Based on the stoichiometry observed at 100 °C (ca. 1.5), the other product, a perthiyl–peroxyl adduct, must decompose to other non-radical products, which we have proposed to be a thiosulfurous acid diester. By comparison, trisulfide-1-oxide and tetrasulfide-1-oxide are better RTAs at ambient temperatures since they are further activated to analogous homolytic substitution (at the sulfinyl sulfur atom), but their activity erodes at elevated temperatures due to the irreversible cleavage of the S1–S2 bond. Still, they retain activity, since this bond cleavage results in formation of reactive polysulfides (tetrasulfide and hexasulfide, respectively). It is anticipated that these findings will prompt researchers to develop olefin sulfurization processes which enrich the product in tetrasulfide, and will result in an expanded role of sulfurized olefins in industrial and/or commercial applications.Experimental section
General
All chemicals obtained from commercial suppliers were used as received unless indicated otherwise. 1-Hexadecene and n-hexadecane were percolated through a column of basic alumina and silica (3:1) twice before use. Column chromatography was carried out with 40–63 μm, 230–400 mesh silica gel. 1H and 13C NMR spectra were recorded on a Bruker AVANCE spectrometer at 400 and 101 MHz, respectively, unless indicated otherwise. High-resolution mass spectra were obtained on a Kratos Concept tandem mass spectrometer (EI) and Micromass Q-TOF (ESI). PBD-BODIPY was synthesized according to our previously reported procedure.22 The hydroperoxide probe38 and the acid probe15 were also synthesized as previously reported. Chlorobenzene was dried over 4 Å molecular sieves before use. UV-visible spectra were measured with a Cary 100 spectrophotometer equipped with a thermostated 6 × 6 multicell holder. Unless indicated otherwise, the reported errors correspond to the standard deviation of at least three independent experimental trials.
Inhibited autoxidations
:4, 215 μL) using the automated reagent dispenser of the microplate reader. Next, 30 μL of a solution containing the fluorogenic coumarin phosphine dye (100 μM) and BODIPY acid probe (100 μM) in acetonitrile was added to each well again using the automated reagent dispenser. The plate, incubated at 37 °C, was stirred for 30 seconds, and after a 5 second delay, the fluorescence of each well was measured every second for 60 seconds (coumarin phosphine dye: absorption 340 nm, emission 425 nm; BODIPY acid probe: absorption 475 nm, emission 515 nm). The concentration of hydroperoxide in each well was determined from the rate of phosphine oxidation using the rate constant for the reaction of the dye with secondary hydroperoxides (k = 5.1 M−1 s−1), assuming pseudo-first-order kinetics.15 The acid concentration in each well was determined from the average fluorescence reading over 10 s (RFU = 359[acid] + 34925).15
Product studies
:1, flow = 125 mL min−1, inlet temperature of 100 °C) on an instrument equipped with an Agilent HP-5 column (30 m × 0.320 mm × 0.25 μm) with a constant helium flow of 2.5 mL min−1 according to the following temperature profile: 100 °C, hold 0 min, 3 °C min−1 to 145 °C, hold 0 min, 34 °C min−1 to 297 °C, hold 0 min, 35 °C min−1 to 100 °C. The detector temperature was set to 300 °C. The method yielded retention times of 2.0, 10.2, 11.5 and 12.6 minutes for hexylbenzene, iPrSSSSiPr, iPrSSSStBu and tBuSSSStBu, respectively.
:1, flow = 125 mL min−1, inlet temperature of 100 °C), on an instrument equipped with an Agilent HP-5 column (30 m × 0.320 mm × 0.25 μm) with a constant Helium flow of 2.5 mL min−1 according to the following temperature profile: 100 °C, hold 0 min, 3 °C min−1 to 145 °C, hold 0 min, 34 °C min−1 to 297 °C, hold 0 min, 35 °C min−1 to 100 °C. The detector temperature was set to 300 °C. The method yielded retention times of 1.5, 7.1 and 12.7 minutes for hexylbenzene, tBuSO2StBu and tBuSSSStBu, respectively.
Computations
Calculations were carried out using the CBS-QB3 (ref. 41) complete basis set method as implemented in the Gaussian 16 suite of programs.42 Except where indicated otherwise, the calculations were performed at standard conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada and the Canada Foundation for Innovation and through generous access to the computational resources of the Centre for Advanced Computing (http://www.cac.queensu.ca). JPC acknowledges the support of the Ontario Graduate Scholarships program.Notes and references
- G. H. Denison Jr, Ind. Eng. Chem., 1944, 36, 477–482 CrossRef.
- G. H. Denison Jr and P. C. Condit, Ind. Eng. Chem., 1945, 37, 1102–1108 CrossRef.
- D. N. Harpp, J. Robertson, K. Laycock and D. Butler, Sulfur Rep., 1985, 4, 195–219 CrossRef CAS.
- K. U. Ingold, Chem. Rev., 1961, 61, 563–589 CrossRef CAS.
- K. U. Ingold and D. A. Pratt, Chem. Rev., 2014, 114, 9022–9046 CrossRef CAS PubMed.
- W. Nehb and K. Vydra, Ullmanns Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 8 edn., 2000, vol. 5, p. 3748 Search PubMed.
- H. E. Westlake, Chem. Rev., 1946, 39, 219–239 CrossRef CAS PubMed.
- E. Block, Angew. Chem., Int. Ed., 1992, 31, 1135–1178 CrossRef.
- E. Block, Garlic and Other Alliums, Royal Society of Chemistry, 2010 Search PubMed.
- T. Ida, T. Sawa, H. Ihara, Y. Tsuchiya, Y. Watanabe, Y. Kumagai, M. Suematsu, H. Motohashi, S. Fujii, T. Matsunaga, M. Yamamoto, K. Ono, N. O. Devarie-Baez, M. Xian, J. M. Fukuto and T. Akaike, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7606–7611 CrossRef CAS PubMed.
- J.-P. R. Chauvin, Z. A. M. Zielinski and D. A. Pratt, Can. J. Chem., 2016, 94, 1–8 CrossRef CAS.
- D. Barnard, L. Bateman, M. E. Cain, T. Colclough and J. I. Cunneen, J. Chem. Soc., 1961, 5339–5346 RSC.
- J. D. Holdsworth, G. Scott and D. Williams, J. Chem. Soc., 1964, 4692–4699 RSC.
- G. E. Zaikov, J. A. Howard and K. U. Ingold, Can. J. Chem., 1969, 47, 3017–3029 CrossRef CAS.
- R. Shah and D. A. Pratt, J. Org. Chem., 2016, 81, 6649–6656 CrossRef CAS PubMed.
- P. Koelewijn and H. Berger, Recl. Trav. Chim. Pays-Bas, 1974, 93, 63–68 CrossRef CAS.
- V. Vaidya, K. U. Ingold and D. A. Pratt, Angew. Chem., Int. Ed., 2008, 48, 157–160 CrossRef PubMed.
- (a) P. T. Lynett, K. Butts, V. Vaidya, G. E. Garrett and D. A. Pratt, Org. Biomol. Chem., 2011, 9, 3320–3330 RSC; (b) G. Derbesy and D. N. Harpp, J. Org. Chem., 1995, 60, 4468–4474 CrossRef CAS.
- R. Amorati, P. T. Lynett, L. Valgimigli and D. A. Pratt, Chem.–Eur. J., 2012, 18, 6370–6379 CrossRef CAS PubMed.
- J.-P. R. Chauvin, E. A. Haidasz, M. Griesser and D. A. Pratt, Chem. Sci., 2016, 7, 6347–6356 RSC.
- An experimental BDE has not been reported for tBuS-SStBu. Therefore, we have given calculated BDEs for tBuS-SStBu and tBuSS-SStBu in the text. However, it should be noted that the former is in good agreement with previously reported experimental values. See: J. A. Hawari, D. Griller and F. P. Lossing, J. Am. Chem. Soc., 1986, 108, 3273–3275 CrossRef CAS.
- E. A. Haidasz, A. T. M. Van Kessel and D. A. Pratt, J. Org. Chem., 2016, 81, 737–744 CrossRef CAS PubMed.
- J.-P. R. Chauvin, M. Griesser and D. A. Pratt, J. Am. Chem. Soc., 2017, 139, 6484–6493 CrossRef CAS PubMed.
- A. J. Bridgewater and M. D. Sexton, J. Chem. Soc., Perkin Trans. 2, 1978, 530–536 RSC.
- The stoichiometry of the di-iso-propyl tetrasulfide was found to be slightly lower than that of the di-tert-butyl tetrasulfide (compare n = 1.0 ± 0.1 to n = 1.4 ± 0.2, respectively) – presumably due to the relative instability of non-tertiary substituted tetrasulfides to self-reaction..
- Alas, application of transition state theory affords a corresponding rate constant of 290 M−1 s−1 at 100 °C, thereby significantly underestimating the observed kinh. Changing the peroxyl radical to a secondary peroxyl radical (iPrOO˙) to better represent the peroxyl radical which propagates 1-hexadecene autoxidations decreased the ΔG‡ and afforded a three-fold increase in the calculated rate constant to 909 M−1 s−1 at 100 °C, but this remains two orders of magnitude lower than the experimental value..
- E. Block and J. O'Connor, J. Am. Chem. Soc., 1974, 96, 3929–3944 CrossRef CAS.
- N. Nakamura, J. Am. Chem. Soc., 1983, 105, 7172–7173 CrossRef CAS.
- A. J. McGrath, G. E. Garrett, L. Valgimigli and D. A. Pratt, J. Am. Chem. Soc., 2010, 132, 16759–16761 CrossRef CAS PubMed.
- R. K. Jensen, S. Korcek, L. R. Mahoney and M. Zinbo, J. Am. Chem. Soc., 1979, 101, 7574–7584 CrossRef CAS.
- J. Igarashi, R. K. Jensen, J. Lusztyk, S. Korcek and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 7727–7736 CrossRef CAS.
- J. A. Howard and E. Furimsky, Can. J. Chem., 1973, 51, 3738–3745 CrossRef CAS.
- J. J. Hanthorn, R. Amorati, L. Valgimigli and D. A. Pratt, J. Org. Chem., 2012, 77, 6895–6907 CrossRef CAS PubMed.
- The difference in the calculated kinh and the experimental kinh indicates the calculated ΔG‡ is about 3.5 kcal mol−1 too high. Assuming this is an overestimation in the entropy of activation, logA ∼ 9..
- Polysulfides, such as trisulfides and tetrasulfides, are known to extrude elemental sulfur at elevated temperature, which could react with olefinic substrates. To rule out any inhibition by sulfur, we carried out 1-hexadecene autoxidations in the presence of up to 100 μM sulfur, wherein no inhibition was observed (see ESI† for kinetic traces)..
- I. Kende, T. L. Pickering and A. V. Tobolsky, J. Am. Chem. Soc., 1965, 87, 5582–5586 CrossRef CAS.
- Attempts to isolate the thiosulfurous acid diester product from a preparative scale reaction were unsuccessful, presumably due to facile subsequent Cope-type elimination of the product..
- J. J. Hanthorn, E. Haidasz, P. Gebhardt and D. A. Pratt, Chem. Commun., 2012, 48, 10141–10143 RSC.
- K. A. Harrison, E. A. Haidasz, M. Griesser and D. A. Pratt, Chem. Sci., 2018, 9, 6068–6079 RSC.
- G. W. Burton, T. Doba, E. J. Gabe, L. Hughes, F. L. Lee, L. Prasad and K. U. Ingold, J. Am. Chem. Soc., 1985, 107, 7053–7065 CrossRef CAS.
- J. A. Montgomery Jr, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822–2827 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data, additional autoxidation data, optimized geometries and energies for computational results. See DOI: 10.1039/c9sc00276f |
| ‡ Harpp proposes formation of tetrasulfides and thiosulfonates (among other products) from trisulfide-1-oxides via an ionic mechanism (see ref. 18b). We have previously investigated the combination of two sulfinyl radicals by calculations and favour a radical pathway (see ref. 18a). |
| This journal is © The Royal Society of Chemistry 2019 |