DOI:
10.1039/C9SC00378A
(Edge Article)
Chem. Sci., 2019,
10, 4684-4691
A modular and concise approach to MIDA acylboronates via chemoselective oxidation of unsymmetrical geminal diborylalkanes: unlocking access to a novel class of acylborons†
Received
23rd January 2019
, Accepted 20th March 2019
First published on 21st March 2019
Abstract
Acylboronates represent a very intriguing and rare class of organoboronates. Synthesis of these compounds from readily available substrates under mild conditions and access to novel classes of acylborons has been challenging. We report a novel and concise route to various MIDA acylboronates from terminal alkynes/alkenes or vinyl boronic esters using unsymmetrical geminal diborylalkanes as key intermediates. The high modularity and mild conditions of this strategy allowed a facile access to acylboronates possessing aliphatic, aromatic as well as the rarer heteroaromatic, alkynyl and α,β-unsaturated scaffolds. To the best of our knowledge, this is the first report of chemoselective oxidation of geminal diborons as well as synthesis of an α,β-unsaturated acylboronate.
Introduction
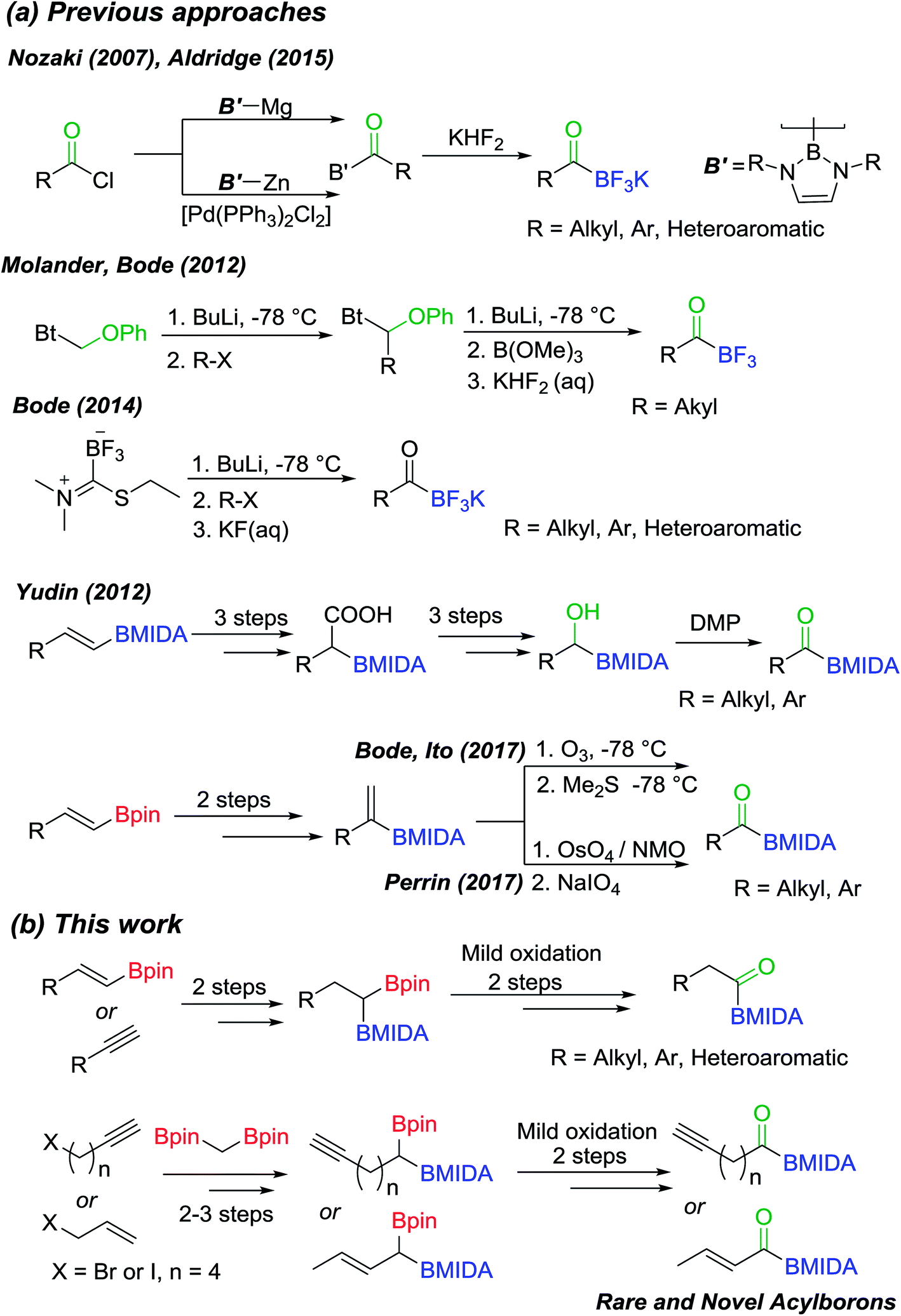
Organoborons are a highly useful class of compounds in organic synthesis and medicinal chemistry. These compounds participate in a variety of C–C and C–X bond forming reactions and serve as versatile building blocks for synthesis of pharmaceuticals, natural products and fine-chemicals.1 Amongst the various organoborons, acylborons represent a rare and very intriguing class of compounds due to their unique reactivity, physical properties and powerful applications in chemical biology and organic synthesis.2 Acylborons were hypothesized to be intermediates in several transformations.3 Until 2007, the synthesis, isolation and characterization of acylboronates eluded chemists due to their presumed instability and tendency for rearrangement.2a Nozaki and co-workers described the first breakthrough, wherein, they reported the preparation of amino stabilized acylboron (Scheme 1(a)) by reacting boryl lithium or boryl magnesium compounds with benzoyl chloride or benzaldehyde.4 Thereafter, some seminal contributions from Molander,5 Bode,6 Yudin7 and others8 have led to creative and useful approaches for preparation of stable acylborons (Scheme 1(a)). Collectively, these reports suggest that decreasing the Lewis acidity of boron by using tetravalent ligands on boron provides enhanced stability to acylborons. In particular, elegant studies by Yudin and Bode have led to the emergence of MIDA acylboronates and potassium trifluoroacyl borates (KATs) as a very attractive class of compounds due to their utility for synthesis of rare borylated heterocycles7a,b and as powerful building blocks for bioorthogonal amide formation and protein ligation.6d,9
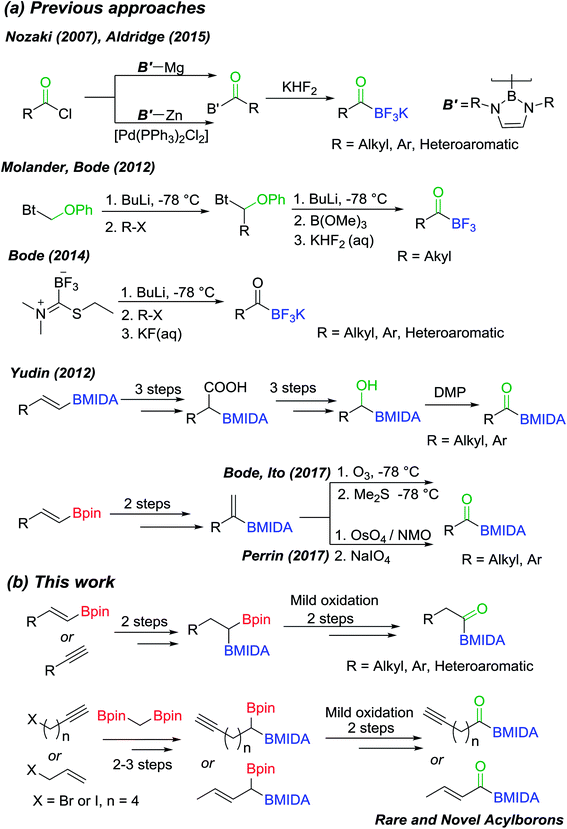 |
| | Scheme 1 (a) Previous synthetic approaches for synthesis of acylborons; (b) the new modular approach to acylboronates via chemoselective oxidation of geminal diborylalkanes. | |
Previous synthetic approaches (Scheme 1(a)) to acylborons involved (i) the reaction of highly nucleophilic boron or organolithiums with carbonyl or boron electrophiles, (ii) oxidative cleavage of alkenyl-2-MIDA boronates using OsO4/NaIO4 or ozonolysis and (iii) oxidation of α-hydroxy MIDA boronates. Despite these notable advances, there are still significant challenges that hamper the broader investigation and application of acylborons. For instance, oxidation of α-hydroxy MIDA boronates is a user-friendly strategy, however, synthetic access to α-hydroxy MIDA boronates is a major challenge as it involves a six-step route from vinyl boronates (Scheme 1(a)).7a Furthermore, many useful α-hydroxy MIDA boronates are inaccessible through conventional methods. Thus, it will be beneficial to develop novel strategies for synthesis of acylborons that combine the following benefits in a single synthetic method: (i) reduce the number of synthetic steps; (ii) use readily available substrates such as alkynes and alkenes (iii) involve mild reaction conditions and avoid the use of highly basic boron/organolithium reagents or harsh oxidizing reagents and ultra-low temperatures and (iv) most importantly, open-up access to rare and novel classes of acylborons.2a,10
In order to address the above challenges, we envisioned that a concise and modular access to MIDA acylboronates could be obtained by chemoselective oxidation of unsymmetrical geminal diborylalkanes (Scheme 1(b)). Geminal diborylalkanes have emerged as attractive building blocks for preparation of a wide range of structurally complex organoborons.11 In comparison to other 1,1-organodimetallic reagents,12 the geminal organodiboron compounds possess several advantages such as enhanced stability, ease of handling and easy accessibility from abundantly available substrates such as alkynes and alkenes. Previous reports have focused on the application of these geminal diborons for C–C bond formation via boron-stabilized carbanion or chemoselective Suzuki coupling.11a–c,g However, chemoselective oxidation of one of the boron in a geminal diboron compound has not been described before.13
Result and discussion
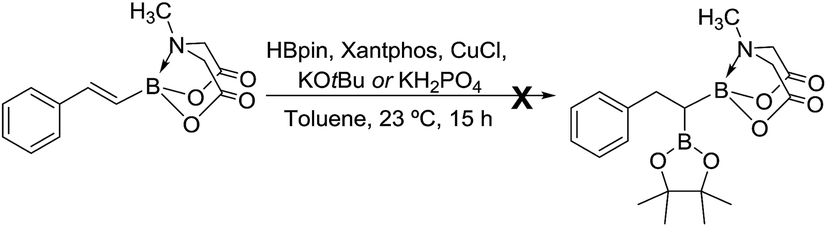
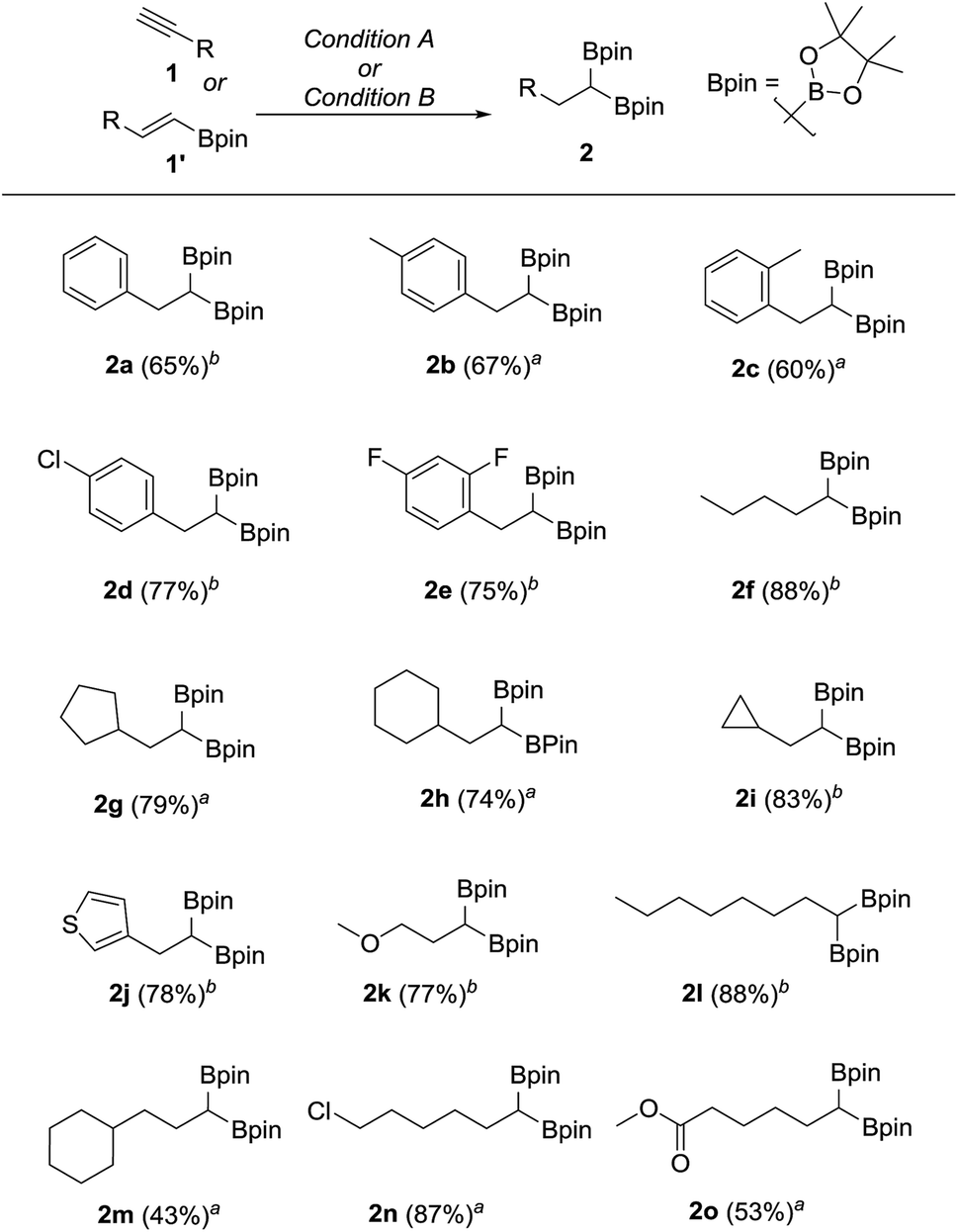
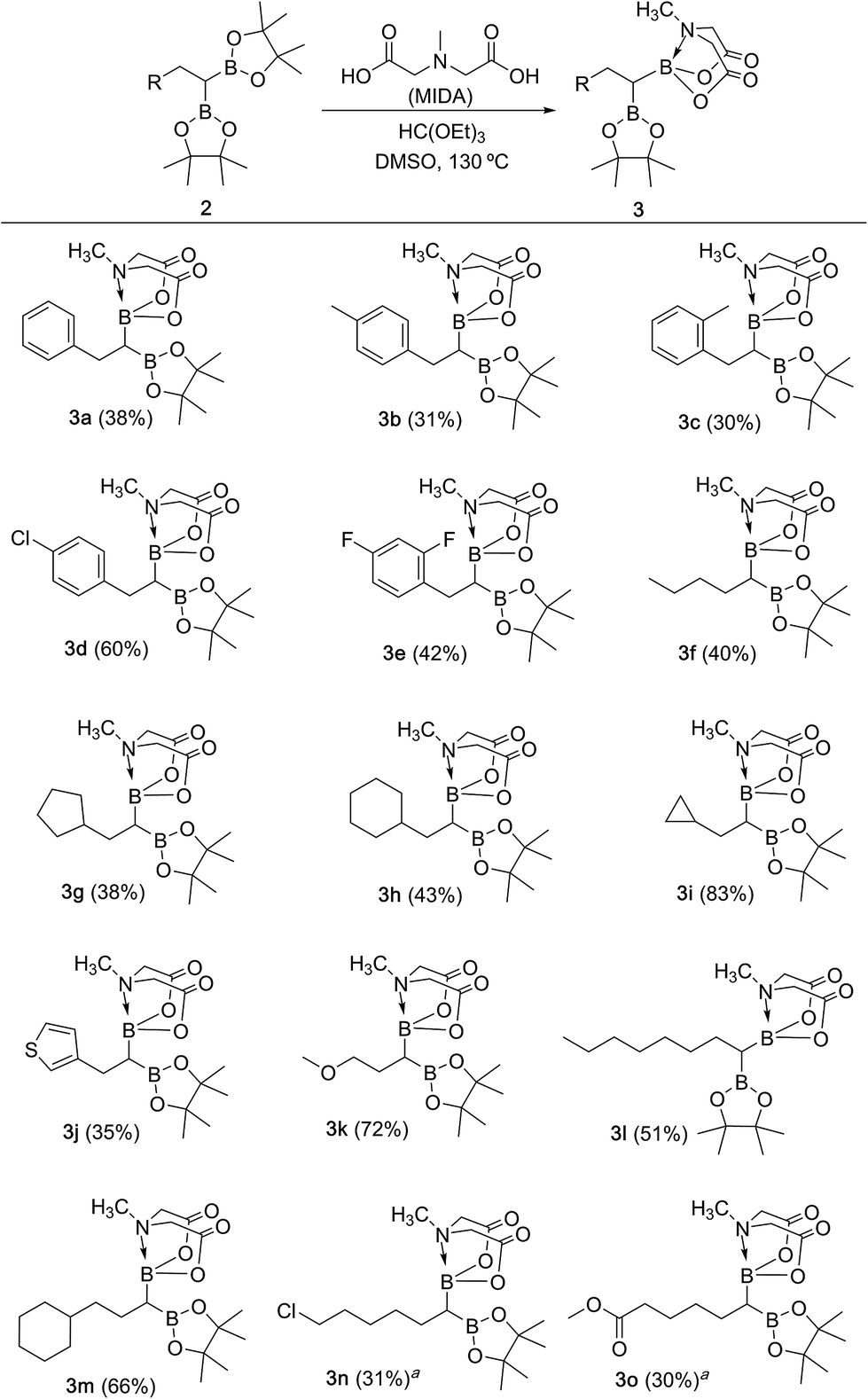
We hypothesized that mild oxidation of an unsymmetrical diborylalkane possessing geminal B(MIDA) and B(Pin) groups should selectively give the α-hydroxy MIDA boronate which can then be further oxidized to acylboronates. This strategy would drastically reduce the number of steps7a required for synthesis of α-hydroxy borons and MIDA acylboronates from vinyl boronic esters or even terminal alkynes, which are more easily available and cheaper. Our initial strategy for synthesis of unsymmetrical geminal diboryl alkanes involved the copper catalyzed regioselective hydroboration14 of vinyl MIDA boronate with HBpin followed by chemoselective oxidation of the more reactive B(Pin) group into the α-hydroxy MIDA boronate. However, this approach proved unsuccessful as the vinyl MIDA boronate was found to undergo MIDA deprotection under the borylation conditions (Scheme 2) using either KOtBu or the milder KH2PO4 as a base. Thereafter, we attempted an alternative strategy that involved regioselective dihydroborylation of readily available terminal alkyne into a symmetrical geminal diborylalkane followed by conversion15 of one of the pinacol boronic ester (B(Pin)) groups into B(MIDA). Thus, reaction of terminal alkynes (1b, c, g, h, m–o) with HBPin (2.4 equiv.) in the presence of CuCl, xantphos and KOtBu provided the corresponding geminal diboron compounds (2b, c, g, h, m–o, Scheme 3). Further, a similar strategy was also applicable on vinyl pinacol boronic esters which are also commercially available (1a, d–f, i–l, Scheme 3). To convert these symmetrical geminal diborylalkanes into unsymmetrical geminal diborons, several reaction conditions were screened (see ESI, Table S1†). Heating the symmetrical diborons with N-methyliminodiacetic acid15 (MIDA, 6 equiv.) in DMSO at 130 °C in the presence of HC(OEt)3 (ref. 6c) provided best conversion (3a–o, Scheme 4). The conversion of B(Pin) into B(MIDA) is known to be a sluggish transformation6c,8a,15 and the yield of this reaction for symmetrical diboron substrates (2a–o, Scheme 4) was comparable to the earlier reports that used monoboron substrates.6c,8a Interestingly, our above method led to the conversion of only one of the B(Pin) groups of symmetrical geminal diborons into B(MIDA) group (3a–o, Scheme 4) as the di-B(MIDA) product was not detected. The unreacted symmetrical diboron substrates (2a–o) from above reaction were recovered and used again. To the best of our knowledge, this is the first example of unsymmetrical geminal diboryl compounds that possess B(Pin)/B(MIDA) groups.11a
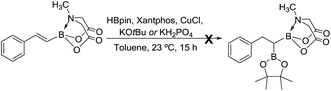 |
| | Scheme 2 Attempted synthesis of unsymmetrical geminal diborylalkane from vinyl MIDA boronate. | |
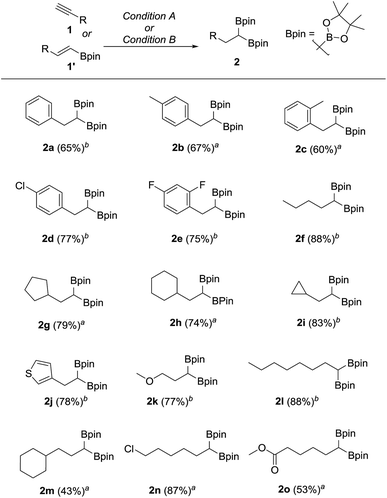 |
| | Scheme 3 Synthesis of symmetrical geminal diborons from alkynes or alkenes. aReaction condition A: terminal alkyne (1.7 mmol), CuCl (5.0 mol%), KOtBu (0.1 equiv.), xantphos (6.0 mol%), HBpin (2.4 equiv.), toluene, 23 °C, 15 h. bReaction condition B: vinyl pinacol boronic ester (2.0 mmol), CuCl (5.0 mol%), KOtBu (0.1 equiv.), xantphos (6.0 mol%), HBPin (1.2 equiv.), toluene, 23 °C, 15 h. | |
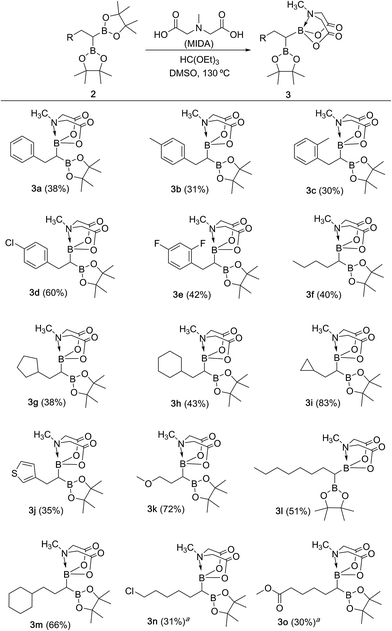 |
| | Scheme 4 Synthesis of unsymmetrical geminal diborylalkanes. Reaction conditions: 1,1-diboryl substrate 2 (1.3 mmol), N-methyliminodiacetic acid (6 equiv.), HC(OEt)3 (4 equiv.), DMSO, 130 °C, 15 h. aReaction condition: (i) 1,1-diboryl substrate (1.3 mmol), NH4OAc (3.1 equiv.), NaIO4 (3.1 equiv.), acetone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (2:1), 80 °C, 2 h; (ii) N-methyliminodiacetic acid (6 equiv.), HC(OEt)3 (4 equiv.), DMSO, 115 °C, 2 h. :water (2:1), 80 °C, 2 h; (ii) N-methyliminodiacetic acid (6 equiv.), HC(OEt)3 (4 equiv.), DMSO, 115 °C, 2 h. | |
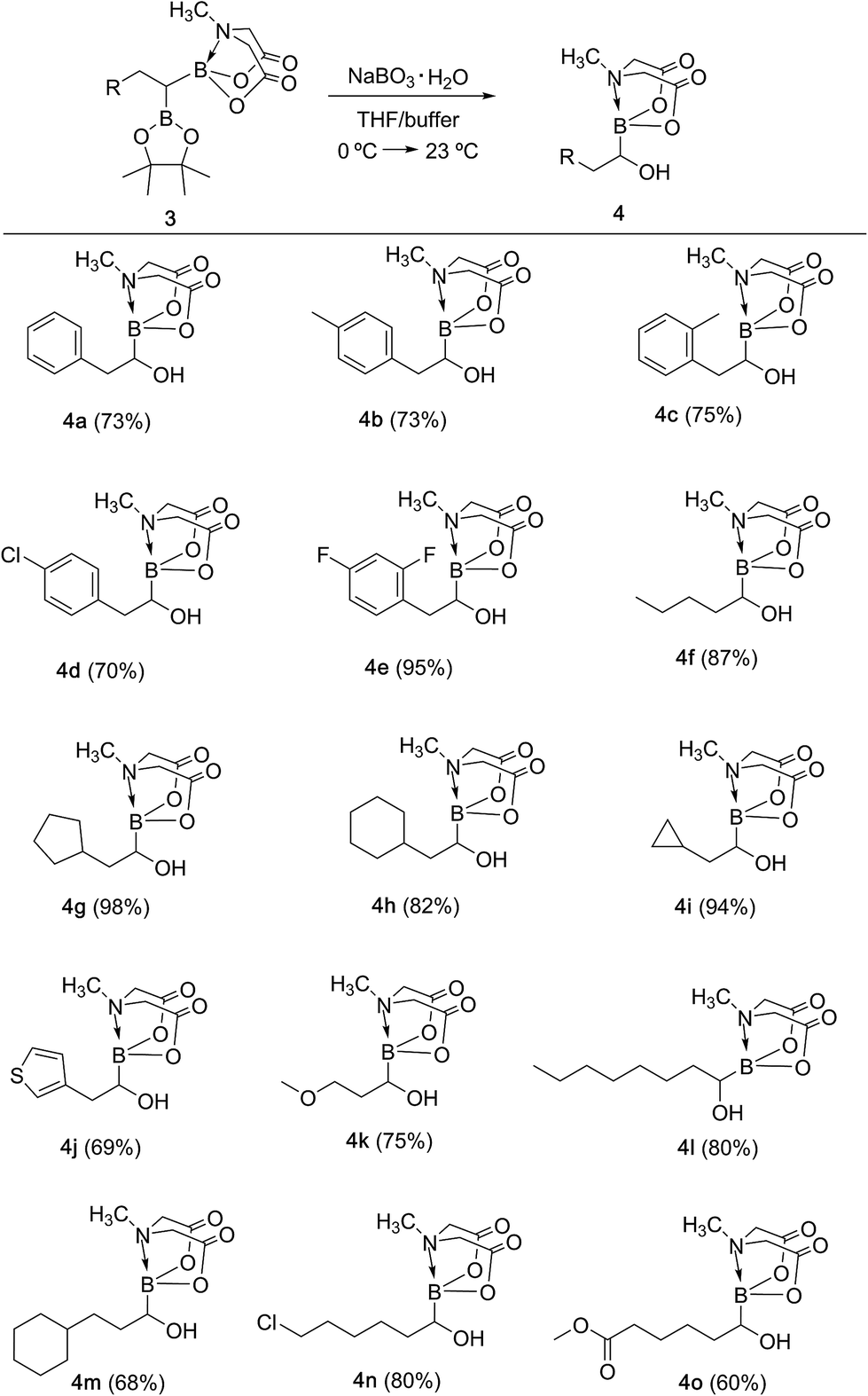
With the unsymmetrical diboron compounds (3) in hand, we screened several conditions (see ESI, Table S2†) for chemoselective oxidation of 3 into α-hydroxy MIDA boronate (4). These investigations revealed that use of sodium perborate16 (1.3 equiv.) in a mixture of THF and buffer solution (KH2PO4/NaOH, pH = 7) afforded better conversion in comparison to Oxone. Importantly, MIDA boronate remained intact during this chemoselective oxidation (Scheme 5). Aqueous solutions of sodium perborate are basic and therefore the use of a buffer solution presumably prevented the deprotection of aq. base labile B(MIDA) group. This optimized protocol was also found to be compatible for mild and efficient oxidation of other diboron compounds (Scheme 5). In general, substrates possessing alkyl side chains provided higher yields than those with aromatic side chains. In particular, substrates possessing heteroaromatic scaffold (3j), alkyl halide (3n) and ester (3o) were also tolerated. Interestingly, a control reaction performed by treating the symmetrical diboron (2h) with NaBO3 (1.3 equiv.) in THF:buffer solution provided mainly the unreacted starting material (2h) after 4 h. This result suggests that differential protection of geminal diborons with suitable boron ligands is critical to enable their chemoselective oxidation into stable α-hydroxy boronates.
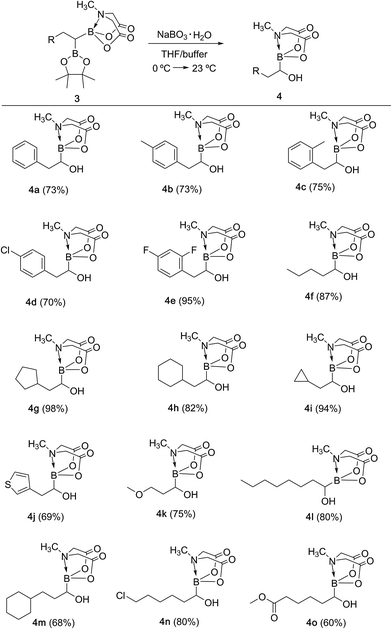 |
| | Scheme 5 Synthesis of α-hydroxy MIDA boronates via chemoselective oxidation of unsymmetrical geminal diborylalkanes. Reaction conditions: 3 (0.6 mmol), sodium perborate monohydrate (1.3 equiv.), THF (2 ml), buffer (KH2PO4/NaOH, pH = 7), 23 °C, 3 h. | |
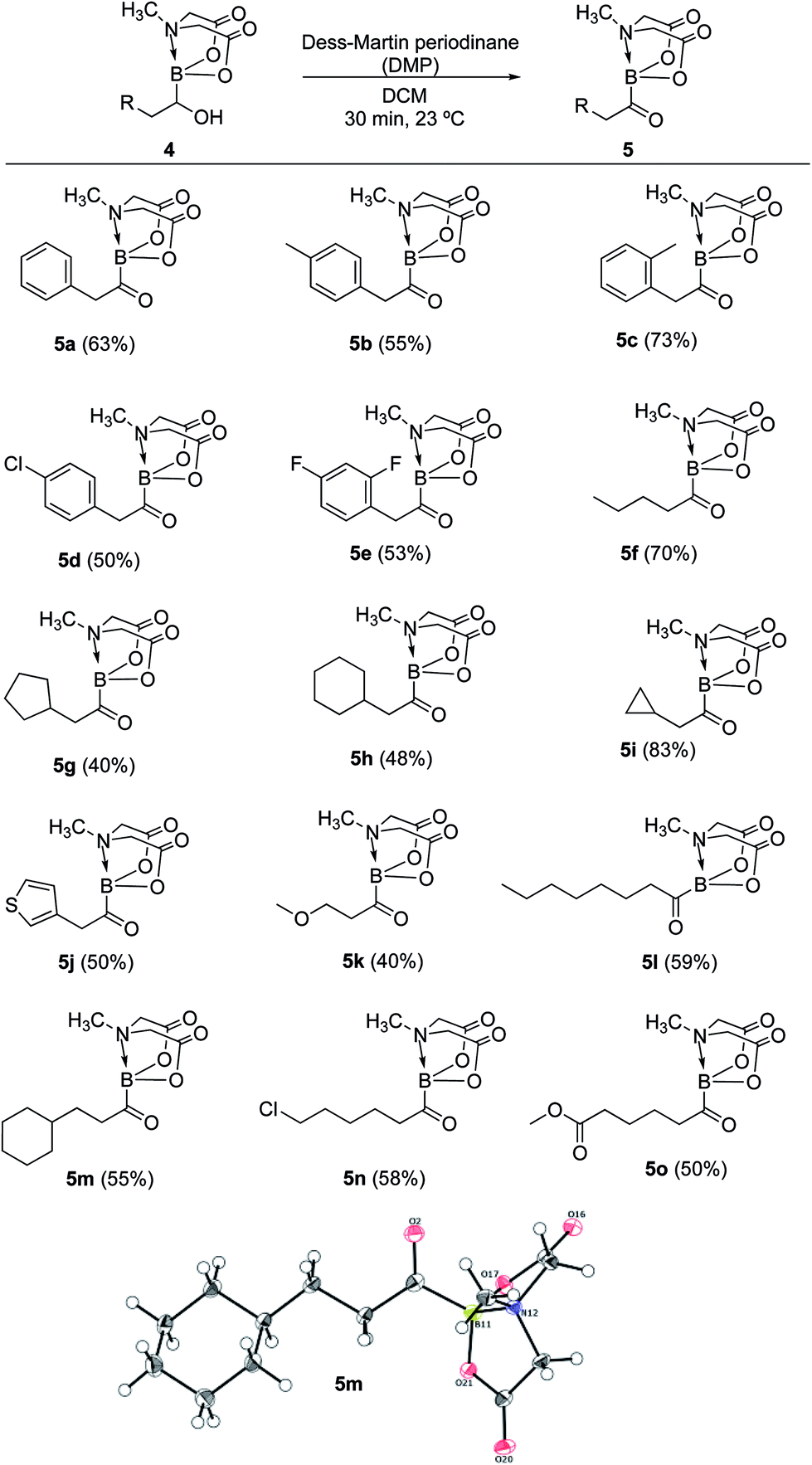
Having established a concise synthetic route to α-hydroxy MIDA boronates, we next turned our attention to their oxidation into acylboronates. All the above synthesized α-hydroxy MIDA boronates were successfully oxidized into the corresponding MIDA acylboronates using 1.1 equiv. of DMP7a in DCM at 23 °C (Scheme 6). The structure of one of the novel acylboronates was confirmed by single crystal X-ray diffraction analysis (5m, Scheme 6). Significantly, this geminal borylation route also afforded a mild and direct access to acylboronate possessing a heteroaromatic scaffold (5j) from corresponding vinyl boronic ester (1j′). This route to heteroaromatic acylboronates is complimentary to previous approaches that involved the reaction of heteroaryl halides or acyl chlorides with organolithium6b or highly basic boron reagents.8c
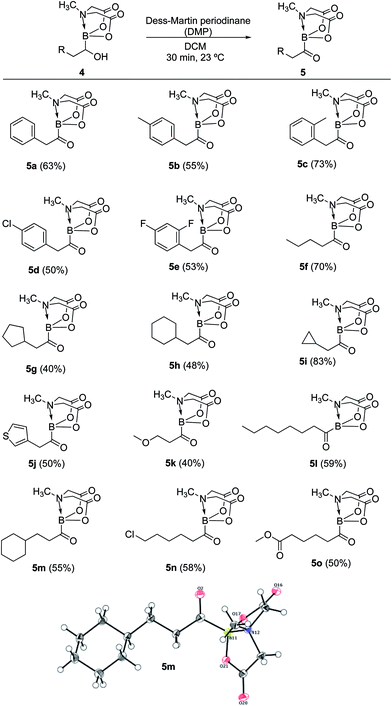 |
| | Scheme 6 Synthesis of MIDA acylboronates. Reaction conditions: 4 (0.4 mmol), DMP (1.1 equiv.), DCM (1.5 ml), 23 °C, 30 min. X-ray crystal structure of one of the novel acylboronates (5m) is also shown. | |
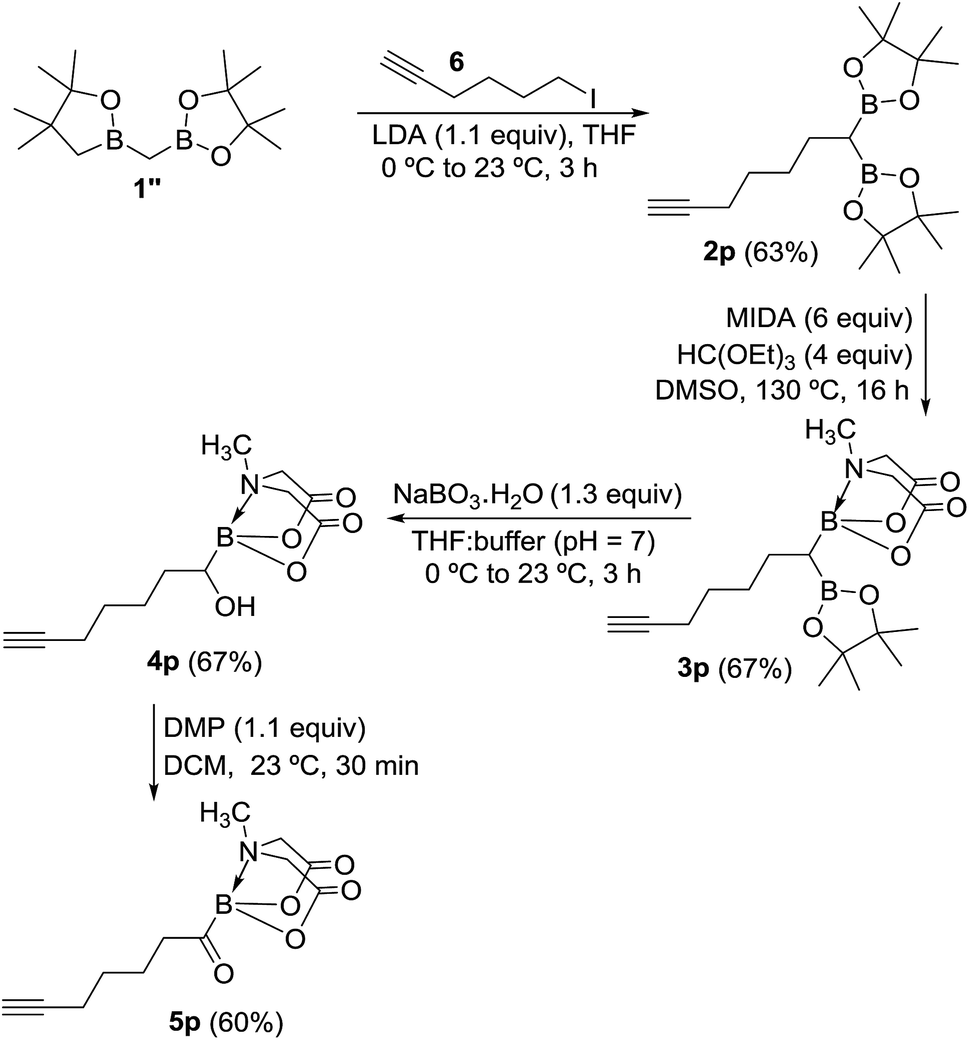
To further demonstrate the utility and unique versatility of diboryl alkane oxidation approach for modular synthesis of multifunctional acylboronates, we envisioned the use of commercially available bis[(pinacolato)boryl]methane (1′′, Scheme 7) as a key precursor for preparation of acylboronates that possess additional functional groups such as alkynes. 1′′ can be easily prepared in the lab at >1 g scale.11b Thus, deprotonation of 1′′ with LDA followed by treatment with 6-iodo-1-hexyne provided the geminal diboryl alkane (2p, Scheme 7) possessing a terminal alkyne group. Conversion of one of the B(Pin) groups of 2p into B(MIDA) followed by chemoselective oxidation of B(Pin) with sodium perborate furnished the corresponding α-hydroxy MIDA boronate (4p). Oxidation of 4p using DMP provided a straightforward access to the first MIDA acylboronate (5p, Scheme 7) possessing a terminal alkyne functional group. The presence of acylboron and alkyne functional groups in the same molecule allows some unique applications in chemical biology and organic synthesis because these compounds can participate in mutually orthogonal10 bioorthogonal chemistry and cascade reactions. The only previous report for an alkynyl acylboron involved the use of BuLi and cryogenic temperature.10
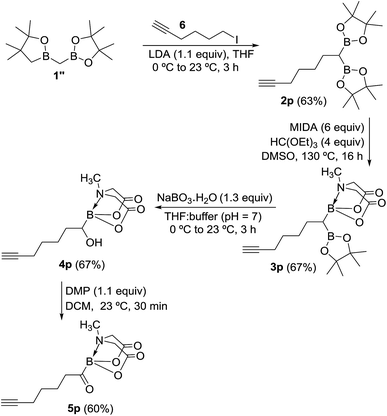 |
| | Scheme 7 Mild and concise route to alkynyl MIDA acylboronate. | |
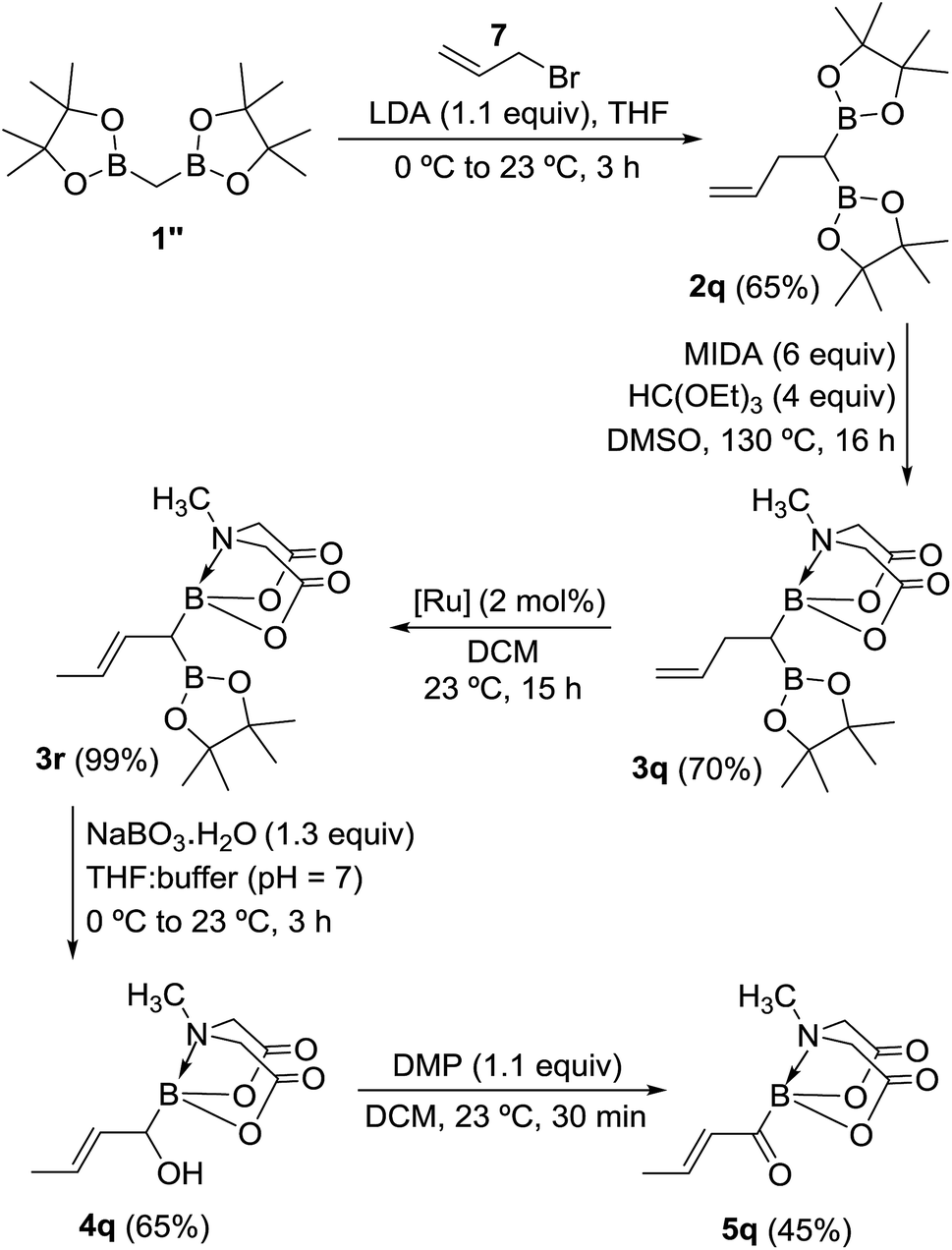
Inspired by the success of above approach, we hypothesized that chemoselective oxidation of B(Pin) in a differentially protected geminal diboron located at the allylic position of an alkene should provide access to a novel class of acylborons: α,β-unsaturated MIDA acylboronates. Thus, allyl bromide (7) was converted into the homoallyl diboron compound (2q, Scheme 8) by treatment with deprotonated diborylmethane (1′′).16a Conversion of one of the B(Pin) groups of 2q into B(MIDA) provided the homoallylic unsymmetrical diboron (3q) which was isomerized to allylic diboron (3r) using a cationic Ru(II) complex.173r was subjected to chemoselective oxidation protocol (Scheme 8) to provide the α-hydroxy boronate (4q). Oxidation of this allylic alcohol using DMP afforded the targeted α,β-unsaturated MIDA acylboronate (5q, Scheme 8) which was found to be stable to silica gel chromatography. To the best of our knowledge, this is the first report of an α,β-unsaturated acylboron. This novel class of acylboronates would be difficult to synthesize using the conventional methods.
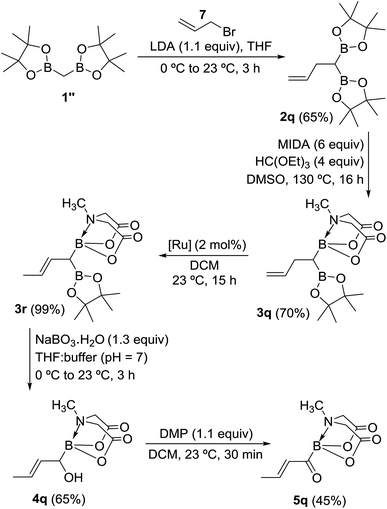 |
| | Scheme 8 Synthesis of a novel acylboron: α,β-unsaturated acylboronate. [Ru] = [CpRu(P–N)(MeCN)]PF6 (P–N = 2-PiPr2-4-tBu-1-Me-imidazole). | |
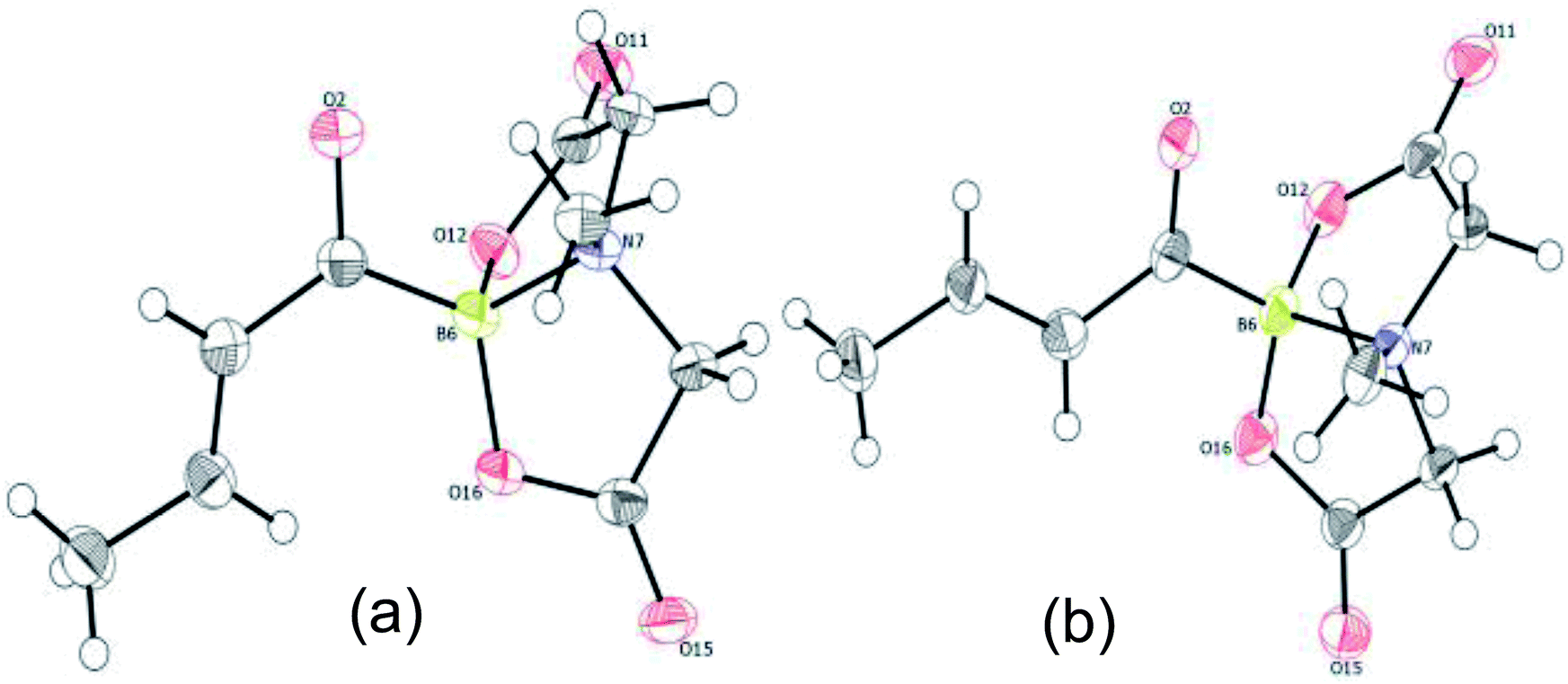
To gain deeper insights into the molecular structure of this novel α,β-unsaturated acylboronate, single crystal X-ray diffraction analysis of 5q was conducted.18 There were two molecules in the asymmetric unit. Interestingly, one of the independent molecules was exclusively s-trans (Fig. 1a) while the second molecule was predominantly s-cis (Fig. 1b). The C(O)C3H5 moiety in this second molecule (Fig. 1b and ESI, Fig. S2†) was disordered in a 9:1 ratio by rotation around the C(sp2)–C(sp2) bond, i.e. a 9:1 mixture of s-cis and s-trans isomers. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length in both of these isomers (1.234 and 1.237 Å) is similar to that of unconjugated acylborons (1.234 Å)6e but slightly longer than α,β-unsaturated ketones/aldehydes (1.222 Å).19 The CC bond length (1.317 Å) of s-cis isomer is slightly shorter than that of s-trans isomer (1.332 Å) which is also shorter than α,β-unsaturated ketones/aldehydes (1.340 Å).19 The C–(CO)–B angle in case of s-trans (125.95°) isomer is larger than that found in typical trigonal planar systems (120°) as well as the s-cis isomer (120.39°, Fig. 1b). Presumably, this is due to increased repulsion between the olefin and boronate groups in case of s-trans isomer.
O bond length in both of these isomers (1.234 and 1.237 Å) is similar to that of unconjugated acylborons (1.234 Å)6e but slightly longer than α,β-unsaturated ketones/aldehydes (1.222 Å).19 The CC bond length (1.317 Å) of s-cis isomer is slightly shorter than that of s-trans isomer (1.332 Å) which is also shorter than α,β-unsaturated ketones/aldehydes (1.340 Å).19 The C–(CO)–B angle in case of s-trans (125.95°) isomer is larger than that found in typical trigonal planar systems (120°) as well as the s-cis isomer (120.39°, Fig. 1b). Presumably, this is due to increased repulsion between the olefin and boronate groups in case of s-trans isomer.
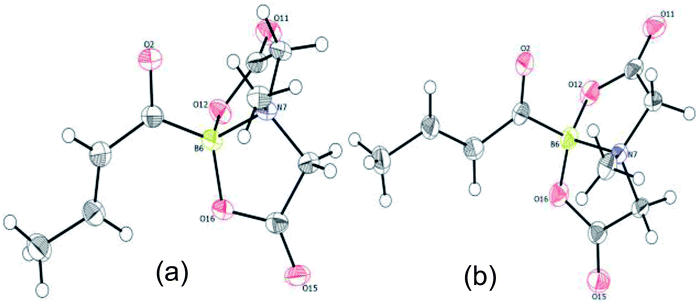 |
| | Fig. 1 X-ray crystal structure of α,β-unsaturated acylboronate (5q). (a) s-trans isomer (b) s-cis isomer (the 10% s-trans isomer has been omitted for better clarity). | |
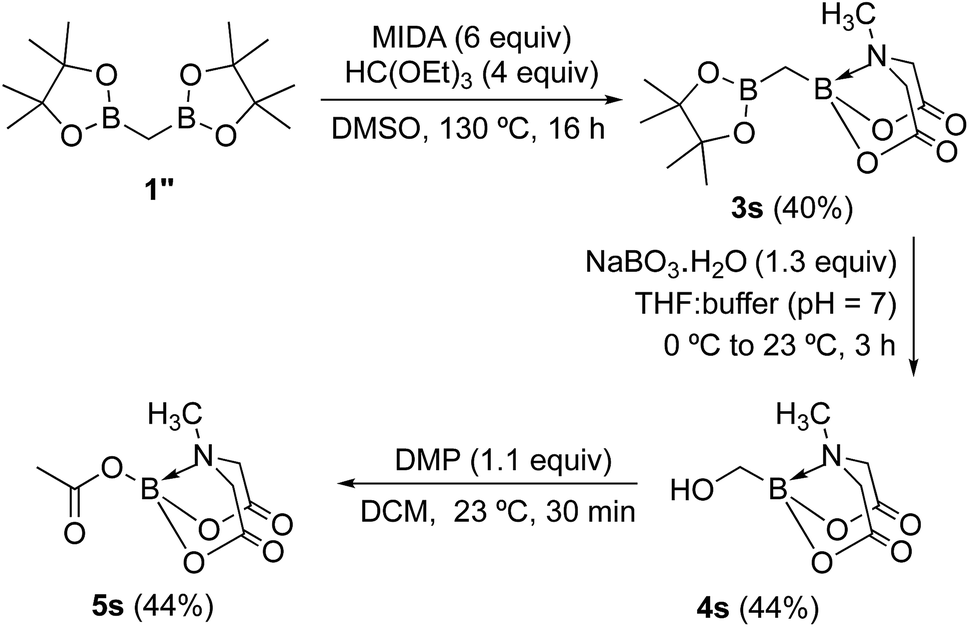
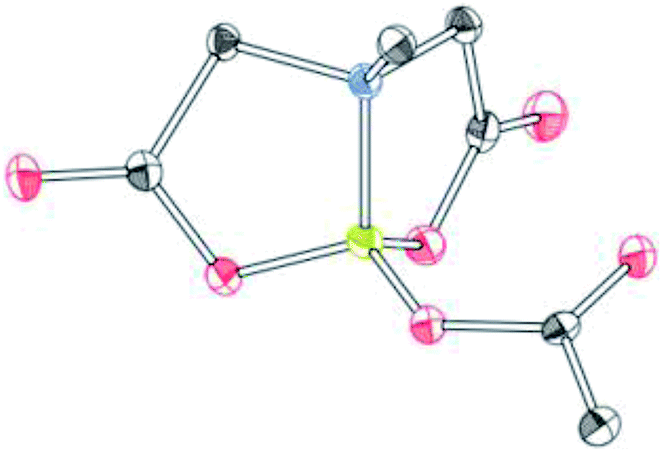
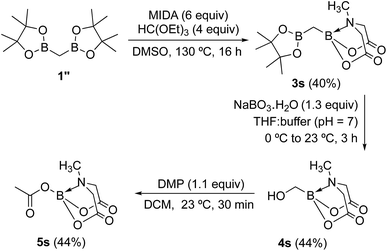
To further investigate the scope of our developed approach for synthesis of unsymmetrical geminal diborylalkanes (Scheme 4), two additional multiborylated substrates: diborylmethane (1′′) and a 1,1,2-triB(Pin) compound (2t) were evaluated. Significantly, 1′′ was also found to be compatible with the reaction conditions as the desired unsymmetrical diborylmethane (3s, Scheme 9) was obtained in 40% yield. 3s was amenable to chemoselective oxidation to afford the hydroxy methyl MIDA boronate (4s). 4s has been previously synthesized by Yudin and coworkers through an alternative approach and this compound is an important building block for construction of biologically important boron-containing heterocyclic compounds.20 Interestingly, oxidation of hydroxy methyl MIDA boronate (4s) using DMP provided the acetoxy MIDA boronate (5s)21 instead of the expected formyl boronate (Scheme 9). The structure of this acetoxy MIDA boronate was confirmed by X-ray diffraction (Fig. 2). Although the detailed mechanism for formation of 5s is not clear at this stage, this unexpected result could be due to the high reactivity/instability of formyl boronate and involvement of acetic acid byproduct generated during DMP mediated oxidation.22
 |
| | Scheme 9 Synthesis and oxidation of hydroxy methyl MIDA boronate. | |
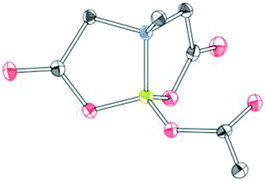 |
| | Fig. 2 X-ray crystal structure of acetoxy MIDA boronate (5s). | |
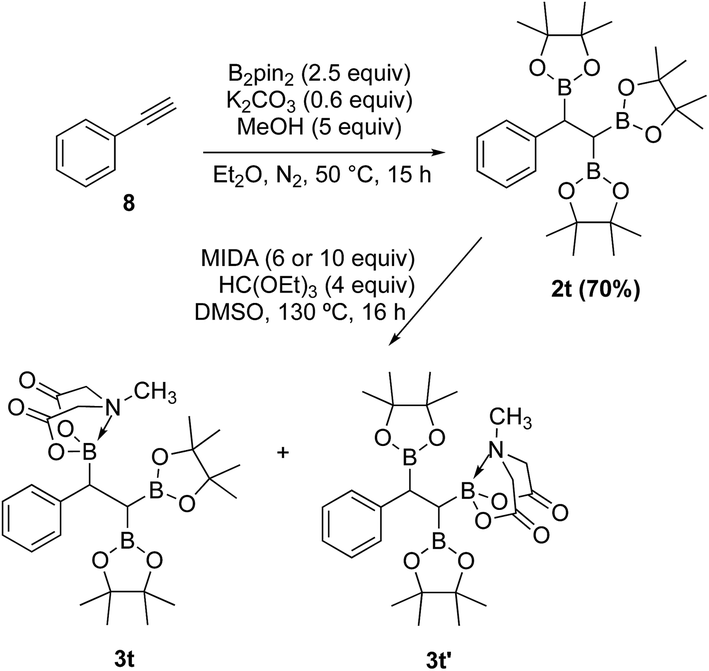
In contrast to the diborylmethane (1′′, Scheme 9) and several other geminal diborylalkanes (2a–o, Scheme 4) that underwent selective conversion into the unsymmetrical geminal diborons, a 1,1,2-triB(Pin) compound was not a suitable substrate. For instance, treatment of 2t (obtained via borylation of alkyne 8)23 with MIDA (Scheme 10) provided a mixture (1:1 ratio, NMR) of the two mono-B(MIDA)–diB(Pin) compounds (3t and 3t′, Scheme 10) possessing B(MIDA) group vicinal to diB(Pin) or geminal to the B(Pin). This mixture was inseparable using silica gel chromatography due to very small Rf difference between the two compounds (3t and 3t′).
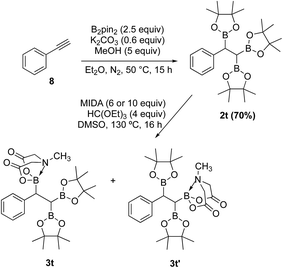 |
| | Scheme 10 Attempted synthesis of an unsymmetrical geminal diborylalkane from a 1,1,2-triB(Pin) compound (2t). | |
Conclusions
In summary, we have developed a novel and modular route to MIDA acylboronates from alkynes/alkenes or vinyl boronic esters. Our strategy involved synthesis of differentially protected geminal diboron compounds as key precursors that were amenable to chemoselective oxidation into MIDA acylboronates. Importantly, this new approach drastically reduces the number of steps required for synthesis of α-hydroxy MIDA boronates and acyl boronates from readily available substrates such as terminal alkynes and vinyl boronic esters. To the best of our knowledge, this is the first report of chemoselective oxidation of geminal diborylalkanes. The methodology provided a concise and mild access to acylboronates possessing aliphatic, aromatic as well as the rarer heteroaromatic and alkynyl scaffolds which have applications in mutually orthogonal bioorthogonal chemistry. Significantly, the unique modularity of this methodology also opened up a facile access to a novel class of multifunctional acylborons: α,β-unsaturated MIDA acylboronate. The availability of α,β-unsaturated MIDA acylboronates is expected to open up exciting opportunities to design novel reactivity patterns and further expand the chemical space of acylborons and organoborons.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Dr Athula Attygalle and Zhaoyu Zheng for HRMS analysis. Financial support from Stevens Institute of Technology is gratefully acknowledged. Dr Daniel Paley performed single-crystal X-ray diffraction studies, prepared the thermal ellipsoid plots in Scheme 6, Fig. 1 and 2, and provided measurements/text concerning the X-ray analysis of compound 5q. Single-crystal diffraction was performed in the Shared Materials Characterization Laboratory at Columbia Nano Initiative (Columbia University).
References
-
(a)
D. G. Hall, Boronic Acids, Wiley-VCH, Weinheim, 2006 Search PubMed;
(b) G. A. Molander, J. Org. Chem., 2015, 80, 7837 CrossRef CAS PubMed.
-
(a) F. K. Scharnagl, S. K. Bose and T. B. Marder, Org. Biomol. Chem., 2017, 15, 1738 RSC;
(b) J. D. St. Denis, Z. He and A. K. Yudin, ACS Catal., 2015, 5, 5373 CrossRef.
- M. R. Ibrahim, M. Bühl, R. Knab and P. V. R. Schleyer, J. Comput. Chem., 1992, 13, 423 CrossRef CAS.
- M. Yamashita, Y. Suzuki, Y. Segawa and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 9570 CrossRef CAS PubMed.
- G. A. Molander, J. Raushel and N. M. Ellis, J. Org. Chem., 2010, 75, 4304 CrossRef CAS PubMed.
-
(a) A. M. Dumas and J. W. Bode, Org. Lett., 2012, 14, 2138 CrossRef CAS PubMed;
(b) G. Erős, Y. Kushida and J. W. Bode, Angew. Chem., Int. Ed., 2014, 53, 7604 CrossRef PubMed;
(c) J. Taguchi, T. Ikeda, R. Takahashi, I. Sasaki, Y. Ogasawara, T. Dairi, N. Kato, Y. Yamamoto, J. W. Bode and H. Ito, Angew. Chem., Int. Ed., 2017, 56, 13847 CrossRef CAS PubMed;
(d) H. Noda and J. W. Bode, Chem. Sci., 2014, 5, 4328 RSC;
(e) H. Noda and J. W. Bode, J. Am. Chem. Soc., 2015, 137, 3958 CrossRef CAS PubMed;
(f) S. M. Liu, D. Wu and J. W. Bode, Org. Lett., 2018, 20, 2378 CrossRef CAS PubMed.
-
(a) Z. He, P. Trinchera, S. Adachi, J. D. St Denis and A. K. Yudin, Angew. Chem., Int. Ed., 2012, 51, 11092 CrossRef CAS PubMed;
(b) S. Adachi, S. K. Liew, C. F. Lee, A. Lough, Z. He, J. D. S. Denis, G. Poda and A. K. Yudin, Org. Lett., 2015, 17, 5594 CrossRef CAS PubMed;
(c) C. F. Lee, A. Holownia, J. M. Bennett, J. M. Elkins, J. D. St. Denis, S. Adachi and A. K. Yudin, Angew. Chem., Int. Ed., 2017, 56, 6264 CrossRef CAS PubMed.
-
(a) M. L. Lepage, S. Lai, N. Peressin, R. Hadjerci, B. O. Patrick and D. M. Perrin, Angew. Chem., Int. Ed., 2017, 56, 15257 CrossRef CAS PubMed;
(b) M. Sajid, G. Kehr, C. G. Daniliuc and G. Erker, Angew. Chem., Int. Ed., 2014, 53, 1118 CrossRef CAS PubMed;
(c) J. Campos and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 14159 CrossRef CAS PubMed.
-
(a) A. M. Dumas, G. A. Molander and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 5683 CrossRef CAS PubMed;
(b) C. J. White and J. W. Bode, ACS Cent. Sci., 2018, 4, 197 CrossRef CAS PubMed;
(c) H. Noda, G. Erős and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 5611 CrossRef CAS PubMed.
- S. M. Liu, D. Mazunin, V. R. Pattabiraman and J. W. Bode, Org. Lett., 2016, 18, 5336 CrossRef CAS PubMed.
-
(a) R. Nallagonda, K. Padala and A. Masarwa, Org. Biomol. Chem., 2018, 16, 1050 RSC;
(b) K. Hong, X. Liu and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 10581 CrossRef CAS PubMed;
(c) B. Potter, A. A. Szymaniak, E. K. Edelstein and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 17918 CrossRef CAS PubMed;
(d) D. J. Blair, D. Tanini, J. M. Bateman, H. K. Scott, E. L. Myers and V. K. Aggarwal, Chem. Sci., 2017, 8, 2898 RSC;
(e) S. A. Murray, J. C. Green, S. B. Tailor and S. J. Meek, Angew. Chem., Int. Ed., 2016, 55, 9065 CrossRef CAS PubMed;
(f) Y. Shi and A. H. Hoveyda, Angew. Chem., Int. Ed., 2016, 55, 3455 CrossRef CAS PubMed;
(g) K. Endo, T. Ohkubo, M. Hirokami and T. Shibata, J. Am. Chem. Soc., 2010, 132, 11033 CrossRef CAS PubMed;
(h) Q. Huang and S. Z. Zard, Org. Lett., 2018, 20, 5304 CrossRef CAS PubMed.
- I. Marek and J.-F. Normant, Chem. Rev., 1996, 96, 3241 CrossRef CAS PubMed.
-
(a) X. Li and D. G. Hall, Angew. Chem., Int. Ed., 2018, 57, 10304 CrossRef CAS PubMed;
(b) J. J. Molloy, T. A. Clohessy, C. Irving, N. A. Anderson, G. C. Lloyd-Jones and A. J. B. Watson, Chem. Sci., 2017, 8, 1551 RSC.
- S. Lee, D. Li and J. Yun, Chem.–Asian J., 2014, 9, 2440 CrossRef CAS PubMed.
- E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716 CrossRef CAS PubMed.
-
(a) D. S. Matteson and R. J. Moody, Organometallics, 1982, 1, 20 CrossRef CAS;
(b) G. W. Kabalka, T. M. Shoup and N. M. Goudgaon, J. Org. Chem., 1989, 54, 5930 CrossRef CAS.
- T. Miura, J. Nakahashi, W. Zhou, Y. Shiratori, S. G. Stewart and M. Murakami, J. Am. Chem. Soc., 2017, 139, 10903 CrossRef CAS PubMed.
- CCDC 1880524 (5m), 1880525 (5q) and 1900591 (5s) contain the supplementary crystallographic data for this paper.†.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, 1, 1 RSC.
- S. Adachi, A. B. Cognetta, M. J. Niphakis, Z. He, A. Zajdlik, J. D. St. Denis, C. C. G. Scully, B. F. Cravatt and A. K. Yudin, Chem. Commun., 2015, 51, 3608 RSC.
- C. F. Lee, D. B. Diaz, A. Holownia, S. J. Kaldas, S. K. Liew, G. E. Garrett, T. Dudding and A. K. Yudin, Nat. Chem., 2018, 10, 1062 CrossRef CAS PubMed.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS.
- G. Gao, J. Yan, K. Yang, F. Chen and Q. Song, Green Chem., 2017, 19, 3997 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures for synthesis of compounds, optimization conditions, spectral and X-ray data. CCDC 1880524, 1880525 and 1900591. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00378a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*