
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single promoter system co-expressing RNA sensor with fluorescent proteins for quantitative mRNA imaging in living tumor cells†
Zhan-Ming
Ying
,
Yue-Yan
Yuan
,
Bin
Tu
,
Li-Juan
Tang
*,
Ru-Qin
Yu
 and
Jian-Hui
Jiang
*
and
Jian-Hui
Jiang
*
Institute of Chemical Biology & Nanomedicine, State Key Laboratory of Chemo/Biosensing & Chemometrics, College of Chemistry & Chemical Engineering, Hunan University, Changsha 410082, China. E-mail: jianhuijiang@hnu.edu.cn; tanglijuan@hnu.edu.cn
First published on 29th March 2019
Abstract
Genetically encoded light-up RNA aptamers afford a valuable platform for developing RNA sensors toward live cell imaging. However, quantitative imaging of intracellular RNAs remains a grand challenge. Here we reported a novel genetically encoded RNA sensor strategy using a plasmid that expresses a splittable fusion of the RNA sensor and the GFP mRNA in an individual transcript using a single promoter system. This splittable fusion design enables synchronous co-expression of the RNA sensor with GFP mRNA while alleviates the interference with correct folding of RNA aptamers due to intramolecular hybridization. This single-promoter system is applied to ratiometric imaging of survivin mRNA in tumor cells. The results reveal that the ratiometric images dynamically correlated with survivin mRNA concentrations and allow quantitative imaging of survivin mRNA in different tumor cells. The RNA sensor strategy may provide a new paradigm for developing a robust imaging platform for quantitative mRNA studies in living cells.
Introduction
“Seeing” the abundance of RNAs as well as their localization and underlying dynamics in living cells is essential for understanding cellular biology, since the behaviours of RNAs drive all sorts of fundamental processes of life.1,2 RNA-targeting imaging probes, including fluorescent protein (GFP)-tagged RNA binding proteins3,4 and hybridization probes5–8 such as molecular beacons9 and spherical nucleic acids (SNA)-based nanoflares,10,11 have significantly contributed to this area. Nevertheless, quantitative RNA imaging of living cells is still largely unexplored because of the perturbations from different analyte-independent factors such as changes in focuses and laser intensity. Ratiometric imaging corrects these factors by providing built-in self-calibration and has the potential to improve the quantitative measurement of cells. Therefore, development of ratiometric RNA imaging approaches is in high demand for live cell studies.Genetically encoded RNA sensors,12,13 based on light-up RNA aptamers,14 have emerged as a valuable platform for fluorescent imaging in living cells. Light-up RNA aptamers, represented by Spinach,15,16 Broccoli17 and Mango,18 are a class of engineered RNA molecules displaying “turn-on” fluorescence upon binding to nonfluorescent small-molecule dyes. When integrated with target-specific sequences as the responsive module, light-up RNA aptamers have enabled detection of varying targets including metabolites,19 proteins20 and RNAs21,22 in varying biological media20 and even in living cells.21,22 We have recently reported a genetically encoded RNA sensor for ratiometric imaging of microRNA via engineering a plasmid with two promoters, which allows stable expression and robust detection of the RNA sensor in living mammalian cells.23 The design of using two promoters separately expressing the RNA sensor and GFP involves two asynchronous transcription events recruiting RNA polymerases II or III, respectively.24 Such asynchronism may result in different concentration ratios of the RNA sensor and GFP mRNA at varying times, leading to asynchronous signals for the GFP reference and the RNA sensor. Therefore, this system only calibrates the variations due to transfection efficiency in different cells, but the asynchronism for the RNA sensor and GFP is not precluded, limiting the ability of the RNA sensor for quantitative RNA imaging. Development of a genetically encoded sensor enabling quantitative ratiometric imaging has not been reported.
Because the GFP fluorescence is largely dependent upon the abundance of GFP mRNA,25 we envisioned that a fusion transcript of the RNA sensor with GFP mRNA had the potential to minimize the asynchronism of GFP and the RNA sensor. Motivated by this hypothesis, we develop a novel genetically encoded RNA sensor strategy using a plasmid that expresses a fusion of the RNA sensor and the GFP mRNA in an individual transcript using a single promoter system, as illustrated in Scheme 1A. The sensor is designed to be fused to the 3′ UTR of GFP mRNA using a two-domain spacer, an mRNA-stabilizing triplex26 in the upstream domain and a dsRNA Dicer substrate27 in the downstream. This fusion design enables a synchronous co-expression of the RNA sensor and GFP mRNA, allowing synchronism of the reference GFP fluorescence with the RNA sensor. However, this fusion can interfere with the folding of the RNA sensor because of the intracellular hybridization of GFP mRNA with the RNA sensor. To avoid this interference, a dsRNA Dicer substrate is incorporated between the RNA sensor and GFP mRNA, which allows the transcript to be cleaved at the dsRNA site by Dicer enzyme that is ubiquitously present in mammalian cells.28 Furthermore, the mRNA-stabilizing triplex is introduced at the 3′ terminal of GFP mRNA to avoid fast degradation of GFP mRNA.
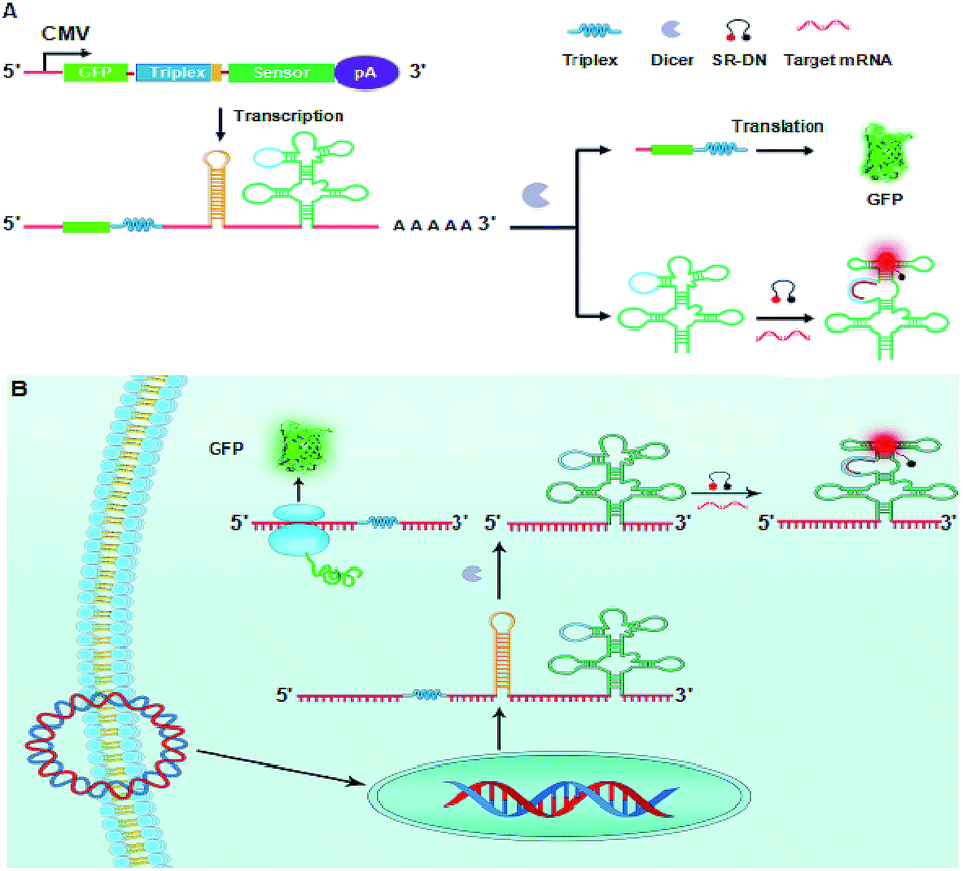 | ||
| Scheme 1 Illustration of plasmid design (A) and the RNA sensor from a splittable fusion transcript with GFP for ratiometric mRNA imaging (B). | ||
According to this design, when the plasmid is transfected into mammalian cells, a splittable RNA fusion can be transcribed from a single CMV promoter in the cells, as illustrated in Scheme 1B. In the cytosol, the transcript is cleaved at the dsRNA site, producing two separate RNAs with one for the stabilized GFP mRNA and the other for the RNA sensor. The RNA sensor is incorporated into a tRNA scaffold, which improves stability of the RNA sensor in mammalian cells.22 On hybridizing with target mRNA, the sensor undergoes a conformational change and recovers the conformation of a light-up aptamer, sulforhodamine B (SR) aptamer. It is recalled that the SR aptamer is capable of lighting up the quenched fluorescence of a conjugate of SR with dinitroaniline (DN).23,29 Therefore, the activated fluorescence from SR gives an indicator for the expression level of target mRNA. On the other hand, the stabilized GFP mRNA still retains its activity to be translated into GFP, which delivers a green fluorescence signal acting as the internal reference for ratiometric imaging. Because the single promoter mediated co-expression of GFP mRNA and the RNA sensor minimizes the asynchronism of GFP fluorescence and the signal of the RNA sensor, the ratiometric imaging afforded the possibility for quantitative imaging of mRNA expression. To demonstrate its potential, this novel RNA sensor has been applied to ratiometric imaging of survivin mRNA, a known biomarker over-expressed in different tumors,30 in living cells. The results revealed that the obtained ratiometric images were dynamically correlated with survivin mRNA concentrations in varying cell lines. Moreover, the results were confirmed by quantification using quantitative reverse transcription PCR (qRT-PCR), implying the promise of the RNA sensor as a robust imaging platform for quantitative mRNA studies in living cells. To our knowledge, there has been no previous study reporting genetically encoded light-up RNA sensors of mRNA imaging in mammalian cells. Hence, our work represents the first time to engineer genetically encoded light-up RNA sensors for quantitative ratiometric imaging of mRNA in living mammalian cells.
Results and discussion
Single promoter mediated co-expression of RNA aptamers and GFP in living tumor cells
A co-expressing RNA sensor with GFP using one plasmid enabled calibrating the variations due to transfection efficiency in different cells.23 Nevertheless, the asynchronism in the transcription efficiency of two different promoters could lead to varied concentration ratios for the RNA sensor to the GFP reference in different cells due to their heterogeneity. As a matter of fact, we found that the fluorescence of the SR aptamer and GFP exhibited obvious differences in their relative intensity in different HeLa cells (Fig. 1A) after the cells were transfected using a plasmid with two promoters for separate expression of GFP and the SR RNA aptamer. The difference revealed significant variations of cells in expression of the RNA aptamer and GFP, evidencing the asynchronism in their relative expression levels and thereby in their fluorescence signals from cell to cell. As the reference fluorescence did not vary in correlation with the SR aptamer signal, there was a limitation for the two-promoter system in quantitative imaging of living cells.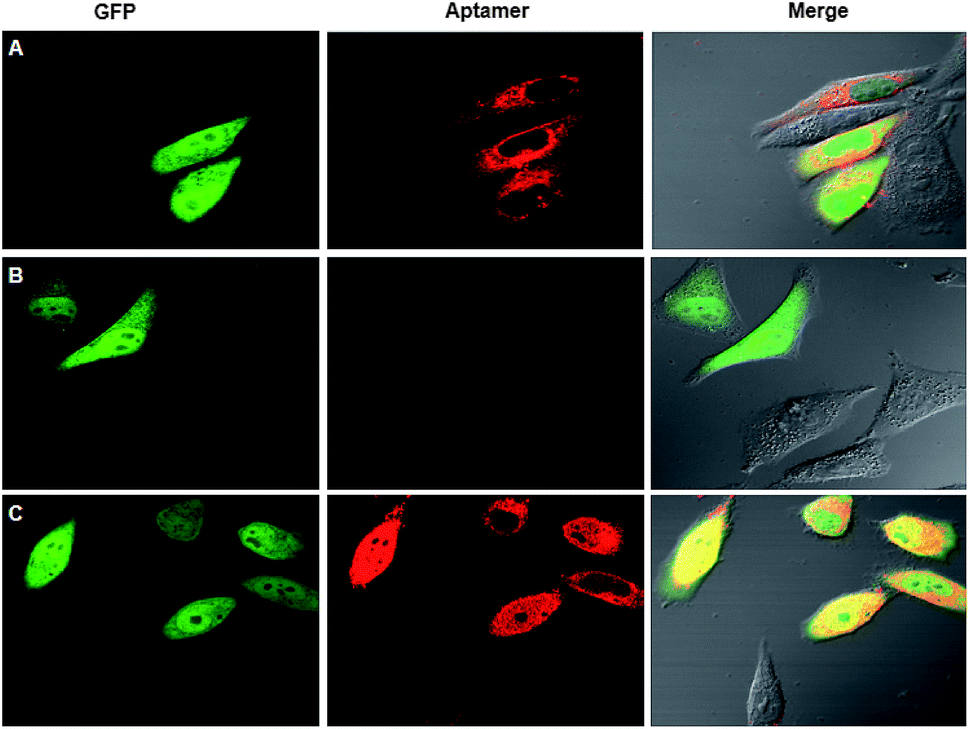 | ||
| Fig. 1 Fluorescence images for HeLa cells transfected using a two-promoter plasmid (A) and a single-promoter plasmid with no Dicer substrate site (B) and a Dicer substrate site (C) for co-expression of the SR aptamer and GFP mRNA. | ||
Co-expressing the RNA aptamer with GFP in the same transcription event had the potential to minimize the asynchronism in the expression of the RNA aptamer and GFP. Based on the hypothesis, we designed a plasmid with a single promoter that could generate a fusion of the SR aptamer and GFP mRNA in an individual transcript. Surprisingly, after transfecting the plasmid into HeLa cells for 24 h, we merely observed green fluorescence of GFP with no detectable red fluorescence for the SR aptamer even in the presence of SR–DN (Fig. 1B), implying misfolding of the SR RNA aptamer. Considering the close proximity of the GFP mRNA located in the upstream of the fusion transcript to the downstream RNA aptamer, we reasoned that the folding of the RNA aptamer was distorted by the GFP mRNA due to the facilitated intramolecular folding of the fusion transcript.
We then envisioned that splitting of the RNA aptamer from the fusion could alleviate the inference with the folding of the SR aptamer from GFP mRNA. Hence, we re-engineered a new plasmid with a Dicer-cleavage site in the fusion between the RNA aptamer and GFP mRNA (Scheme S1†). After the fusion transcript was expressed in mammalian cells, Dicer enzyme split the fusion at the dsRNA site in the cytosol into the GFP mRNA with a stabilizing triplex tail and the RNA aptamer in a tRNA scaffold.23 To confirm the success of this design, the total RNA extracts of HeLa cells after transfection with the plasmid were detected using RT-PCR with gel electrophoresis using three pairs of primers specific to the GFP mRNA, Dicer substrate site and the tRNA-scaffolded RNA aptamer, respectively. The bright gel bands that appeared at 171 bp, 109 bp and 138 bp, respectively, were consistent with the design (Fig. S1†). To testify the cleavage of the RNA transcript, qRT-PCR analysis was performed separately for the plasmids with or without the Dicer substrate site. A much lower concentration was found for the RNA transcript with the Dicer substrate site than for that without the site (Fig. S2†). This result confirmed the efficient cleavage of the RNA transcript with the Dicer substrate site in cells. Additionally, confocal microscopy images of cells transfected with the plasmid with the Dicer substrate site showed bright red fluorescence for the SR aptamer in the presence of SR–DN and green fluorescence for GFP (Fig. 1C). The images indicated successful expression and correct folding of the SR RNA aptamer and GFP reference in the cells. The appearance of red fluorescence actually verified our previous hypothesis that the correct conformation of the SR aptamer was distorted by GFP mRNA due to intramolecular folding, considering that cells transfected with the plasmid without the Dicer substrate site did not display the red fluorescence of the SR aptamer. Moreover, we observed that the red fluorescence signals in different cells changed in a consistent manner with the corresponding green fluorescence, indicating the synchronism of the red and the green fluorescence signals. Taken together, the results demonstrated the feasibility of co-expressing the correctly folded RNA aptamer with the GFP reference using a single promoter that afforded synchronous fluorescence signals in living mammalian cells.
To further testify the synchronism of fluorescence signals for the RNA aptamer and GFP, qRT-PCR analysis for the total RNA extracts from cells transfected with a single-promoter plasmid revealed that the relative concentrations of the RNA aptamer were approximate to those of GFP mRNA at different times (Fig. S3†). This quantification result supported our design that a single promoter plasmid allowed synchronous expression of the RNA aptamer and GFP mRNA. In addition to the RNA level, we then investigated the fluorescence signals for GFP and the RNA aptamer using confocal images. It was observed that the fluorescence signals of GFP appeared very bright after 24 or 48 h transfection, and the intensity decreased slightly after 60 h transfection (Fig. 2). Similar time-dependent changes were also found for fluorescence signals of the RNA aptamer. This finding suggested that a single promoter plasmid afforded synchronous fluorescence intensities for the RNA aptamer and GFP in cells, implying its potential for developing quantitative ratiometric genetically encoded RNA sensors for imaging of living tumor cells.
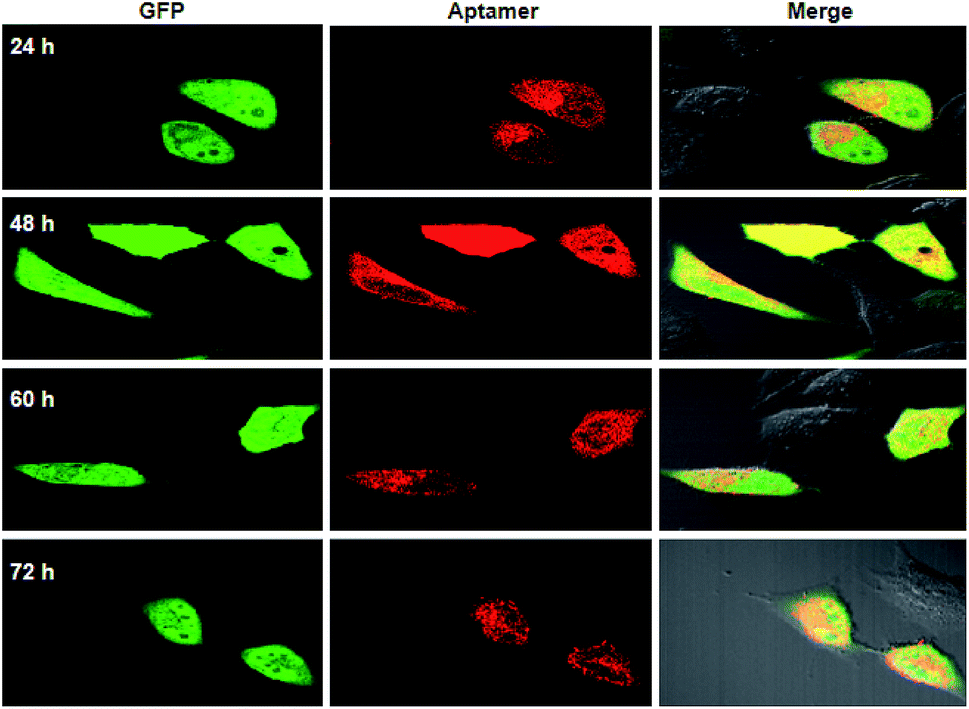 | ||
| Fig. 2 Fluorescence images for HeLa cells transfected using a single-promoter plasmid expressing splittable fusion of the RNA aptamer and GFP at different times. | ||
Single promoter mediated co-expression of the RNA sensor and GFP for quantitative mRNA imaging in living cells
Next, we utilized a single promoter plasmid to develop an RNA sensor for quantitative imaging of mRNA in living cells. It is known that mRNA is critical in regulating cellular polarity, migration and development, and aberrant expression of mRNA is highly implicated in the progression of various diseases including cancer. In this study we chose mRNA survivin31 as the model target and designed the SR aptamer into an RNA sensor for its detection in living cells (Scheme 1A). The sensor was engineered by fusing a stem–loop motif in response to survivin mRNA in the terminal stem of the SR aptamer in light of our previous design.23 The presence of target mRNA allowed re-folding of the SR-binding loop, activating the fluorescence signals that illuminated the expression of survivin.The as-prepared RNA sensor was firstly examined via in vitro assays using a synthetic DNA template for the RNA sensor with a T7 promoter for in vitro transcription. The sensor was found to display remarkable fluorescence activation in response to survivin with an enhancement ratio of ∼11 in peak intensities (Fig. S4†). In addition, we found that the tRNA-scaffolded sensor has comparable performance with its prototype RNA sensor without the tRNA scaffold (Fig. S5†). The sensor also exhibited high selectivity to survivin in contrast to other interferents including other RNA sequences, proteins and a lysate of C166 cells, a cell line with negative expression for survivin32 (Fig. S6†). Gel electrophoresis analysis gave further evidence for stable hybridization and fluorescence activation of the RNA sensor in response to survivin in the presence of SR–DN (Fig. S7†). Moreover, the RNA sensor displayed enhanced fluorescence responses to survivin in a dose dependent manner in a concentration range of 1 nM to 5 μM (Fig. S8†). The detection limit was estimated to be 0.4 nM according to the 3σ rule, suggesting the high sensitivity of the sensor for survivin detection.
On the basis of the preliminary results, we then used the single-promoter plasmid for transfection of living cells for ratiometric imaging of survivin in the cells (Scheme S2†). After transfecting HeLa cells using the plasmid in the presence of SR–DN for 24 h, very bright green fluorescence signals from GFP appeared relatively evenly in the nuclei and in the cytosol. Bright red fluorescence was also observed in the cytosol of HeLa cells, a cell line with overexpressed survivin, which indicated activated fluorescence of the RNA sensor in response to survivin (Fig. 3A). It was noticed that the fluorescence response of the RNA sensor was appreciably weaker than the signals generated using the RNA aptamer obtained in the preliminary experiment (Fig. 2). This result was ascribed to the low abundance of survivin in HeLa cells, which did not restore the conformation of all RNA aptamers expressed in the cells. Additionally, we observed fluorescence signals arising from the RNA aptamer both in the nuclei and in the cytosol, indicating that the tRNA scaffolded RNA aptamer were localized throughout the cells. In contrast, the fluorescence of the RNA sensor was only found in the cytosol, which is ascribed to the cytosol localization of target survivin mRNA. To validate the selectivity of the fluorescence response, we also performed a control experiment using the survivin-negative cell line C166 (ref. 32) (Fig. 3B). The dim red fluorescence displayed in C166 cells suggested that the fluorescence of the RNA sensor was specifically activated in the presence of survivin. In addition, we also did not observe an enhanced fluorescence response in HeLa cells transfected using a single-promoter plasmid with no Dicer-substrate site inserted between the GFP mRNA and the RNA sensor (Fig. S9†), confirming the necessity of the Dicer-substrate site for the single-promoter system.
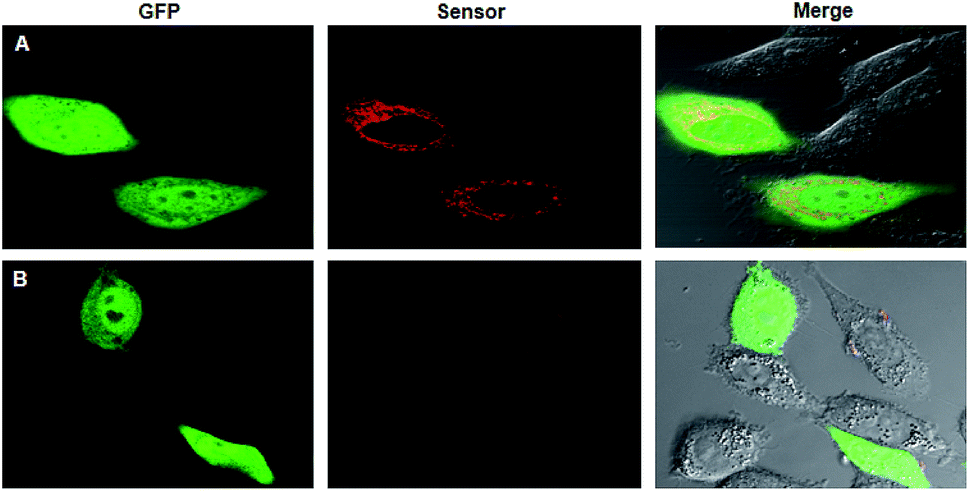 | ||
| Fig. 3 Fluorescence images of survivin mRNA in HeLa cells (A) and C166 cells (B). | ||
Next, we explored the RNA sensor for ratiometric imaging of survivin in different cell lines, MDA-MB-435S, HeLa, MCF-7 and MCF-10A (Fig. 4A). After transfecting these cells using the plasmid for 24 h, bright green fluorescence signals of GFP were observed in different cells, affording a robust reference for the RNA sensor. By contrast, we observed red fluorescence images of varying brightness and ratiometric images of different colors in these cells. MDA-MB-435S cells gave the highest expression level of survivin, whereas MCF-10A cells had the lowest expression, and HeLa cells gave a little higher expression than MCF-7 cells. These results were in good agreement with previous studies,33,34 implying that the fluorescence responses of the RNA sensor had the potential for quantitative detection of survivin expression. To further demonstrate its ability for quantitative imaging, we performed quantitative determination of survivin expression in these cell lines using qRT-PCR (Fig. 4B). As anticipated, the average values of the red-to-green fluorescence ratio in the regions of interest (ROIs), ten ROIs in each image, dynamically correlated with the relative concentrations of survivin (Fig. S10†). Moreover, the values of red-to-green fluorescence ratio were in a linear correlation with the relative concentrations of survivin (Fig. 4C), giving direct evidence of the potential of the RNA sensor in quantifying mRNA in living cells. The copy numbers of survivin in different cells calculated according to the average values of the red-to-green fluorescence ratio in ROIs also highly agreed with those as-determined by qRT-PCR, which further confirmed the ability of the RNA sensor for quantitative imaging of mRNA (Fig. S11†).
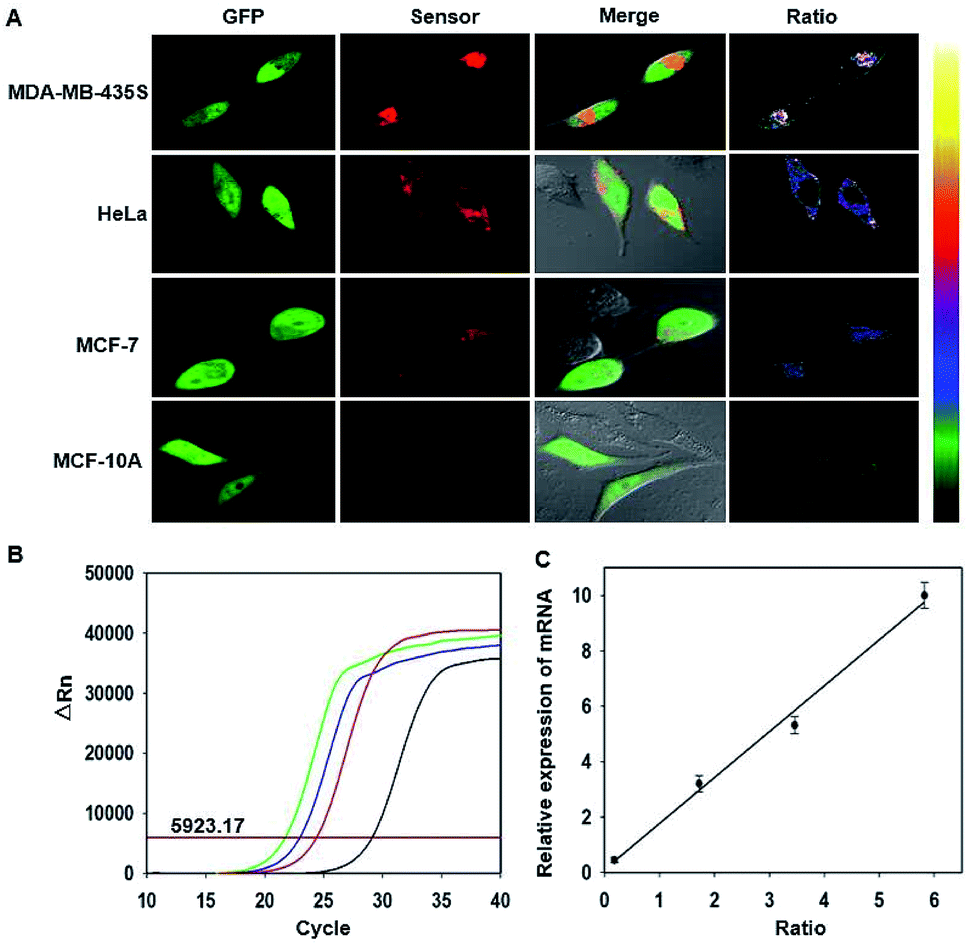 | ||
| Fig. 4 (A) Fluorescence and ratiometric images for different cells. (B) qRT-PCR analysis for the cells. Green: MDA-MB-435S. Blue: HeLa. Red: MCF-7. Black: MCF-10A. (C) Relationship of average values of the red-to-green fluorescence ratio in ROIs with relative concentrations of survivin mRNA in different cells. | ||
We further demonstrated the RNA sensor for quantitative imaging of survivin expression in drug-treated HeLa cells. The survivin expression in HeLa was altered by two small molecules of docetaxel and YM155.35 Docetaxel treatment led to docetaxel-induced cell apoptosis with a concomitant upregulation of survivin, while YM155 was a survivin suppressant.36 During plasmid transfection, HeLa cells were co-incubated with docetaxel or YM155 of a given dose for 24 h. We observed that red signals from the RNA sensor were brighter in the cells treated using docetaxel but darker in the YM155-treated ones as compared to non-treated cells (Fig. 5A), indicating changed survivin expression in cells after drug treatment. Moreover, a higher dose of the upregulating drug resulted in higher red fluorescence, while a higher dose of the downregulating drug delivered lower red fluorescence. Additionally, the ratiometric images displayed changing colors obviously depending on the doses of docetaxel and YM155 used for HeLa cell treatment. Furthermore, we found the average ratio values in ROIs had a linear relationship with the amount of survivin in the treated cells as determined by qRT-PCR (Fig. 5B and C). These results confirmed the ability of the RNA sensor for quantitative imaging of survivin in living cells.
 | ||
| Fig. 5 (A) Fluorescence and ratiometric images of HeLa cells treated with 100 nM docetaxel (a), 30 nM docetaxel (b), 0 nM (c), 5 nM YM155 (d), and 20 nM YM155 (e). (B) qRT-PCR analysis for the treated HeLa cells. Red: a, blue: b, green: c, black: d, and pink: e. (C) Relationship of average values of the red-to-green fluorescence ratio in ROIs with survivin mRNA concentrations in the treated HeLa cells. | ||
Conclusions
In summary, we have developed a novel genetically encoded RNA sensor strategy based on a single-promoter system that co-expressed an RNA sensor with GFP for quantitative imaging of mRNAs in living tumor cells. We demonstrated that a direct fusion of the SR RNA aptamer with GFP mRNA in an individual transcript using a single promoter distorted the correct folding the RNA aptamer, which failed to show the light-up property in living cells. Splitting of the RNA aptamer from the fusion by incorporating a Dicer substrate site could avoid the misfolding of the RNA aptamer, retaining fluorescence for both the RNA aptamer and the GFP reference. To our knowledge, this is the first time that co-expression of light-up RNA aptamers with fluorescent proteins using a single promoter has been realized in mammalian cells. Moreover, we demonstrated that this single-promoter system enabled highly synchronous co-expression of the RNA aptamer and GFP, affording a desirable platform for ratiometric imaging. This system was successfully applied to ratiometric imaging of survivin mRNA in tumor cells. The results revealed that the ratiometric images were dynamically correlated with survivin mRNA concentrations. Moreover, the values of the red-to green fluorescence ratio in different cell lines and drug-treated HeLa cells exhibited a linear relationship with the concentration of survivin as determined by qRT-PCR. Our design may open the possibility for developing a genetically encoded ratiometric imaging platform for quantitative studies of living tumor cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21874040, 21527810, 21575036 and 21521063).Notes and references
- M. Carmo-Fonseca, RNA, 2015, 21, 580–581 CrossRef CAS PubMed.
- M. Baker, Nat. Methods, 2012, 9, 787–790 CrossRef CAS.
- E. Bertrand, P. Chartrand, M. Schaefer, S. M. Shenoy, R. H. Singer and R. M. Long, Mol. Cell, 1998, 2, 437–445 CrossRef CAS PubMed.
- H. Y. Park, H. Lim, Y. J. Yoon, A. Follenzi, C. Nwokafor, M. Lopez-Jones, X. Meng and R. H. Singer, Science, 2014, 343, 422–424 CrossRef CAS PubMed.
- P. J. Santangelo, A. W. Lifland, P. Curt, Y. Sasaki, G. J. Bassell, M. E. Lindquist and J. E. Crowe Jr, Nat. Methods, 2009, 6, 347–351 CrossRef CAS PubMed.
- R. J. Deng, K. X. Zhang, Y. P. Sun, X. J. Ren and J. H. Li, Chem. Sci., 2017, 8, 3668–3675 RSC.
- F. Hövelmann, I. Gaspar, J. Chamiolo, M. Kasper, J. Steffen, A. Ephrussi and O. Seitz, Chem. Sci., 2016, 7, 128–135 RSC.
- N. Li, C. Y. Chang, W. Pan and B. Tang, Angew. Chem., Int. Ed., 2012, 51, 7426–7430 CrossRef CAS PubMed.
- J. Zheng, R. H. Yang, M. L. Shi, C. C. Wu, X. H. Fang, Y. H. Li, J. S. Li and W. H. Tan, Chem. Soc. Rev., 2015, 44, 3036–3055 RSC.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376–1391 CrossRef CAS PubMed.
- T. L. Halo, K. M. McMahon, N. L. Angeloni, Y. Xu, W. Wang, A. B. Chinen, D. Malin, E. Strekalova, V. L. Cryns, C. Cheng, C. A. Mirkin and C. S. Thaxton, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17104–17109 CrossRef CAS PubMed.
- W. J. Song, R. L. Strack and S. R. Jaffrey, Nat. Methods, 2013, 10, 873–875 CrossRef CAS PubMed.
- M. X. You, J. L. Litke and S. R. Jaffrey, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E2756–E2765 CrossRef CAS PubMed.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–644 CrossRef CAS PubMed.
- R. L. Strack, M. D. Disney and S. R. Jaffrey, Nat. Methods, 2013, 10, 1219–1224 CrossRef CAS PubMed.
- C. A. Kellenberger, S. C. Wilson, J. S. Lee and M. C. Hammond, J. Am. Chem. Soc., 2013, 135, 4906–4909 CrossRef CAS PubMed.
- G. S. Filonov, J. D. Moon, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 16299–16308 CrossRef CAS PubMed.
- E. V. Dolgosheina, S. C. Y. Jeng, S. S. S. Panchapakesan, R. Cojocaru, P. S. K. Chen, P. D. Wilson, N. Hawkins, P. A. Wiggins and P. J. Unrau, ACS Chem. Biol., 2014, 9, 2412–2420 CrossRef CAS PubMed.
- J. S. Paige, T. Nguyen-Duc, W. Song and S. R. Jaffrey, Science, 2012, 335, 1194 CrossRef CAS PubMed.
- Y. Su, S. F. Hickey, S. G. L. Keyser and M. C. Hammond, J. Am. Chem. Soc., 2016, 138, 7040–7047 CrossRef CAS PubMed.
- Z. J. Wang, Y. Luo, X. D. Xie, X. J. Hu, H. Y. Song, Y. Zhao, J. Y. Shi, L. H. Wang, G. Glinsky, N. Chen, R. Lal and C. H. Fan, Angew. Chem., Int. Ed., 2018, 130, 984–988 CrossRef.
- M. D. E. Jepsen, S. M. Sparvath, T. B. Nielsen, A. H. Langvad, G. Grossi, K. V. Gothelf and E. S. Andersen, Nat. Commun., 2018, 9, 18 CrossRef PubMed.
- Z. M. Ying, Z. Wu, B. Tu, W. H. Tan and J. H. Jiang, J. Am. Chem. Soc., 2017, 139, 9779–9782 CrossRef CAS PubMed.
- M. Jinek, A. East, A. Cheng, S. Lin, E. B. Ma and J. Doudna, elife, 2013, 2, e00471 CrossRef PubMed.
- T. Maier, M. Güell and L. Serrano, FEBS Lett., 2009, 583, 3966–3973 CrossRef CAS PubMed.
- J. E. Wilusz, C. K. JnBaptiste, L. Y. Lu, C. D. Kuhn, L. T. Joshua and P. A. Sharp, Genes Dev., 2012, 26, 2392–2407 CrossRef CAS.
- G. Hutvágner, J. McLachlan, A. E. Pasquinelli, E. Bálint, T. Tuschl and P. D. Zamore, Science, 2001, 293, 834–838 CrossRef PubMed.
- P. J. Paddison, A. A. Caudy, E. Bernstein, G. J. Hannon and D. S. Conklin, Genes Dev., 2002, 16, 948–958 CrossRef CAS PubMed.
- M. Sunbul and A. Jäschke, Angew. Chem., Int. Ed., 2013, 52, 13401–13404 CrossRef CAS PubMed.
- D. C. Altieri, Oncogene, 2003, 22, 8581–8589 CrossRef CAS PubMed.
- W. Pan, T. Zhang, H. Yang, W. Diao, N. Li and B. Tang, Anal. Chem., 2013, 85, 10581–10588 CrossRef CAS PubMed.
- D. S. Seferos, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477–15479 CrossRef CAS PubMed.
- X. L. Li, Z. L. Zhang, W. Zhao, X. H. Xia, J. J. Xu and H. Y. Chen, Chem. Sci., 2016, 7, 3256–3263 RSC.
- W. Pan, Y. L. Yan, M. M. Wang, H. J. Y. Yang, N. Li and B. Tang, Chem. Commun., 2016, 52, 4569–4572 RSC.
- X. H. Peng, Z. H. Cao, J. T. Xia, G. W. Carlson, M. M. Lewis, W. C. Wood and L. Yang, Cancer Res., 2005, 65, 1909–1917 CrossRef CAS PubMed.
- K. Yamanaka, T. Nakahara, T. Yamauchi, A. Kita, M. Takeuchi, F. Kiyonaga, N. Kaneko and M. Sasamata, Clin. Cancer Res., 2011, 17, 5423–5431 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00458k |
| This journal is © The Royal Society of Chemistry 2019 |