
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTracking the pyrolysis process of a 3-MeOsalophen-ligand based Co2 complex for promoted oxygen evolution reaction†
Bingxin
Pan
a,
Xu
Peng
 *a,
Yifan
Wang
a,
Qi
An
a,
Xu
Zhang
a,
Yuexing
Zhang
a,
Thomas S.
Teets
c and
Ming-Hua
Zeng
*ab
*a,
Yifan
Wang
a,
Qi
An
a,
Xu
Zhang
a,
Yuexing
Zhang
a,
Thomas S.
Teets
c and
Ming-Hua
Zeng
*ab
aHubei Collaborative Innovation Center for Advanced Organic Chemical Materials, Ministry-of-Education Key Laboratory for the Synthesis and Application of Organic Functional Molecules, College of Chemistry & Chemical Engineering, Hubei University, Wuhan, 430062, P. R. China. E-mail: pengxu@hubu.edu.cn; zmh@mailbox.gxnu.edu.cn
bDepartment of Chemistry and Pharmaceutical Sciences, Guangxi Normal University, Key Laboratory for the Chemistry and Molecular Engineering of Medicinal Resources, Guilin, 541004, P. R. China. E-mail: zmh@mailbox.gxnu.edu.cn
cDepartment of Chemistry, University of Houston, 3585 Cullen Boulevard, Room 112, Houston, Texas 77204-5003, USA
First published on 12th March 2019
Abstract
Efficient oxygen evolution reaction catalysts can be prepared via controlled pyrolysis of molecular platforms, and there is still minimal mechanistic understanding of such pyrolysis processes. Here, we introduce a 3-MeOsalophen-ligated cobalt complex as a precursor to obtain a Co-based OER electrocatalyst via controlled pyrolysis under an inert atmosphere. In our case, the unique N, O chelation mode of the 3-MeOsalophen ligand (bis[3-methoxysalicylydene]-1,2 iminophenylenediamine) was used to synthesis a Co2 complex [Co2(3-MeOsalophen)(Cl)3(CH3OH)2]. By regulating the pyrolysis conditions, we successfully obtained a N-doped carbon Co/CoOx core–shell nanostructure. More importantly, TG-MS was first adopted for tracking the decomposition products of the complex in the pyrolysis process, further finding out the evolution mechanism from Co2 to the core–shell nanostructure. As an electrocatalyst for the oxygen evolution reaction, the core–shell Co/CoOx@NC-800 nanostructure achieves an ultralow overpotential of 288 mV at 10 mA cm−2 in 1 M KOH solution. This work offers guiding insight into controlled pyrolysis via TG-MS analysis, using a novel complex precursor for precise regulation of heteroatom-doped (3d) transition metal-based electrocatalysts.
Introduction
Finding an efficient, stable and environmentally friendly material system for electrocatalytic reactions has long been a research target pursued by scientists.1,2 The electrochemical splitting of water offers a promising process for green energy utilization. In this process, the oxygen evolution reaction (OER) half reaction directly determines the overall efficiency of water splitting.3 Although traditional OER catalysts such as precious metal oxides (e.g., RuO2 and IrO2) are used with good catalytic performance, new low-cost catalysts are needed for large-scale implementation.4–7 Of note, cobalt-based inorganic materials have become promising catalysts because of their lower cost, easier synthesis methods, and higher catalytic activity.8–10 Most precursors for Co-based electrocatalysts are cobalt oxides, cobalt nitrides, cobalt chalcogenides, or Co-based MOFs.11–17 Despite the fact that this variety of Co-based precursors bring synergic advantages for OER electrocatalysts, there is still impetus to design new precursors to prepare OER catalysts with superior electrochemical performance.Metal–ligand coordination complexes are attractive as precursors for OER catalysts, with compositions and reactivity that can be precisely controlled.18,19 The concept is similar to the use of MOF precursors, which via simple pyrolysis can convert into the M/MOx@NC core–shell structure with catalytic properties.8,20,21 The chemical state of nitrogen in the core–shell structure can be adjusted by selecting a suitable nitrogen-containing precursor and regulating the pyrolysis temperature, and there is some benefit of precise regulation of heteroatom-doped (3d) transition metal-based catalysts with a core–shell structure.22 With these precedents in mind, we turned our attention to Schiff base metal complexes, an important class of compounds in many applications. Among Schiff base derivatives, salen complexes are widely used in catalytic reactions due to the advantages of the N, O coordination mode and ability to stabilize 3d metal ions. These ligands support well-defined coordination complexes and offer the potential to form materials with precise compositions under reductive pyrolysis.23 However, reports of pyrolysis reactions of salen to prepare electrocatalytic materials are rare, inspiring us to investigate the pyrolysis of a complex supported by a 3-MeOsalophen ligand to access 3d metal-based nanostructures embedded in N-doped carbon networks.
Here, we introduce a simple 3-MeOsalophen-ligand based Co complex as a precursor to obtain a Co-based electrocatalyst via controlled pyrolysis under an inert atmosphere. In our case, we use the unique N, O coordination chelation mode of the 3-MeOsalophen ligand to synthesis Co2 [Co2(3-MeOsalophen)(Cl)3(CH3OH)2]. By regulating the pyrolysis conditions, we successfully obtained an N-doped carbon Co/CoOx core–shell nanostructure. Our work transforms the 3-MeOsalophen ligand outside the Co metal core into N-doped carbon with controlled pyrolysis, and the TG-MS technique is introduced to study the progress of pyrolysis from the Co2 molecular precursor to N-doped carbon Co/CoOx core–shell nanostructures.
Results and discussion
The typical synthesis procedure of the resulting catalysts from the 3-MeOsalophen ligand to N-doped carbon Co/CoOx core–shell nanostructures is illustrated in Scheme 1. Single-crystal X-ray diffraction analysis reveals that [Co2(3-MeOsalophen)(Cl)3(CH3OH)2] crystallizes in the monoclinic space group P21/c (Table S1†). The binuclear complex is bridged by two μ2-phenolate moieties from the salen ligand. The Co(III) ion in the complex is in a six-coordination mode, in a nearly octahedral geometry. Meanwhile, the Co(II) ion is in a five-coordination mode with a nearly trigonal bipyramidal geometry. It should be noted that the observation that the ligand OCH3 substituents do not participate in coordination has been rarely reported before. Co(III) is coordinated with the deprotonated phenol oxygens O1 and O2 (Co(III)–O1 = 1.905 Å, Co(III)–O2 = 1.890 Å), the ligand nitrogen atoms N1 and N2 (Co(III)–N1 = 1.871 Å, Co(III)–N2 = 1.883 Å) and anions Cl1 and Cl2 (Co(III)–Cl1 = 2.276 Å, Co(III)–Cl2 = 2.270 Å), where N1, N2, O1, and O2 are coplanar. Co(II) is coordinated with deprotonated phenol oxygen atoms O1 and O2 (Co(II)–O1 = 2.231 Å, Co(II)–O2 = 2.119 Å), two CH3OH molecules and the anion Cl3 (Co(II)–Cl3 = 2.330 Å) (Fig. S3 and Table S2†). In addition, two N and two O atoms from the 3-MeOsalophen ligand complete the peripheral ligation around the metallic Co core. In the context of pyrolysis (see below), we propose that the (N, N, O, O) coordination mode of the salen prevents cobalt leaching at low temperature and provides control over the pyrolysis process. In addition, we anticipate that the aromatic core of the ligand and the ligand C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds provide the template for the N-doped carbon that surrounds the cobalt following pyrolysis. Based on these features, we initiated TG-MS studies to understand the decomposition and recombination mechanism of the 3-MeOsalophen ligand-based complexes during pyrolysis, with the goal of preparing highly active OER electrocatalysts.
N bonds provide the template for the N-doped carbon that surrounds the cobalt following pyrolysis. Based on these features, we initiated TG-MS studies to understand the decomposition and recombination mechanism of the 3-MeOsalophen ligand-based complexes during pyrolysis, with the goal of preparing highly active OER electrocatalysts.
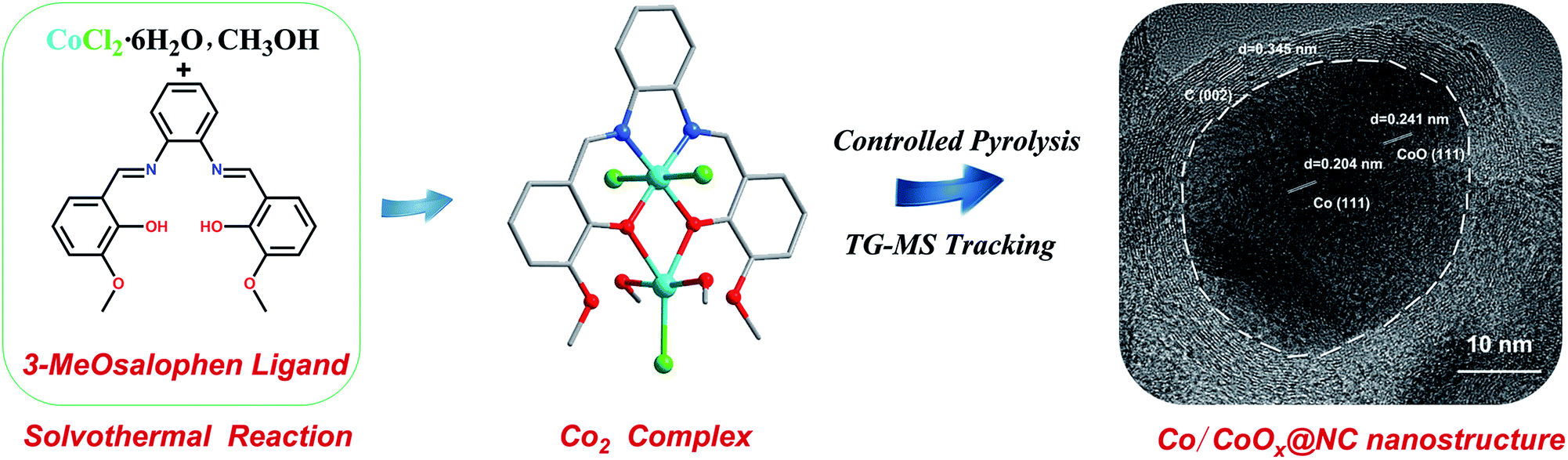 | ||
| Scheme 1 The schematic illustration of the synthesis strategy from the 3-MeOsalophen ligand to Co-based complex and the finally obtained controlled pyrolysis Co/CoOx@NC nanostructures. | ||
The as-synthesized Co2 was pyrolysed under Ar-annealing conditions to prepare the hybrid porous Co/CoOx@NC-T (T represents the pyrolysis temperature). Of note, pyrolysis temperature plays a key role in determining the structures of the resulting samples, thus leading to different electrocatalytic activities, so we analyzed the sample composition obtained at different pyrolysis temperatures by powder X-ray diffraction (PXRD) and high-resolution transmission electron microscopy (HRTEM) (Fig. 1). The X-ray diffraction (XRD) patterns of the Co/CoOx@NCs are shown in Fig. 1a. It can be found that Co/CoOx@NC-500 exists only in the form of elemental cobalt, and the peaks at 44.2°, 51.6° and 75.9° can be assigned to the (111), (200) and (220) facets of structured metallic cubic Co (JCPDS 15-0806). Meanwhile, the peaks at 41.5° and 47.4° belong to the (100) and (101) facets of hexagonal Co (JCPDS 05-0727). When the pyrolysis temperature was increased from 500 °C to 600 °C, the diffraction peak of CoO first appeared, corresponding to the diffraction peak at 36.5°, 42.4° and 61.5°. It should be noted that the peak of hexagonal Co disappears, and this fact suggests the transformation of small particles of hexagonal cobalt into cubic cobalt, and the agglomeration grows into cubic cobalt due to the increase in temperature. Furthermore, the peaks at 31.3°, 59.4° and 65.2° can be assigned to Co3O4 when the temperature increases further to 700 °C.24,25 Interestingly, when the pyrolysis temperature increases to 800 °C, we hardly find the diffraction peaks of Co3O4, which may be due to the reduction gases produced during the pyrolysis process, which would be systematically analyzed as described in Fig. 3. Of note, the stronger diffraction peak of Co3O4 appears again in Co/CoOx@NC-900. The specific phase changes and facet information of Co/CoOx@NCs are summarized in Table S3.† In addition, carbon peaks were negligible in all the XRD patterns of Co/CoOx@NCs, presumably because of their relatively weak intensities. To further confirm its phase composition, the HRTEM images of Co/CoOx@NC-800 were obtained and are shown in Fig. 1b, demonstrating that the nanoparticle is encapsulated in a carbon layer, where the Co (111) crystal plane and CoO (111) are clearly seen with a lattice spacing of 0.204 nm and 0.241 nm, respectively, consistent with the XRD results of Co/CoOx@NC-800. Meanwhile, the 0.345 nm spacing on the carbon layer can be attributed to the (002) plane of the graphitic carbon. The carbon layer provides good protection for the nanoparticles and improves the catalytic stability.26–28 Furthermore, high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) imaging and elemental mapping were also conducted, in which we can find that metallic cobalt is located in the center of the core–shell structure, while C and N elements are uniformly distributed around, as shown in Fig. 1c and d.29,30 In addition, the distribution of Co and O indicates that Co atoms may be bonded with O to form amorphous CoOx parts which are favorable for electrocatalytic activity.31 Therefore, the aforementioned results clearly demonstrate that the Co/CoOx@NC-800 sample was successfully prepared. The TEM images of other Co/CoOx@NCs are shown in Fig. S4,† and the particle size increases with increasing the pyrolysis temperature (600–900 °C).
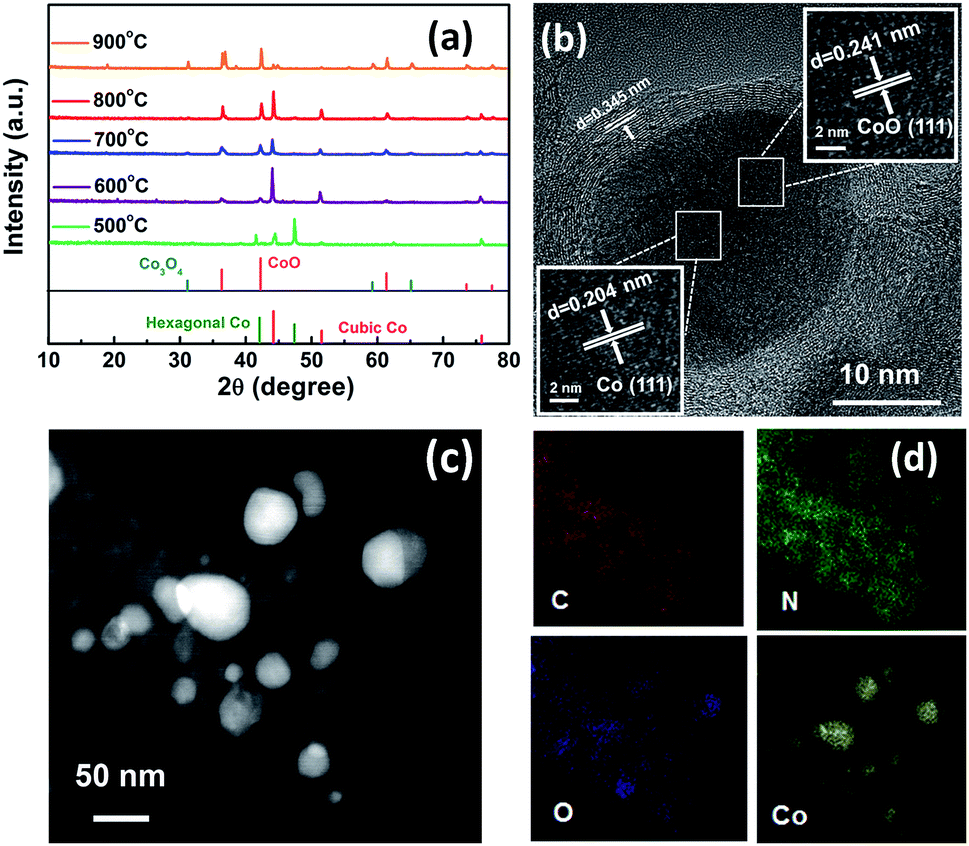 | ||
| Fig. 1 Structural information of the Co/CoOx@NC nanostructures. (a) Powder XRD patterns of Co/CoOx@NCs synthesized at 500–900 °C; (b) HRTEM and (c) HAADF images of Co/CoOx@NC-800; (d) elemental mapping images of Co/CoOx@NC-800. | ||
To further explore the chemical bonding environment of Co/CoOx@NC nanostructures, X-ray photoelectron spectroscopy (XPS) is effective in determining the chemical bonding properties of N, C, O and Co species in catalysts, as shown in Fig. 2a–d. The specific element details are listed in Table S4,† and XPS data for other Co/CoOx@NCs are shown in Fig. S5, S6, S8 and S9.† The XPS survey spectrum in Fig. 2a shows main peaks at 284.6, 398.4, 531.1, and 777.9 eV, which could be assigned to C 1s, N 1s, O 1s, and Co 2p, respectively.30 The deconvoluted high-resolution C 1s spectrum exhibited three bands, and the bands were related to the sp2 C (284.6 eV), C–N or C–O (285.6 eV), and CN or CO (287.5 eV), respectively. We observed asymmetric signals at 284.3 eV and peaks related to C–N and C–O, which confirmed the successful bonding of N and O to the carbon structure. When the pyrolysis temperature was increased, there was no significant change in the percentages of the three bands of the material. This suggests that the composition of the surrounding carbon is basically stable (Fig. S5, S7 and Table S5†).32Fig. 2b shows the Co 2p spectra of Co@CoOx/NC-800, which indicate the simultaneous presence of Co0 (778.6 eV), Co2+ (782.0 eV) and Co3+ (780.1 eV), which has a synergistic effect on the electrocatalytic processes.33,34 Furthermore, we find that Co2+ is dominant compared with Co3+ in Co/CoOx@NC-800 (Fig. 2c), which may be due to the reductive gases produced during the pyrolysis process, and this is consistent with the data in Fig. 3 and S8.† In addition, the O 1s spectra also confirmed the coexistence of oxygen defects (531.6 eV), C–O (533.5 eV) and surface oxidation of the metallic cobalt at 529.8 eV, respectively (Fig. S6†), indicating that there are abundant O vacancies in the Co/CoOx@NC-800 nanostructures.35,36 When the pyrolysis temperature reaches 700 °C and 800 °C, the percentage of Co–O in the materials significantly decreased (Fig. S7 and Table S6†). Meanwhile, the N 1s spectrum of Co/CoOx@NC-800 was also collected (Fig. 2d). Four main peaks associated with pyridinic-N (398.5 eV), pyrrolic-N (400.0 eV), Co–Nx (399.1 eV) and graphitic-N (401.3 eV) species were observed (Fig. S9 and Table S7†).32 The Co content in Co/CoOx@NC-600, Co/CoOx@NC-700, Co/CoOx@NC-800 and Co/CoOx@NC-900 was determined by inductively coupled plasma optical emission spectroscopy and found to be 35.75, 50.42, 40.49 and 68.42 wt%, respectively (Table S8†). These results demonstrate the formation of Co/CoOx@NC core–shell nanostructures.25
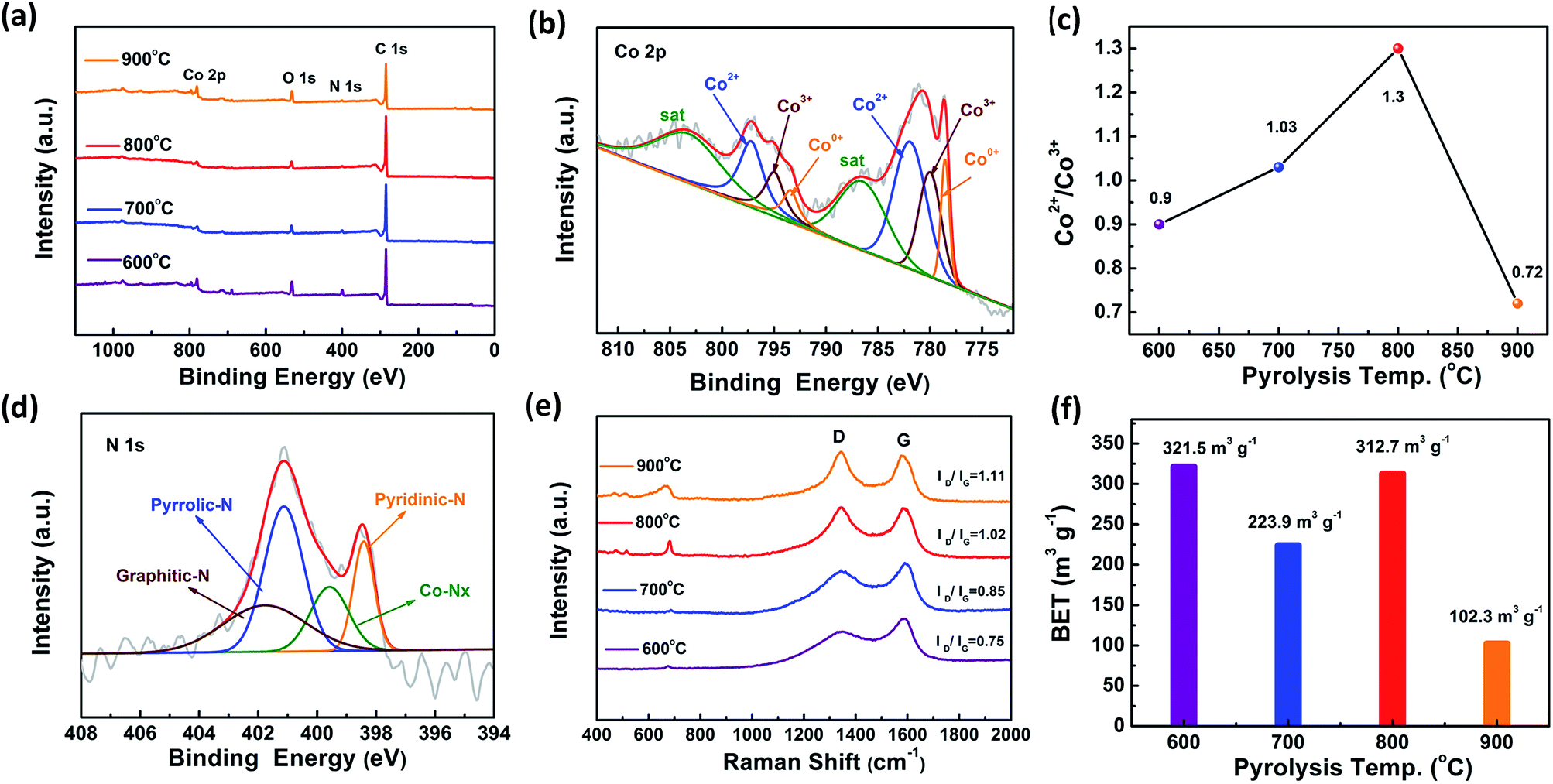 | ||
| Fig. 2 The chemical bonding environment of Co/CoOx@NC nanostructures. (a) XPS survey spectra. (b) XPS survey spectrum of Co/CoOx@NC-800 for Co 2p. (c) XPS analysis-based Co2+/Co3+ atomic ratios of Co/CoOx@NCs. (d) XPS survey spectrum of Co/CoOx@NC-800 for N 1s. (e) Raman spectroscopy of Co/CoOx@NCs. (f) Brunauer–Emmett–Teller (BET) values of Co/CoOx@NCs. | ||
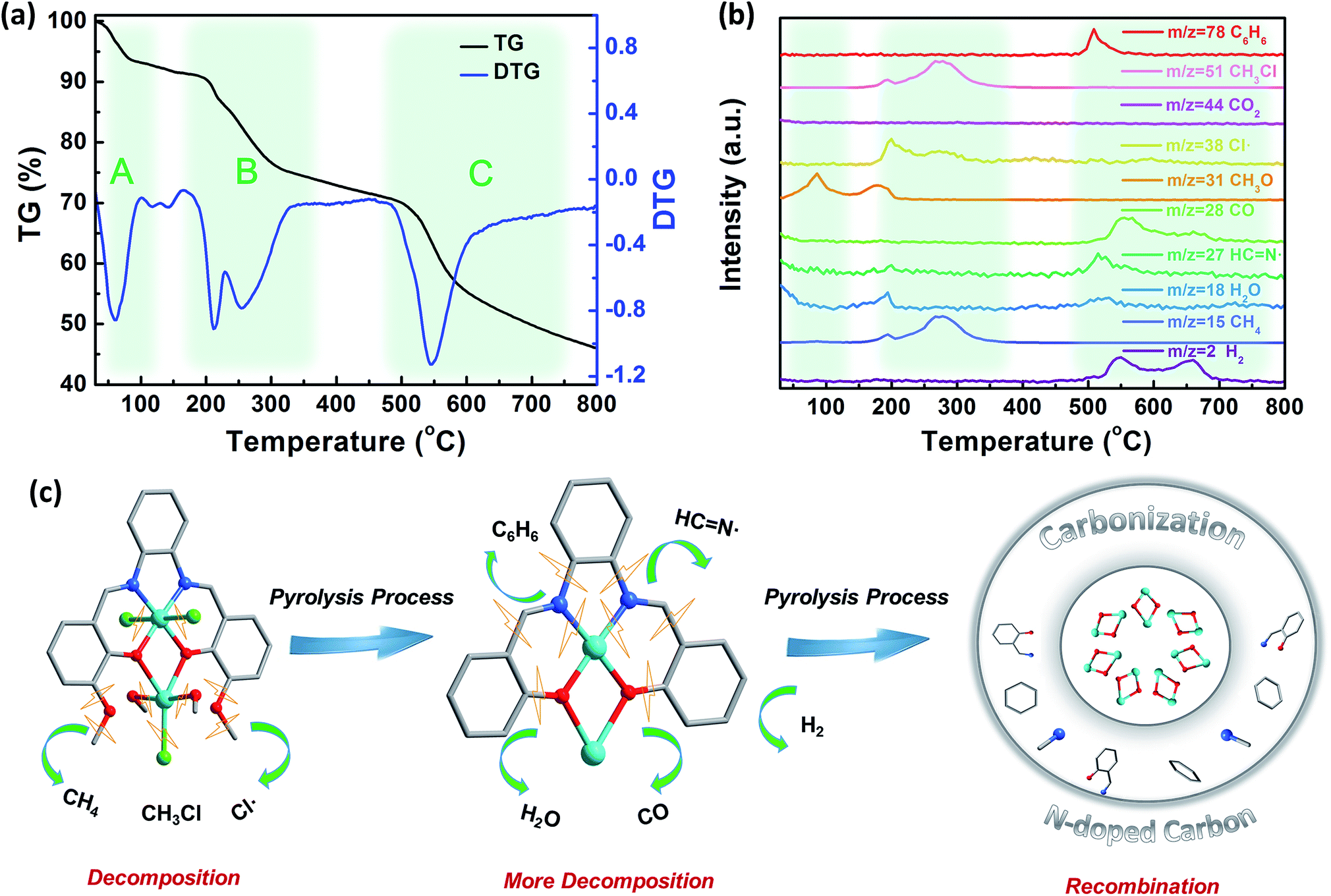 | ||
| Fig. 3 Controlled pyrolysis tracking analysis. (a) TG and DTG profiles of Co2. (b) TG-MS curves of the corresponding Co2 pyrolysis tracking process. (c) Possible decomposition and polymerization reaction occurring in the pyrolysis process from Co2 to Co/CoOx@NCs. | ||
Raman spectroscopy was further used to characterize the degree of graphitization of the Co/CoOx@NCs (Fig. 2e). In the spectra, two sharp Raman peaks were observed at 1354 and 1584 cm−1 which are characteristic of carbon materials and can be ascribed to the disordered structures/structural defects (or D band) and the graphitic G band, respectively. The intensity ratio of the D band to G band (ID/IG) increases from 0.75 to 1.11 for Co/CoOx@NC-600–900, indicating that the resulting defects increase at higher pyrolysis temperature. In addition, peaks at 470.6, 515.6 and 680.9 cm−1 are classical vibration modes of Co–O.37,38 Furthermore, the surface area along with pore size distribution of the Co/CoOx@NCs was evaluated. As shown in Fig. 2f, it was found that the BET surface area was 321.5 m2 g−1 for Co/CoOx@NC-600, 223.9 m2 g−1 for Co/CoOx@NC-700, 312.7 m2 g−1 for Co/CoOx@NC-800 and 102.319 m2 g−1 for Co/CoOx@NC-900. Usually, the BET surface areas of complexes that undergo the pyrolysis process would gradually decrease as pyrolysis temperature increases,39 yet the Co/CoOx@NC-800 sample is nearly similar to Co/CoOx@NC-600, which may be due to the reduction gases produced during the pyrolysis process, as described further in Fig. 3. Meanwhile, the presence of mesopores and the pore size distribution determined by the QSDFT method are displayed in Fig. S10 and S11,† respectively. Therefore, the chemical bonding environment of Co, O and C was systematically analyzed by XPS, Raman spectroscopy and BET surface area measurement, showing that the Co/CoOx@NC-800 sample has higher Co2+/Co3+ and a reasonable ID/IG ratio as well as a relatively higher surface area compared with other Co/CoOx@NCs.
It is essential to trace the decomposition products in the pyrolysis process, and further to find out the mechanism of the pyrolysis process and the evolution of the core–shell structure. Hence, thermogravimetric analysis combined with mass spectrometry (TG-MS) was used to track the pyrolysis process. As shown in Fig. 3a, the TG and DTG curves of Co2 can be divided into three areas from 30–800 °C. Generally, the mass decrease between 30 °C and 100 °C is attributed to the loss of CH3OH or H2O guest molecules (namely region A); moreover, the decrease between 150–350 °C is largely due to the partial decomposition of the ligand (namely region B); and finally between 480 °C and 800 °C we observe complete decomposition of Co2 to form the core–shell structure of Co/CoOx@NC (namely region C).40–42
According to the structure of Co2 and the fragments detected by TG-MS as shown in Fig. 3b, the whole pyrolysis process can be divided into several processes: (i) the process from room temperature to 100 °C only detects fragments of CH3OH, where the escape of guest molecules occurs. As the temperature increases from 100 °C to 200 °C, CH3OH is detected again, assigned to the CH3OH molecules coordinated to Co which escape the coordination sphere of the Co2 complex. (ii) When the temperature is raised from 150 °C to 300 °C, two peaks appear in the temperature region as seen from the DTG curve. In this temperature range, several fragments are detected by TG-MS. Characteristic fragments include CH4 (m/z = 15), Cl˙ (m/z = 38), and CH3Cl (m/z = 52). Of note, CH4 can be attributed to the ligand centered ether bond cleavage. In addition, the chlorine atom on Co can be combined with a methyl group on a similar ligand to form CH3Cl. And the generation of CH3Cl is further increased when the temperature is increased. It may be related to the increase of CH4 and the escape of more chlorine atoms, and the extra Cl atoms escape as free radicals (m/z = 38). The loss of coordinated methanol and chloride ligands will lead to Co becoming an unusually active center, which may facilitate the aggregation of cobalt atoms and the association with the partially decomposed fragments to form a stable core–shell nanostructure. (iii) Further increase in temperature leads to complete rupture of the whole Co2 complex, resulting in C6H6 (m/z = 78) fragment formation and ether C–O, aromatic C–C, and C–N bond cleavage processes, producing an active site that promotes ligand polymerization and release of H2.43 On the other hand, HCN˙ formed by the simultaneous cleavage of the Co–N coordination bond and C6H6 in this process participates in the formation of a carbon network, and finally forms N-doped carbon. Prominently, while the ligand undergoes decomposition and polymerization, the metal oxide formed by Co and phenol oxygen is also reduced to some extent by adjacent small molecule fragments such as H2 and CH4 to form metallic Co with H2O and CO escaping. Since H2 is generated in this process, no CO2 (m/z = 44) is detected. Similarly, we tracked the pyrolysis process of the 3-MeOsalophen ligand (Fig. S12 and S13†). It is found that the decomposition of the ligand produces more fragments than that of Co2, and a little sample remained after pyrolysis (2.32%), suggesting that the Co active site produced by the decomposition of Co2 may accelerate the recombination of organic fragments, and then rapidly forms a stable carbon network, which is consistent with the aforementioned phenomena. In addition, H2 was not detected in the process of ligand decomposition, suggesting that the Co active site is crucial for the production of H2. To summarize, the whole pyrolysis process from Co2 to Co/CoOx@NC nanostructures is depicted in Fig. 3c, indicating that the pyrolysis process of Co2 occurs in a reductive atmosphere between 520–700 °C, accounting for the distinct nanostructure of Co/CoOx@NC-800 formed via this process.
The catalytic ability of Co/CoOx@NCs for the oxygen evolution reaction was also assessed in 1 M KOH solution using a typical three-electrode configuration. The loading of catalyst on carbon paper is 1.68 mg cm−2. For comparison, commercial RuO2 was also examined under the same conditions. The Co/CoOx@NCs and commercial RuO2 electrodes were first oxidized in alkaline medium saturated with O2, and then measured at 5 mV s−1 to obtain the polarization curves shown in Fig. 4a. The polarization curves obtained for Co/CoOx@NC-800 show that the material has significant electrocatalytic properties for the OER. The Co/CoOx@NC-800 electrode displayed the smallest overpotential of 288 mV at 10 mA cm−2. In contrast, Co/CoOx@NC-600, Co/CoOx@NC-700, Co/CoOx@NC-900 and commercial RuO2 possessed larger overpotentials of 433 mV, 356 mV, 413 mV and 339 mV, respectively. Meanwhile, the Tafel slope for Co/CoOx@NC-800 is 98 mV dec−1 (Fig. 4b). The results indicated that Co/CoOx@NC-800 exhibits excellent electrocatalytic performance, which is comparable to that of the state-of-the-art Co-based electrocatalysts (Table S9†). To gain further insight into the electrocatalytic activity of the above electrocatalysts, the electrochemical impedance spectra (EIS) and the Cdl (double layer capacitance) of the electrocatalysts were obtained.36,44 As shown in Fig. 4d, the Cdl value of the Co/CoOx@NC-800 electrode is calculated to be 35.04 mF cm−2, which is larger than the 31.72, 26.55, 17.2 mF cm−2 of Co/CoOx@NC-700, Co/CoOx@NC-900 and Co/CoOx@NC-600, respectively (Fig. S14†). This suggests that Co/CoOx@NC-800 has more catalytically active centers in the electrolysis process. In addition, electrochemical impedance spectroscopy (EIS) was conducted to explore the electrode kinetics under OER conditions. In the Nyquist plots (Fig. 4e) of Co/CoOx@NCs, Co/CoOx@NC-800 could be clearly seen to have the smallest semicircle (with the smallest radius) or the smallest charge transfer resistance (Rct, 7.40 Ω) compared with the other electrocatalysts (Rct = 13.61 Ω for Co/CoOx@NC-600, Rct = 11.09 Ω for Co/CoOx@NC-700 and Rct = 12.79 Ω for Co/CoOx@NC-900). The measured impedance spectra data were fitted and evaluated using Zview software. As shown in Fig. 4e, the symbols refer to the experimental data and the lines denote the corresponding fitting results. The inset is the equivalent circuit in the form of Rs (CPE1, Ro) (CPE2, Rct), where Rs represents the solution resistance, CPE1 and CPE2 are the constant phase elements which describe electron transport at the GCE/catalyst interface and the catalyst/electrolyte interface, respectively, Ro is the oxide film resistance and Rct denotes the charge transfer resistance at the catalyst/electrolyte interface.45 This suggested that the electron transfer rate during the OER was faster in Co/CoOx@NC-800. The results of electrochemical impedance spectroscopy (EIS) and electrochemical double-layer capacitance (Cdl) measurements of electrocatalysts confirm the excellent OER performance of Co/CoOx@NC-800 among Co/CoOx@NCs. More importantly, high durability is of great significance for electrocatalysts in practical applications. Chronopotentiometry (CP) was conducted to study the electrocatalytic durability of Co/CoOx@NC-800 for the OER in O2-saturated 1.0 M KOH solutions. As shown in Fig. 4f, Co/CoOx@NC-800 shows negligible potential fluctuation at 50 mA cm−2 for 15 h. Furthermore, the phase and the corresponding peaks of XRD patterns of the Co/CoOx@NC-800 sample before and after the catalysis were compared (Fig. S15†), showing that the main phase peak of the sample is well preserved. At the same time, as confirmed by the TEM image, the core–shell nanostructure morphology is retained after long-term OER operation (inset of Fig. 4f). The above indicates that the Co/CoOx@NC-800 structure from Co2 has excellent stability for the OER in alkaline media. Besides, the influence of pyrolysis temperature on final catalytic performance is discussed in Fig. S4.† In other words, the excellent electrochemical performance of Co/CoOx@NC-800 is attributed to the synergic effects of the designable 3-MeOsalophen-based Co complex as the precursor and controlled pyrolysis conditions, forming such a distinct nanostructure.
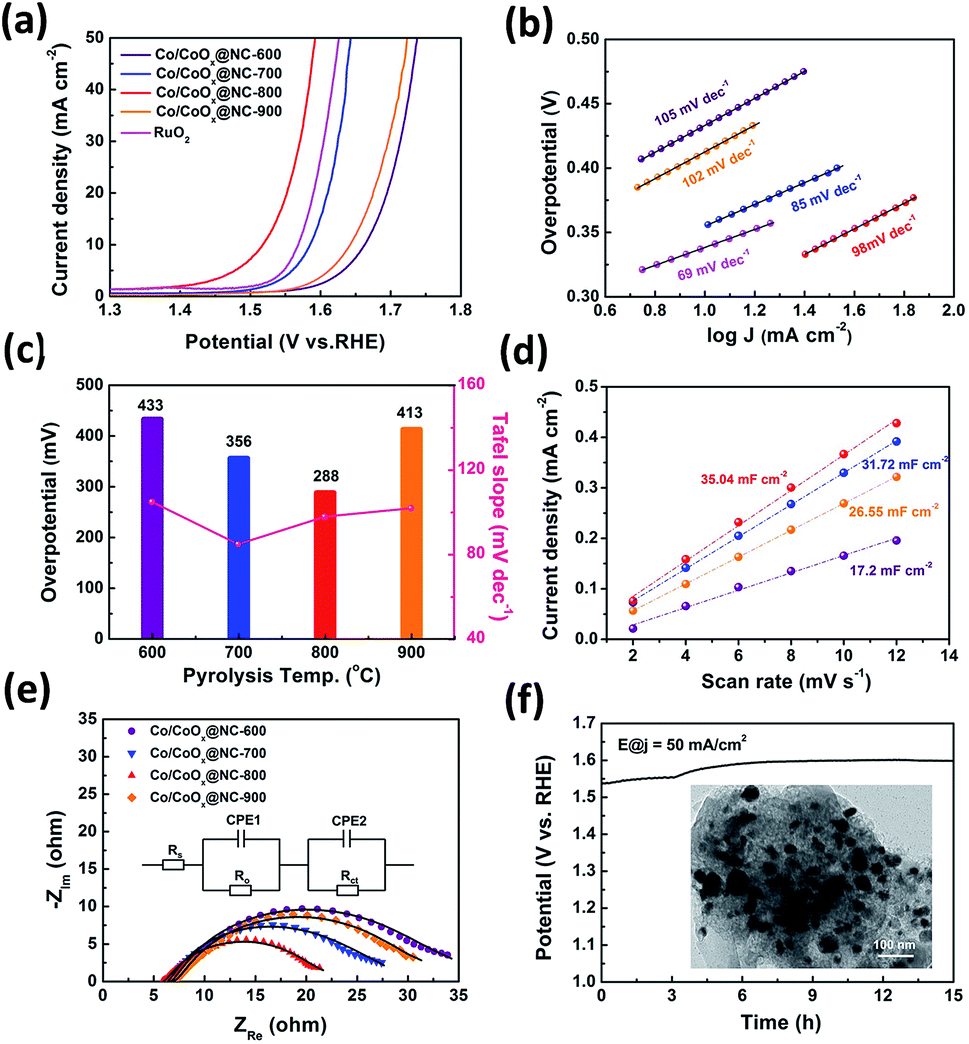 | ||
Fig. 4 OER activity of Co/CoOx@NC nanostructures and commercial RuO2. (a) IR-corrected polarization LSV curves. (b) Corresponding Tafel plots. (c) Comparison of overpotentials at j = 10 mA cm−2 corresponding to Tafel slopes. (d) Current density as a function of the scan rates for Co/CoOx@NCs. (e) Nyquist plots of catalysts at the biased potential of 1.6 V vs. RHE over the frequency range from 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 0.01 Hz. (f) Chronopotentiometric measurements of long-term stability of Co/CoOx@NC-800. The inset shows the TEM image after the OER chronopotentiometric test, scale bar is 100 nm. 000 to 0.01 Hz. (f) Chronopotentiometric measurements of long-term stability of Co/CoOx@NC-800. The inset shows the TEM image after the OER chronopotentiometric test, scale bar is 100 nm. | ||
Conclusions
In summary, we have shown that the 3-MeOsalophen-ligand based Co2 complex undergoes controlled pyrolysis to form distinct core–shell Co/CoOx@NC nanostructures which are efficient OER electrocatalysts. TG-MS analysis reveals the pyrolysis process and indicates that the decomposition of the 3-MeOsalophen-ligated Co2 provides a reductive atmosphere, and the possible decomposition and polymerization mechanism of Co2 was elucidated, which facilitates the formation of core–shell nanostructures. Notably, the core–shell Co/CoOx@NC-800 nanostructure achieves an ultralow overpotential of 288 mV at 10 mA cm−2 in 1 M KOH solution, which is comparable to that of the state-of-the-art Co-based electrocatalysts. This work offers a general route for designing synthetically derived core–shell nanostructures by designing selected complexes and controlling pyrolysis conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Fund for Distinguished Young Scholars (Grant 21525101), the BAGUI Talent and Scholar Program (2014A001), NSFC (Grants 81773775, 21805074 and 21661088), NSFC of Hubei (Grants 2017CFA006, 2018CFB151), and NSFGX (Grants 2014GXNSFFA118003, 2017GXNSFDA198040).Notes and references
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. D. Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- S. L. Zhao, Y. Wang, J. C. Dong, C. T. He, H. J. Yin, P. F. An, K. Zhao, X. F. Zhang, C. Gao, L. J. Zhang, J. W. Lv, J. X. Wang, J. Q. Zhang, A. M. Khattak, N. A. Khan, Z. X. Wei, J. Zhang, S. Q. Liu, H. J. Zhao and Z. Y. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- X. Kong, K. Xu, C. Zhang, J. Dai, S. Norooz Oliaee, L. Li, X. Zeng, C. Wu and Z. Peng, ACS Catal., 2016, 6, 1487–1492 CrossRef CAS.
- C. Hu and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 11736–11739 CrossRef CAS PubMed.
- C. McCrory, S. Jung, J. Peters and T. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- E. Hu, Y. Feng, J. Nai, D. Zhao, Y. Hu and X. W. Lou, Energy Environ. Sci., 2018, 11, 872–880 RSC.
- S. Dou, C. L. Dong, Z. Hu, Y. C. Huang, J. l. Chen, L. Tao, D. Yan, D. Chen, S. Shen, S. Chou and S. Wang, Adv. Funct. Mater., 2017, 27, 1702546 CrossRef.
- K. Yan, J. Chi, J. Xie, B. Dong, Z. Liu, W. Gao, J. Lin, Y. Chai and C. Liu, Renewable Energy, 2018, 119, 54–61 CrossRef CAS.
- K. Yan, X. Shang, Z. Li, B. Dong, X. Li, W. Gao, J. Chi, Y. Chai and C. Liu, Appl. Surf. Sci., 2017, 416, 371–378 CrossRef CAS.
- Y. Tong, P. Chen, T. Zhou, K. Xu, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 7121–7125 CrossRef CAS PubMed.
- P. Chen, K. Xu, Z. Fang, Y. Tong, J. Wu, X. Lu, X. Peng, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 14710–14714 CrossRef CAS PubMed.
- M. Zhang, Q. Dai, H. Zheng, M. Chen and L. Dai, Adv. Mater., 2018, 30, 1705431 CrossRef PubMed.
- Y. Lu, L. Yu, M. Wu, Y. Wang and X. W. Lou, Adv. Mater., 2018, 30, 1702875 CrossRef PubMed.
- X. Wang, L. Dong, M. Qiao, Y. Tang, J. Liu, Y. Li, S. Li, J. Su and Y. Lan, Angew. Chem., Int. Ed., 2018, 57, 9660–9664 CrossRef CAS PubMed.
- K. Yan, J. Chi, Z. Liu, B. Dong, S. Lu, X. Shang, W. Gao, J. Chi, Y. Chai and C. Liu, Inorg. Chem. Front., 2017, 4, 1783–1790 RSC.
- K. Yan, J. Qin, J. Lin, B. Dong, J. Chi, Z. Li, F. Dai, Y. Chai and C. Liu, J. Mater. Chem. A, 2018, 6, 5678–5686 RSC.
- M. H. Zeng, Z. Yin, Z. H. Liu, H. B. Xu, Y. C. Feng, Y. Q. Hu, L. X. Chang, Y. X. Zhang, J. Huang and M. Kurmoo, Angew. Chem., Int. Ed., 2016, 55, 11407–11411 CrossRef CAS PubMed.
- J. Wang, L. Gan, W. Zhang, Y. Peng, H. Yu, Q. Yan, X. Xia and X. Wang, Sci. Adv., 2018, 4, eaap7970 CrossRef PubMed.
- L. Jiao and H. L. Jiang, Chem, 2019 DOI:10.1016/j.chempr.2018.12.011.
- H. Zhang, J. Nai, L. Yu and X. W. Lou, Joule, 2017, 1, 77–107 CrossRef CAS.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- C. Baleizao and H. Garcia, Chem. Rev., 2006, 106, 3987–4043 CrossRef CAS PubMed.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef CAS PubMed.
- A. Aijaz, J. Masa, C. Rçsler, W. Xia, P. Weide, A. J. R. Botz, R. A. Fischer, W. Schuhmann and M. Muhler, Angew. Chem., Int. Ed., 2016, 55, 4087–4091 CrossRef CAS PubMed.
- G. Xu, G. C. Xu, J. J. Ban, L. Zhang, H. Lin, C. L. Qi, Z. P. Sun and D. Z. Jia, J. Colloid Interface Sci., 2018, 521, 141–149 CrossRef CAS PubMed.
- Y. Z. Chen, C. Wang, Z. Yu. Wu, Y. Xiong, Q. Xu, S. H. Yu and H. L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS PubMed.
- X. Zhang, R. Liu, Y. Zang, G. Liu, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Chem. Commun., 2016, 52, 5946–5949 RSC.
- S. Li, C. Cheng, X. Zhao, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2018, 57, 1856–1862 CrossRef CAS PubMed.
- K. Xu, H. Cheng, L. Liu, H. Lv, X. Wu, C. Wu and Y. Xie, Nano Lett., 2016, 17, 578–583 CrossRef PubMed.
- S. Huang, Y. Meng, S. He, A. Goswami, Q. Wu, J. Li, S. Tong, T. Asefa and M. Wu, Adv. Funct. Mater., 2017, 27, 1606585 CrossRef.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., Int. Ed., 2016, 128, 2270–2274 CrossRef.
- X. Yan, Y. Jia, J. Chen, Z. Zhu and X. Yao, Adv. Mater., 2016, 28, 8771–8778 CrossRef CAS PubMed.
- B. Chen, X. He, F. Yin, H. Wang, D. J. Liu, R. Shi, J. Chen and H. Yin, Adv. Funct. Mater., 2017, 27, 1700795 CrossRef.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- H. Xu, Z. X. Shi, Y. X. Tong and G. R. Li, Adv. Mater., 2018, 30, 1705442 CrossRef PubMed.
- S. H. Johnson, C. L. Johnson, S. J. May, S. Hirsch, M. W. Cole and J. E. Spanier, J. Mater. Chem., 2010, 20, 439–443 RSC.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- Y. Ding, Y. Niu, J. Yang, L. Ma, J. Liu, Y. Xiong and H. Xu, Small, 2016, 12, 5414–5421 CrossRef CAS PubMed.
- R. A. Schäfer, D. Dasler, U. Mundloch, F. Hauke and A. Hirsch, J. Am. Chem. Soc., 2016, 138, 1647–1652 CrossRef PubMed.
- R. Risoluti, M. A. Fabiano, G. Gullifa, L. W. Wo and S. Materazzi, Russ. J. Gen. Chem., 2017, 87, 564–568 CrossRef CAS.
- T. K. Kim, K. J. Lee, J. Y. Cheon, J. H. Lee, S. H. Joo and H. R. Moon, J. Am. Chem. Soc., 2013, 135, 8940–8946 CrossRef CAS PubMed.
- L. Zhang and Y. H. Hu, J. Phys. Chem. C, 2010, 114, 2566–2572 CrossRef CAS.
- T. Tang, W. J. Jiang, S. Niu, N. Liu, H. Luo, Y. Y. Chen, S. F. Jin, F. Gao, L. J. Wan and J. S. Hu, J. Am. Chem. Soc., 2017, 139, 8320–8328 CrossRef CAS PubMed.
- J. Zhang, F. Li, W. Chen, C. Wang and D. Cai, Electrochim. Acta, 2019, 300, 123–130 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1882936. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00505f |
| This journal is © The Royal Society of Chemistry 2019 |