
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRenewing accessible heptazine chemistry: 2,5,8-tris(3,5-diethyl-pyrazolyl)-heptazine, a new highly soluble heptazine derivative with exchangeable groups, and examples of newly derived heptazines and their physical chemistry†
Laurent
Galmiche
a,
Clémence
Allain
a,
Tuan
Le
a,
Régis
Guillot
 b and
Pierre
Audebert
*a
b and
Pierre
Audebert
*a
aPPSM, ENS Cachan, CNRS, Université Paris-Saclay, 94235 Cachan, France. E-mail: pierre.audebert@ens-paris-saclay.fr
bICMMO, Université Paris Sud, CNRS, Université Paris-Saclay, 91405 Orsay, France
First published on 25th April 2019
Abstract
We have prepared 2,5,8-tris(3,5-diethyl-pyrazolyl)-heptazine, the first highly soluble heptazine derivative possessing easily exchangeable leaving groups. We present its original synthesis employing mechanochemistry, along with a few examples of its versatile reactivity. It is, in particular, demonstrated that the pyrazolyl leaving groups can be replaced by several secondary or primary amino substituents or by three aryl- or benzyl-thiol substituents. In addition to being a synthetic platform, 2,5,8-tris(3,5-diethyl-pyrazolyl)-heptazine is fluorescent and electroactive, and its attractive properties, as well as those of the derived heptazines, are briefly presented.
Introduction
Heptazines (Fig. 1) are a fascinating family of high nitrogen aromatic compounds that have already aroused the special interest of Linus Pauling 80 years ago.1 Indeed, there are still few heptazines fully described in the literature,2 but those known to date already present desirable properties.3–5 They have a high nitrogen to carbon ratio, making them strongly electron-deficient rings. Because of this reason, heptazines can be reversibly reduced at relatively high potentials,6 which makes them interesting compounds in view of applications for photovoltaic devices. They are also fluorescent,7 and heptazines displaying delayed fluorescence have been described.8,9 Most of them display a very high thermal stability,2 which is highly desirable for the development of light-emitting or photovoltaic devices. A recent review on their synthesis highlights the interest in these molecules.10 | ||
| Fig. 1 General chemical structure of heptazines. | ||
Finally, even more attractively, heptazine polymers have demonstrated outstanding photocatalytic properties11–13 for hydrogen evolution14,15 and for CO2 reduction16 and might be one of the rare molecular platforms which could give rise to molecular catalysts able to perform water splitting. They now constitute an extremely interesting class of materials.17,18
However, a major drawback in heptazine synthetic chemistry lies in the fact that almost all derivatives that have already been reported are prepared starting from the only heptazine known to date to have exchangeable side groups, 2,5,8-trichloro-heptazine, (sometimes designated as cyameluric chloride). The Kroke group and other groups have indeed shown that the chlorides could be efficiently replaced by various nucleophiles, like amines,6,7 thiols,19 selenols19 and phosphines.20 This very useful synthon has, however, two major drawbacks. (1) It is only sparingly soluble in most common solvents and (2) more challenging, the original synthetic route reported by Kroke21 requires heating the intimate solid mixture of the tris potassium salt of cyameluric acid and phosphorus pentachloride, with cautious multi-step extraction of the gaseous hydrochloric acid formed (along with traces of chlorine gas), followed by sublimation or Soxhlet extraction with dry toluene; variants of this synthesis are slightly better but do not really solve the problem.15,22 This synthetic route is delicate and dangerous to some extent, which probably explains the relatively low number of publications on heptazines except for the discovery (again by Kroke) that amino groups could be exchanged for hydrazino groups,23 which can be converted to azide, and then iminophosphorane groups.23 However, again 2,5,8-triazidoheptazine is an unstable compound. We felt that 2,5,8-trihydrazinoheptazine would be a better starting point than cyameluric acid salts, and we decided to convert it into another derivative bearing easily exchangeable pyrazolyl groups.
Results and discussion
Synthesis
We describe in this article the facile synthesis of a heptazine derivative possessing exchangeable diethylpyrazolyl leaving groups (Scheme 1), along with several examples of new heptazines derived by nucleophilic exchange of these groups, to illustrate the versatility of this new synthetic pathway.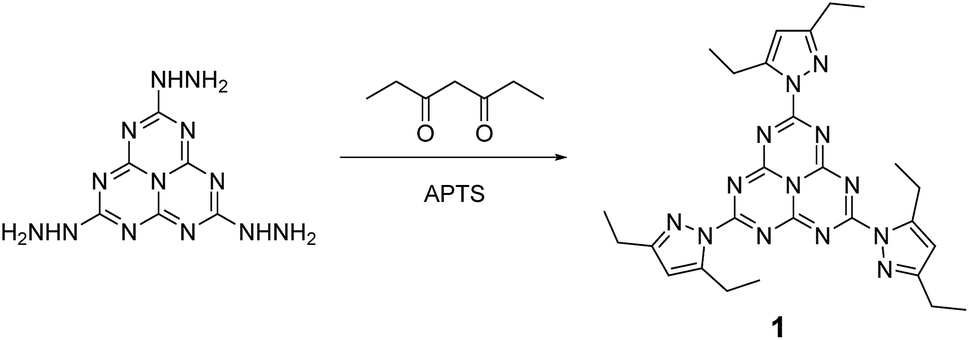 | ||
| Scheme 1 Synthesis of 2,5,8-tris(3,5-diethyl-pyrazolyl)-heptazine 1 bearing three pyrazolyl leaving groups. | ||
As previously mentioned, we preferred trihydrazinoheptazine to the weakly reactive potassium salt of cyameluric acid, as a starting point for new heptazines. We were able to prepare it according to Kroke's method.23 After careful drying in a desiccator in the presence of phosphoric anhydride, the trihydrazinoheptazine is processed without further purification. We found out that, although elegant, the purification method published by Kroke et al. was tedious (Caution: Kroke's procedure uses hydrazine hydrate, a compound which is stable but toxic. We recommend the use of a cautiously tightly sealed Teflon-lined pressure reaction vessel inserted into a very resistant shielded stainless steel fitted mantle).
Therefore, we found it more convenient to use the carefully dried crude trihydrazinoheptazine. Since mechanochemistry has been recently demonstrated to improve the facileness and the yields in organic synthesis,24–27 especially in heterocyclic synthesis,28,29 we developed a mechanochemical route for preparing 1. The trihydrazinoheptazine is reacted with 3,5-heptanedione to provide the desired 2,5,8-tris(3,5-diethyl-pyrazolyl)-heptazine 1 using mechanochemistry (see Synthesis details in the ESI section†). As a quick summary, the crude trihydrazinoheptazine is placed with 50% excess diketone in an 80 mL agate mixer with fifteen 1 cm diameter agate balls, and crushed for 8 min (2 times, 4 min) at 500 rpm. The resulting paste is extracted three times with 30 mL dichloromethane and subjected to chromatography. Although the yields are average, the work-up and purification processes are extremely simple and scalable, and allow routine preparation of 1–2 g batches of the compound, after simple flash chromatography.
We have investigated to some extent the influence of a few (empirical) parameters for mechanical milling (Table 1), which leads to 3 tendencies. (1) A time of 8 min is sufficient to reach the appropriate yield; increasing the milling time to 16 min does not lead to better efficiency (entry 4). However, we did not try shorter times. (2) Addition of silica30 slightly improves the yields, though it is not obvious if this is a mechanical effect, or if it works as a co-catalyst (entry 5). (3) It is better if the product load is not in excess (e.g. over 1.5–2 g, for an 80 mL milling bowl). Otherwise the yields progressively decrease (entry 3). We checked that the reaction can be scaled-up using a 250 mL milling bowl (entry 6) and probably further.
We have been able to obtain an X-ray structure of heptazine 1 (Fig. 2). Details of refinement data and crystal packing are provided in the ESI section.† In the crystal, the heptazine core is almost coplanar with the pyrazole substituents. The molecules are organized in shifted columnar packing, with stacking interactions between pyrazole substituents.
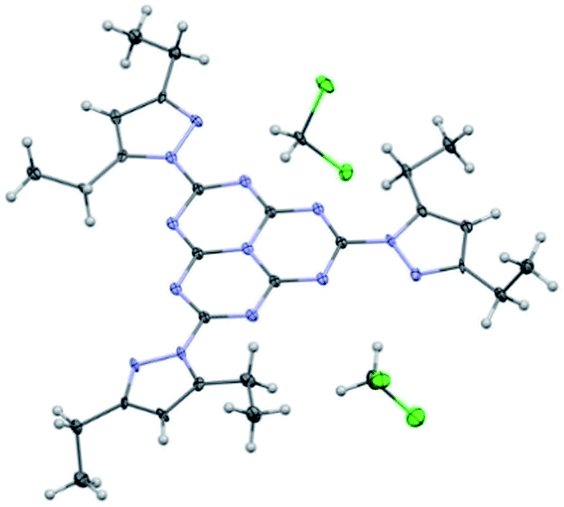 | ||
| Fig. 2 ORTEP diagram of 2,5,8-tris(3,5-diethyl-pyrazolyl)-heptazine 1, showing 30% probability ellipsoids. N atoms are depicted in blue and Cl atoms (from the DCM molecules co-crystallized with heptazine 1) in green. | ||
We have tested the facileness of exchanging the pyrazolyl groups on heptazine 1. Actually, we found out that exchange with mild nucleophiles like amines and thiols was straightforward. Concerning amines, cyclic amines such as morpholine, piperidine and pyrrolidine or primary amines such as 2-ethylhexylamine can be used as nucleophiles to give the corresponding tris-substituted heptazines 2a–d in good yields after three hours at 60 °C or under gentle reflux and simple recrystallization for purification. For the substitution with thiols, adding a non-nucleophilic base in the reaction mixture, such as 2,4,6-trimethylpyridine, is necessary. Under reflux in acetonitrile, the reaction proceeds to completion using benzylthiol as a nucleophile (compound 3c), while with arylthiols (compounds 3a–b) partial substitution products are also observed but can be separated by flash chromatography. Thus, the reactivity of heptazine 1 appears to be similar to that of 3,6-bis(3,5-dimethyl-pyrazolyl)-tetrazine, a major synthon of tetrazine chemistry.31Scheme 2 summarizes the reaction sequence.
 | ||
| Scheme 2 Synthetic pathways for the new heptazines prepared. | ||
It should, however, be noted that pyrazolyl groups in 1 do not exactly mimic the reactivity of chlorine in cyameluric chloride, while the substitution chemistry of 1 is quite similar (and possibly wider); on the other hand electrophilic activated substitution, like with cyameluric trichloride, is not possible, a feature which distinguishes the reactivity of these two remarkable compounds.
Electrochemical studies
We have briefly investigated the electrochemical and photophysical properties of the compounds prepared. All heptazines are electroactive, and can be reduced almost reversibly to their anion-radical at a relatively high potential, which stems from their high nitrogen content (Table 2). As an example, the CV of 2,5,8-tris(3,5-diethyl-pyrazolyl)-heptazine is represented in Fig. 3. It shows the almost reversible first reduction of this compound around −1.3 V (Ag/AgCl ref.) A second system likely corresponding to the dianion can be observed at around – 1.9 V but it is completely irreversible under our conditions (DCM). Other heptazines all display a reduction peak, sometimes weakly reversible (thiol-substituted) or irreversible; this is in accordance with the reports of L. Dubois for related compounds.6| Compound | λ abs max (nm) | ε, M−1 cm−1 | λ em max (nm) | Stokes shift (cm−1) | Φ f | E° (vs. SCE) (V) |
|---|---|---|---|---|---|---|
| a Reference: quinine sulphate in 0.5 N H2SO4; sh = shoulder. | ||||||
| 1 | 302(sh), 314, 384(sh) | 83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
459 | 4300 | 0.15 | −1.33 |
| 2a | 251(sh), 273, 315(sh) | 92500 |
386 | 5800 | 0.15 | — |
| 2b | 249(sh), 272, 310(sh) | 92100 |
384 | 6200 | 0.10 | — |
| 2c | 249(sh), 272, 313(sh) | 86000 |
373 | 5100 | 0.10 | — |
| 2d | 237(sh), 263, 314(sh) | 83400 |
372 | 5000 | 0.19 | — |
| 3a | 301, 319(sh), 396(sh), | 36300 |
615 | 7700 | 0.03 | −1.20 |
| 3b | 301, 316(sh), 396(sh) | 52700 |
565 | 7600 | 0.07 | −1.00 |
| 3c | 300(sh), 320, 392(sh) | 66400 |
450 | 3300 | 0.16 | −1.24 |
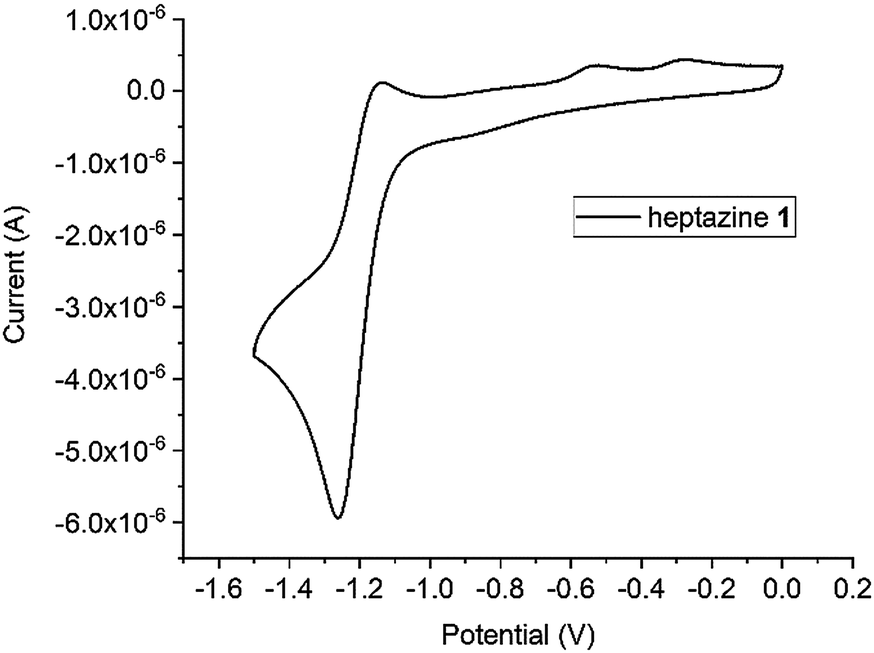 | ||
| Fig. 3 Cyclic voltammogram for the reduction of 2,5,8-tris(3,5-diethyl-pyrazolyl)-heptazine 1; electrolyte DCM/TBFP, El. C 2 mm diameter, scan rate 100 mV s−1, C = 2 × 10−3 mol L−1, Ag/AgCl ref., checked ca. −0.40 V vs. Fc/Fc+. | ||
Photophysical studies
The photophysical properties of heptazine derivatives 1, 2a–d and 3a–c were investigated in dichloromethane (Fig. 4 and Table 2). All heptazines prepared present a large UV band absorption: molar extinction coefficients are very high (8 × 104 to 9 × 104 M−1 cm−1) for heptazines substituted by pyrazole or amines, and slightly lower for heptazines substituted by thiols (Table 2). The position of the absorption maximum varies notably according to the electron-rich character of the substituent: while heptazines substituted with amino group have absorption maxima below 280 nm, the introduction of a thiol group shifts the absorption maximum to 300 nm (aryl thiol), or 320 nm (benzyl thiol). Interestingly, all heptazines studied feature a low intensity band at lower energy (see ESI Fig. S18†), which according to TD-DFT calculations (see below) corresponds to a transition between a NTO orbital located on the heptazine core to the LUMO. The absorption maxima do not vary significantly with the polarity of the solvent (ESI section, spectroscopy†).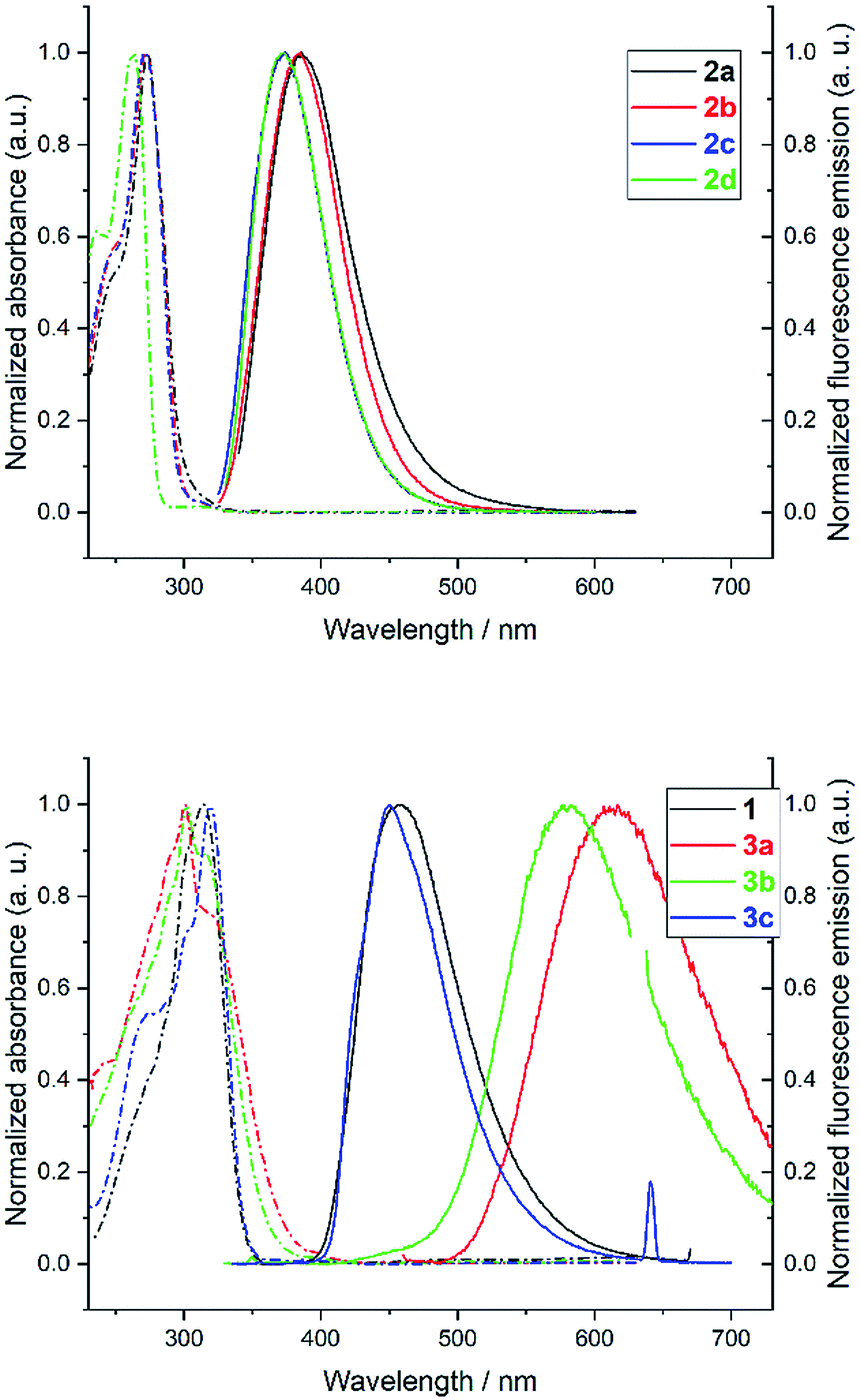 | ||
| Fig. 4 Normalized absorption (dashed line) and fluorescence emission (plain line) of heptazines 1–3 in dichloromethane. Emission spectra were recorded at OD < 0.1 at the excitation wavelength (310 to 320 nm) and below; this corresponds to concentrations between 1 and 3 μM for heptazines 1 and 3a–c, and between 35 and 45 μM for heptazines 2a–d. | ||
| Compound | τ/ns (aerated) | τ/ns (degassed) |
|---|---|---|
| a Excitation: 300 nm for heptazines 1 and 2a–d, 320 nm for heptazines 3a–c. Fluorescence decays were recorded at the emission maximum for each compound. | ||
| 1 | 62.0/292 | 66.8/314 |
| 2a | 18.6/88.3 | 19.4/198 |
| 2b | 12.4/78.2 | 14.3/138 |
| 2c | 14.5/65.0 | 18.1/106 |
| 2d | 19.8/88.6 | 23.7/218 |
| 3a | <10/135 | <10/148 |
| 3b | <10/367 | <10/673 |
| 3c | 21/90 | 26/101 |
All heptazines display fluorescence in the near UV to visible region, stemming from the previously mentioned low energy transition. The photophysical properties of heptazines 2a–d are similar to those of the 2,5,8-tris(diisopropylamino)-heptazine provided by Dubois et al.6 Within the aminoheptazines, a small hypsochromic shift (−9 nm) is noted going from cyclic amine substituents 2a–c to primary amine 2d. For aminoheptazines 2a–d, the maximum emission varies between 372 nm (2d) and 386 nm (2a) with fluorescence quantum yields between 0.10 (2c) and 0.19 (2d). Heptazines 1 and 3a–c display fluorescence emission in the visible region. Heptazines 1 and 3c display fluorescence emission quantum yields of 0.15 and 0.16, respectively, while the quantum yield is much lower for heptazine 3a (0.03), and is intermediary for heptazine 3b (0.07); it is also accompanied by a larger Stokes shift. The solvatochromic behaviour of heptazines 1, 2a, 3b and 3c was then investigated (see the ESI†). Heptazine 2a does not show significant solvatochromism. Moderate solvatochromism is observed for heptazines 1 and 3c except in ethanol where a strong hypsochromic shift can be noted, in absorption (−29 nm compared to DCM for 3c) and in emission (−94 nm compared to DCM for 3c) spectra. This shift could be attributed to specific interactions with the solvent; detailed studies are in progress. Heptazine 3b also displays this hypsochromic shift in ethanol, and shows, in addition, strong positive solvatochromism in non protic solvents (see the ESI†).
All the compounds studied are also fluorescent in the solid state, unlike many other fluorophores. For heptazine 1, 2a–d, and 3c the fluorescence emission spectra of thin films (drop-cast on a glass slide from the DCM solution) are relatively similar to the ones in DCM solution: the fwhm are similar and small shifts of the emission maxima are observed (from −16 to +25 nm, see ESI Fig. S19 to S26†). Contrariwise, heptazines 3a and 3b display large hypsochromic shifts when going from the solution to the solid state (−152 nm for 3a and −129 nm for 3b). Additionally, the emission spectrum of 3a is significantly broadened in the solid state, revealing the coexistence of several emissive species.
The time-resolved fluorescence emission of the eight heptazines prepared was investigated, in DCM solutions, either aerated or degassed by Ar bubbling (Table 3). To our delight, all compounds display prompt and delayed fluorescence, the longest lifetimes being observed for heptazine 3b. Further investigations on the TADF properties of these molecules are underway in our group, since they appear to be very promising blue (for heptazine 1) or UV (for heptazines 2a–d) TADF emitters which could find applications in OLED devices.
DFT and TD-DFT calculations
DFT calculations were performed on heptazine derivatives 1, 2a–c, 2d′ (an analog of compound 2d with an ethyl chain), 3′ (an analog of compound 3c where the butyl chain is replaced by a methyl), and 3b–c. Geometry optimizations (B3LYP, 6-31G(d) – see Fig. 5 and the ESI†) show that in 2,5,8-tris(3,5-diethyl-pyrazolyl)-heptazine 1 the three pyrazole groups are coplanar to the heptazine core. The geometry computed in the gas phase is similar to the one in the crystal structure for 2,5,8-tris(3,5-diethyl-pyrazolyl)-heptazine. Interestingly, in heptazines substituted by cyclic amines 2a–c, the three carbons substituting the nitrogen of the amino substituents are also coplanar. In contrast, in 2,5,8-tris(p-tolylthio)heptazine 3′ and in 2,5,8-tris(phenylthio)heptazine 3b, the phenyl groups are almost perpendicular to the heptazine core. In heptazine 3c, the three SCH2 moieties are coplanar with the heptazine core, while the three phenyl rings are orthogonal to the heptazine core.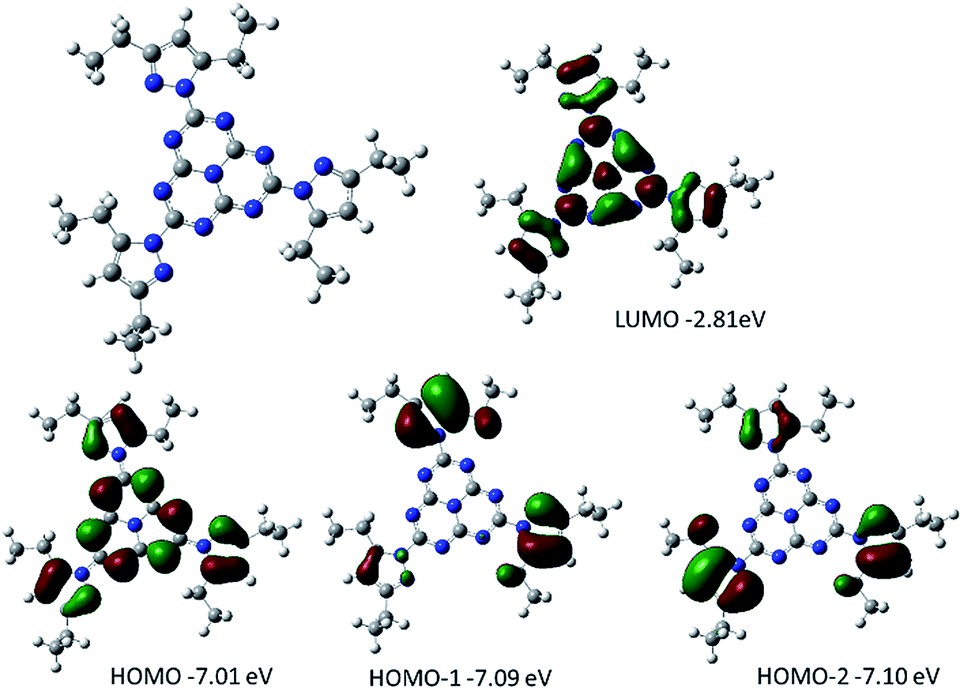 | ||
| Fig. 5 Optimized geometry together with the representation and energy levels of HOMO−2, HOMO−1, HOMO and LUMO orbitals of 2,5,8-tris(3,5-diethyl-pyrazolyl)-heptazine 1 (B3LYP, 6.311G+(d,p)). | ||
Orbital energy levels were then computed at the B3LYP, 6-311G+(d,p) level, with an implicit solvent model (iefpcm, dichloromethane). In all cases the LUMO presents a contribution of the heptazine ring and of the heteroatoms linked to it (N or S, see Fig. 5 and the ESI†). The HOMO, HOMO−1 and HOMO−2 are close in energy (ΔE < 0.15 eV), except for the heptazine 2′ (ΔE = 0.33 eV). It is worth noting that in heptazine 1 the HOMO is located both on the heptazine and the pyrazole while the HOMO−1 and HOMO−2 are located only on the pyrazole substituents. In heptazines 2a–c and 2′, the HOMO−1 (2a), HOMO−2 (2b) or HOMO (2c and 2′) is located purely on the heptazine core. In heptazine 3′, similarly to heptazine 1, the HOMO, HOMO−1 and HOMO−2 are located mostly on the thiophenol substituents. It is worth noting that this is not the case in heptazine 3b in which the HOMO is located on the heptazine core. This indicates that introducing substituents on the thiophenol ring allows modulating the electronic properties of the phenylthio heptazines.
In terms of orbital energy levels, going from an diethylpyrazole substituent to an amino substituent increases the LUMO level (from −2.81 eV for 1 to −1.36 eV for 2b) and to a smaller extent the HOMO level also (from −7.01 eV for 1 to −6.41 eV for 2b). In contrast, HOMO and LUMO energy levels are similar for heptazines 1 and 3a–c (see ESI, Fig. S34 to S40†). The optical bandgaps correlate well as expected with the calculated HOMO–LUMO differences, as shown in Fig. 6 (each point corresponds to a specific heptazine).
 | ||
| Fig. 6 Correlation between measured bandgaps and calculated HOMO–LUMO differences. | ||
TD-DFT calculations were performed at the same level of theory. In all cases, computed transitions are in very good agreement with the experimental ones (see ESI Table S2 to S8†) and in most cases involve several transitions between occupied orbitals and the LUMO. It is worth noting that in all cases, transitions of very low or zero oscillator strengths can be found at low energies, and are followed by an intense transition which is in agreement with the tail in absorption observed experimentally. NTO analysis32 was then performed, which revealed that for the eight heptazines studied, for the first transition the hole is located purely on the heptazine core. Interestingly in heptazines 1 and 3′ a manifold of the three optical transitions very close in energy (ΔE < 450 cm−1) imply NTO orbitals that are spatially separated (see the ESI†) which is in good agreement with the delayed fluorescence observed. Similarly, TD-DFT calculations reveal a small energy gap (≤1200 cm−1) between the S1 state and the closest triplet state, which is in good agreement with the TADF properties observed experimentally.
Conclusions
We have described in this article the second heptazine known to date bearing easily exchangeable leaving groups, and have given examples of its versatility, as a new platform for this highly interesting family. In particular, pyrazolyl-substituted heptazines are very easy to prepare at the multigram scale, with reasonable yields and only one easy chromatography step. Further work is ongoing to explore new examples of heptazines, along with their applications in various fields of physical chemistry, like photovoltaics and delayed fluorescence.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. A. would like to acknowledge the IUF (Institut Universitaire de France) for personal and financial support. C. A. would like to thank Arnaud Brosseau (PPSM) for his help with the TGA and the fluorescence lifetime measurements. This work was granted access to the computing resources of CINES (Montpellier, allocation A0050810547 awarded by GENCI).Notes and references
- L. Pauling and J. H. Sturdivant, Proc. Natl. Acad. Sci. U. S. A., 1937, 23, 615–620 CrossRef CAS PubMed.
- A. Schwarzer, T. Saplinova and E. Kroke, Coord. Chem. Rev., 2013, 257, 2032–2062 CrossRef CAS.
- V. W. Lau, M. B. Mesch, V. Duppel, V. Blum, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2015, 137, 1064–1072 CrossRef CAS PubMed.
- J. Li, H. Nomura, H. Miyazaki and C. Adachi, Chem. Commun., 2014, 50, 6174–6176 RSC.
- E. J. Rabe, K. L. Corp, A. L. Sobolewski, W. Domcke and C. W. Schlenker, J. Phys. Chem. Lett., 2018, 9, 6257–6261 CrossRef CAS PubMed.
- A. Zambon, J. M. Mouesca, C. Gheorghiu, P. A. Bayle, J. Pecaut, M. Claeys-Bruno, S. Gambarelli and L. Dubois, Chem. Sci., 2016, 7, 945–950 RSC.
- I. Bala, H. Singh, V. R. Battula, S. P. Gupta, J. De, S. Kumar, K. Kailasam and S. K. Pal, Chem.–Eur. J., 2017, 23, 14718–14722 CrossRef CAS PubMed.
- J. Li, Q. Zhang, H. Nomura, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2014, 105, 013301 CrossRef.
- J. Li, T. Nakagawa, J. MacDonald, Q. Zhang, H. Nomura, H. Miyazaki and C. Adachi, Adv. Mater., 2013, 25, 3319–3323 CrossRef CAS PubMed.
- S. Kumar, N. Sharma and K. Kailasam, J. Mater. Chem. A, 2018, 6, 21719–21728 RSC.
- G. Zhang, G. Li, T. Heil, S. Zafeiratos, F. Lai, A. Savateev, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 3433–3437 CrossRef CAS PubMed.
- G. Zhang, L. Lin, G. Li, Y. Zhang, A. Savateev, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 9372–9376 CrossRef CAS PubMed.
- A. Savateev, S. Pronkin, J. D. Epping, M. G. Willinger, C. Wolff, D. Neher, M. Antonietti and D. Dontsova, ChemCatChem, 2017, 9, 167–174 CrossRef CAS.
- V. W. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- K. Kailasam, J. Schmidt, H. Bildirir, G. Zhang, S. Blechert, X. Wang and A. Thomas, Macromol. Rapid Commun., 2013, 34, 1008–1013 CrossRef CAS PubMed.
- F. Goettmann, A. Thomas and M. Antonietti, Angew. Chem., Int. Ed., 2007, 46, 2717–2720 CrossRef CAS PubMed.
- Q.-Q. Dang, Y.-F. Zhan, X.-M. Wang and X.-M. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 28452–28458 CrossRef CAS PubMed.
- Y. Ke, D. J. Collins, D. Sun and H.-C. Zhou, Inorg. Chem., 2006, 45, 1897–1899 CrossRef CAS PubMed.
- C. Posern, U. Bohme, J. Wagler, C. C. Hohne and E. Kroke, Chem.–Eur. J., 2017, 23, 12510–12518 CrossRef CAS PubMed.
- C. Posern, U. Böhme and E. Kroke, Z. Anorg. Allg. Chem., 2018, 644, 121–126 CrossRef CAS.
- E. Kroke, M. Schwarz, E. Horath-Bordon, P. Kroll, B. Noll and A. D. Norman, New J. Chem., 2002, 26, 508–512 RSC.
- I. Siva Kumar and S. Kumar, Chem. Commun., 2017, 53, 11445–11448 RSC.
- T. Saplinova, V. Bakumov, T. Gmeiner, J. Wagler, M. Schwarz and E. Kroke, Z. Anorg. Allg. Chem., 2009, 635, 2480 CAS.
- A. Stolle, T. Szuppa, S. E. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317–2329 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC.
- J. L. Do and T. Friscic, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- R. M. Claramunt, C. López, D. Sanz and J. Elguero, in Advances in Heterocyclic Chemistry, ed. A. R. Katritzky, Academic Press, 2014, vol. 112, pp. 117–143 Search PubMed.
- P. K. Sahoo, C. Giri, T. S. Haldar, R. Puttreddy, K. Rissanen and P. Mal, Eur. J. Org. Chem., 2016, 2016, 1283–1291 CrossRef CAS.
- X. Zhu, Z. Li, C. Jin, L. Xu, Q. Wu and W. Su, Green Chem., 2009, 11, 163–165 RSC.
- M. D. Coburn, G. A. Buntain, B. W. Harris, M. A. Hiskey, K. Y. Lee and D. G. Ott, J. Heterocycl. Chem., 1991, 28, 2049–2050 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic protocols, spectroscopic properties, DFT optimization, and X-ray diffraction analyses CCDC. CCDC 1843875. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00665f |
| This journal is © The Royal Society of Chemistry 2019 |