DOI:
10.1039/C9SC00861F
(Edge Article)
Chem. Sci., 2019,
10, 4015-4024
Organogelating and narcissistic self-sorting behaviour of non-preorganized oligoamides†
Received
19th February 2019
, Accepted 2nd March 2019
First published on 8th March 2019
Abstract
Several series of structurally flexible, non-preorganized oligoamides with carboxylic acid, hydride, N-propargyl amide and N-propyl amide end groups were synthesized and characterized. They were found to be strong organogelators for aromatic solvents. Both the number and density of the amide units, and the nature of the end group were found to influence the gel strength as well as the kinetics of gel formation. Oligomers with a higher number and density of amide units were found to form stronger gels. Rather unexpectedly, a slight change of the end group could significantly affect the enthalpy and entropy of formation of the resulting self-assembly. Oligomers with the N-propargyl amide end group were found to be kinetically trapped in the melted state, which devoid them of forming crystalline zone upon cooling. On the other hand, the corresponding saturated N-propyl amide analog did not show the same effect. Upon mixing of these oligomers, narcissistic self-sorting could be observed in both the wet gel and xerogel states if (i) the difference in the number of amide units is greater than one or (ii) the amide unit densities are different. A semi-quantitative assessment of the extent of self-sorting could be achieved by constructing the corresponding phase diagram of the mixed gel systems. It is concluded that narcissistic self-sorting does not just prevail in mixtures making up of structurally different molecules, but appears to be fairly general even in mixtures of structurally similar molecular homologs.
Introduction
Molecular self-sorting1–4 is a phenomenon which describes the high fidelity recognition between molecules in a complex mixture. If strong affinity for others is exhibited then the self-assembly process is called social self-sorting, while the affinity for itself will be named narcissistic self-sorting. Generally, the driving forces which lead to the self-sorting process are hydrogen bonding,5–7 metal–ligand interaction,8–13 fluorophobic,14 host–guest recognition15,16 and/or dynamic covalent bonding.17–20 Controlling the outcome of self-sorting is therefore of pivotal importance in the design and construction of complex functional and responsive soft materials for various applications.
In synthetic systems, careful design and precise control over the placements of the interacting functionalities inside the various components are required before any high fidelity self-sorting process can be realized. In general, high fidelity self-sorting is commonly observed in two scenarios. In the first case, the molecules can be structurally very similar to each other, but they all possess a higher degree of structural rigidity with preorganized interacting units. Self-sorting is manifested via geometry controlled, such that slight changes in ligand biting angles, metal coordination geometries, complementarity of hydrogen bonding motifs, and shape complementarity in host–guest binding can dedicate the outcome. In the second scenario, self-sorting is non-geometry controlled. The molecular ingredients are either structurally very similar or very different from each other. Rather, the overall molecular properties, such as solubility and miscibility, are much more important in dedicating the outcome. In general, functionally different systems tend to produce narcissistic self-sorting, while social self-sorting is expected from structurally similar systems. The formation of immiscible (phase segregation) or miscible polymer blends actually falls into this category.21
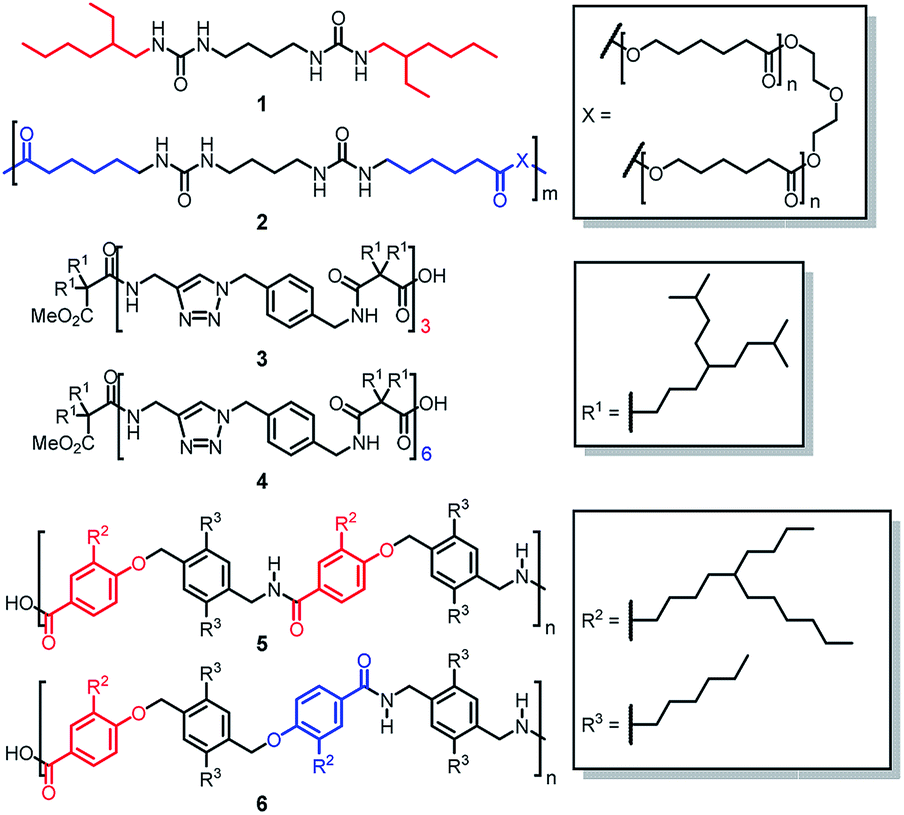
Thus far most self-sorting studies have been focused on small, structural rigid and preorganized molecular systems having dissimilar molecular structures. However, there is one uncharted area in self-sorting that remains relatively unexplored. This involves structurally similar and flexible molecules such as oligomeric and polymeric homologs. Although Nature provide the perfect example of social self-sorting involving the pairing of complementary DNA strands, systematic studies of self-sorting phenomenon on synthetic oligomers22,23 and polymers24 are rare. Normally one would expect that social self-sorting to be the outcome for homologs of oligomers and polymers because of their structural similarity. However, we and others have reported a few counter examples showing that narcissistic self-sorting could actually occur even in structurally similar compounds.25–29 For examples, Meijer reported that narcissistic self-sorting was found between a mixture of bis(urea)butylene molecule 1 and bis(urea)butylene polymer 2 when the amount of 1 was greater than 23 mol% (Fig. 1).25 Kumaki showed that when two narrowly dispersed isotactic poly(methyl methacrylate) (it-PMMA) chains of different lengths were mixed, only chains of the same length formed double-stranded helices, while the formation of heterogeneous double-stranded helices comprising two it-PMMA of different lengths was not observed.26 Narcissistic self-sorting can also be manifested in the form of gel formation in multi-component gel system.30 In this context, we earlier showed that self-sorting occurred in a mixture of oligoamide gels 3 and 4,27 and also in polyamide gels 5 and 6.28 In the case of the oligamides, self-sorting was attributed to the difference in the number of amide units of the two structurally homologous hexamide 3 and dodecaamide 4. In the case of the polymers 5 and 6, the origin of self-sorting was ascribed to the different alignment patterns of the dipole units on the polymer chain.
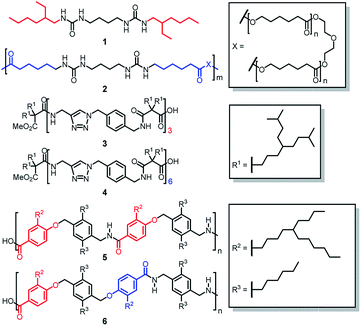 |
| | Fig. 1 Structural flexible bisurea compound 1 and bisurea polymer 2, hexaamide 3 and dodecaamide 4, and polyamides 5 and 6 showing narcissistic self-sorting when mixed. Their structural differences are highlighted in red and blue. | |
In the above examples, except for polymers 5 and 6, narcissistic self-sorting appeared to be due to the very significant difference in the number of amide units in the two oligomers or polymers. Recent studies31–34 by Hunter on hydrogen bonded duplexes formed from the dimerization of two structural flexible oligoamides prompted us to speculate that the observed self-sorting behaviour may be originated from the significant difference of their self-association strengths. In this paper we would like to address several specific questions that are pertaining to our understanding of self-sorting in multi-component gel systems involving structurally flexible oligoamides. First, how much difference in the number of amide units is needed for two different oligoamides to exhibit self-sorting? Second, is it possible to address the degree of self-sorting semi-quantitatively? If so, does the degree of self-sorting has a strong correlation to the difference in the number of amide motifs? Third, can a difference in the density of the interacting amide units (i.e. number of unit per mass) of two oligomer chains lead to self-sorting? Fourth, can self-sorting be observed in ternary systems involving three different oligoamides? It should be emphasized here that the self-sorting behaviour between structurally similar, non-preorganized molecules are not very well understood. This kind of system is of general interest to our understanding of the underlying mechanisms of self-sorting and will help to extend the scope of applications of multi-component gel systems. This ‘mix and match’ approach to create multi-component gel systems also represents a highly robust way to create and fine tune the gel properties, and is far more versatile than single component gel systems.
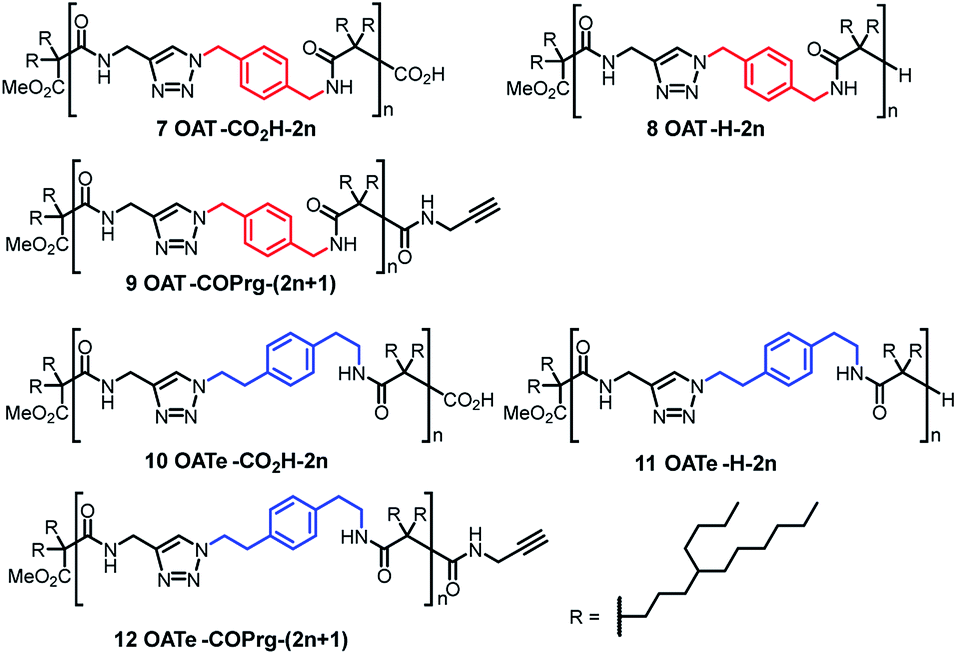
In the present work, we first disclosed the synthesis of three homologous series of oligo(amide-triazole)s 7–9 featuring a p-xylylene spacer in the backbone but with three different types of end group, namely, carboxylic acid 7 (OAT-CO2H-2n), hydride 8 (OAT-H-2n) and N-propargyl amide 9 [OAT-COPrg-(2n+1)], where 2n and (2n + 1) signified the number of CONH units in the oligomers (Fig. 2). To address the effect of the density of the amide units on the self-sorting behaviour, we also prepared the corresponding elongated analogous series of oligomers 10 (OATe-CO2H-2n), 11 (OATe-H-2n) and 12 [OATe-COPrg-(2n+1)] bearing a longer p-ethylenylphenyleneethylene spacer. Herein we reported that (i) these oligomers form strong organogels and their gel strength is dependent not only on the number and density of the CONH units, but also on the nature of the end group; (ii) the small end group can exert strong kinetic trapping effects on the enthalpy and entropy of the formation of the resulting self-assembly; (iii) narcissistic self-sorting does exist between these structurally related, non-preorganized homologs in the wet gel and also xerogel states; (iv) the extent of self-sorting can be correlated to the difference in the number and density of the amide units, and is also affected by the nature of the end group and (v) significant difference in the self-sorting behaviour could be found between the samples prepared in the diluted wet gel state vs. melted state, and this was mainly controlled by the nature of the end group.
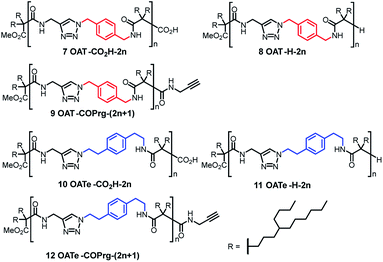 |
| | Fig. 2 Structures of oligo(amide-triazole)s synthesized and studied in this research. | |
Results and discussion
Synthesis
The oligo-controlled syntheses of OAT-CO2H-2n, OAT-COPrg-(2n+1), OATe-CO2H-2n, and OATe-COPrg-(2n+1) involved an iterative procedure, which was similar to that reported in our previous work.27 On the other hand, OAT-H-2n and OATe-H-2n compounds were prepared in situ during annealing (200 °C for 5 min) inside a differential scanning calorimeter. These oligomers all possess a bottlebrush-shaped architecture which minimize the back folding of the backbone. Instead of using a hyperbranched dendron as previously disclosed, a racemic 4-butyldecyl (i.e. R) group was used as the side chain. This strategy avoided the tedious preparation of the dendron and also led to an enhancement of the solubility of the target compounds. Furthermore, in addition to the (n + 1) iterative approach, a (n + 2) approach was also adopted to grow the higher oligomers. The latter approach allowed the addition of four additional CONH units in one iterative cycle and improved the synthetic efficiency significantly.
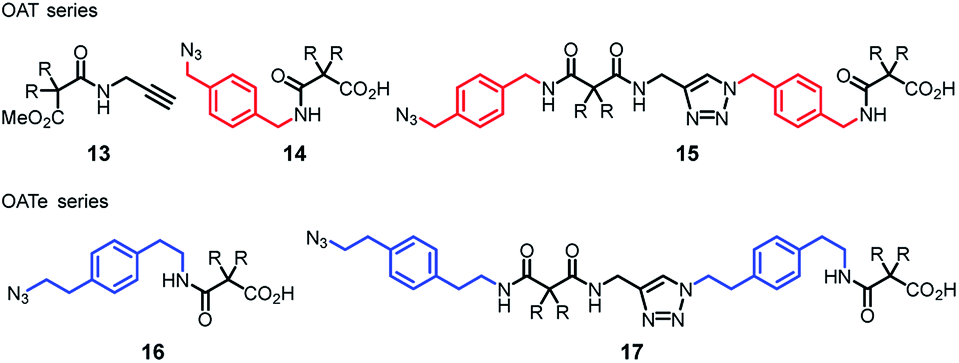
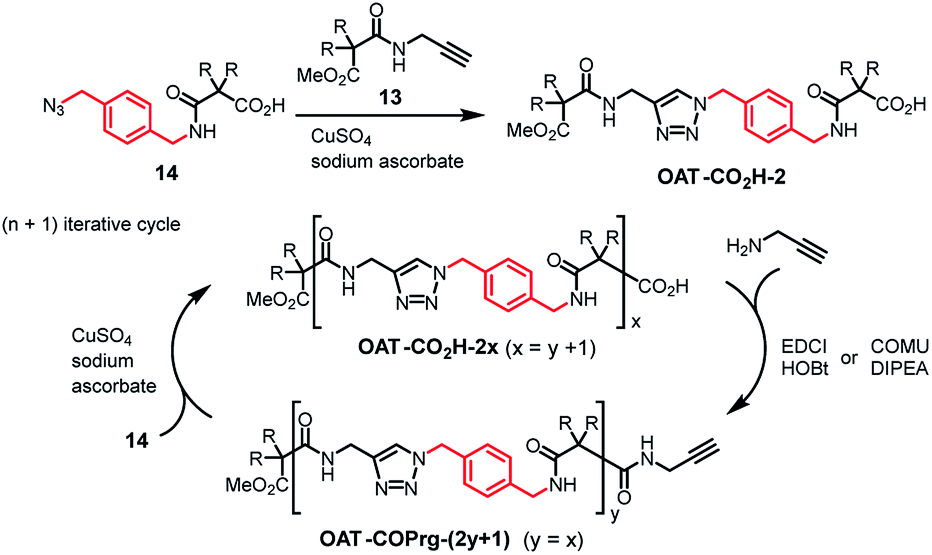
To begin with, several key intermediates 13–17 were firstly prepared from commercially available materials (Fig. 3).35 Compound 13 is an acetylenic partner that was used to couple with various propagating azido units 14–17 to furnish the target oligomers. Hence, diamide OAT-CO2H-2 was prepared via CuAAC click coupling (CuSO4 and sodium ascorbate)36 between compounds 13 and 14 in 86% yield (Scheme 1). Employing the (n + 1) iterative synthetic cycle, amidation of OAT-CO2H-2 then gave the triamide OAT-COPrg-3 in 65% yield in the presence of 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide methiodide (EDCI) and 1-hydroxybenzotriazole (HOBt). Further click coupling of OAT-COPrg-3 and the propagating unit 14 then produced the tetramide OAT-CO2H-4 in 79% yield. Repetition of the same reaction cycle then afforded OAT-COPrg-5, OAT-CO2H-6 and OAT-COPrg-7 in 78, 89 and 80% yield, respectively. Significantly difference in the chromatographic mobilities of the OAT-CO2H-2n and OAT-COPrg-(2n+1) ensured that no cross-contamination product was carried down in the subsequent synthesis, thus exerting full oligo-controlled synthesis of the target oligomers.
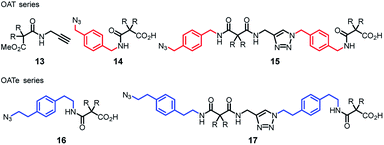 |
| | Fig. 3 Key compounds 13–17 required for the synthesis of oligo(amide-triazole)s OAT and OATe. | |
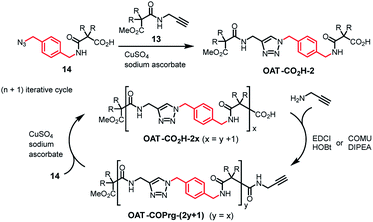 |
| | Scheme 1 Synthesis of oligomers OAT-CO2H-2x and OAT-COPrg-(2y+1) using an (n + 1) iterative approach. | |
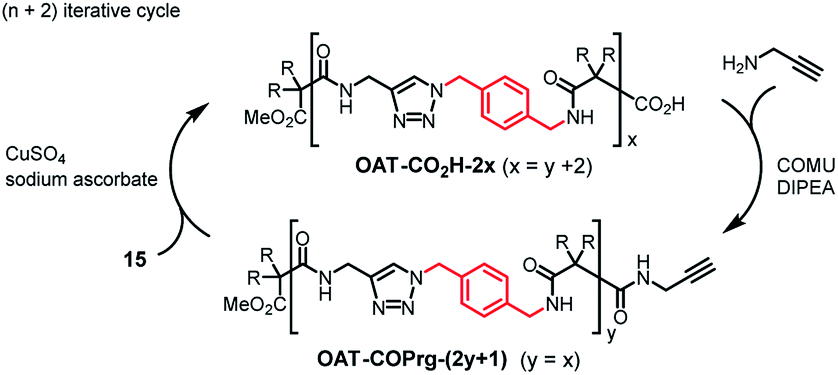
For the synthesis of the longer oligomers, the (n + 2) approach was adopted by using the longer propagating unit 15 (Scheme 2). For example, click reaction between OAT-COPrg-5 and 15 gave OAT-CO2H-8 in 96% yield. Subsequent amidation with propargylamine in the presence of N,N-diisopropylethylamine (DIPEA) and 1-cyano-2-ethoxy-2-oxoethylidenaminooxy-dimethylamino-morpholinocarbenium hexafluorophosphate (COMU) then afforded OAT-COPrg-9 in 79% yield. In a similar manner, OAT-CO2H-10, OAT-COPrg-11 and OAT-CO2H-12 could be obtained. In the synthesis cycles, the yields of click reaction were around 66–96% and for the coupling reactions were 65–80%.
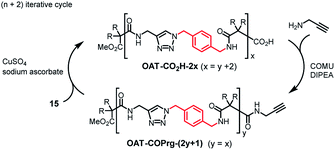 |
| | Scheme 2 Synthesis of oligomers OAT-CO2H-2x and OAT-COPrg-(2y+1) using an (n + 2) iterative approach. | |
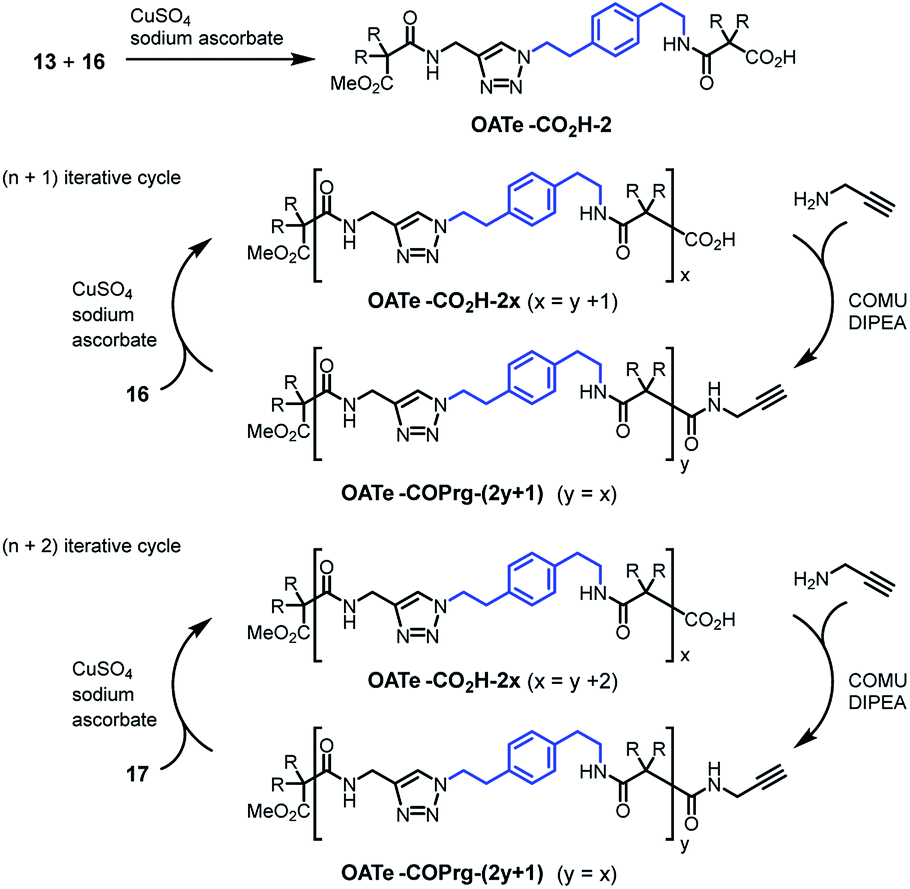
For the OATe series, propagating units 16 and 17 were used for the (n + 1) and (n + 2) iterative cycles, respectively (Scheme 3). The elongated diamide OATe-CO2H-2 was prepared by click coupling between compounds 13 and 16 in 82% yield. Using the (n + 1) route, OATe-COPrg-3, OATe-CO2H-4, OATe-COPrg-5, OATe-CO2H-6 and OATe-COPrg-7 were synthesized, while OATe-CO2H-8, OATe-COPrg-9 and OATe-CO2H-10 were prepared via the (n + 2) approach by using compound 17 as the propagating unit. In the synthesis cycles, the yields of click reactions were 77–88% and for amide formation reactions were 66–92%.
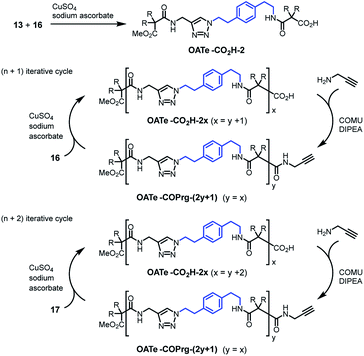 |
| | Scheme 3 Synthesis of oligomers OATe-CO2H-2x and OATe-COPrg-(2y+1) using the (n + 1) and (n + 2) approaches. | |
Structural characterizations
The structural identities of all prepared compounds were characterized by 1H and 13C NMR spectroscopy, and the results were consistent with the proposed structure.35 The degree of oligomerization could be confirmed by mass spectrometry and 1H NMR end group analysis.35 Hence, all the target oligomers were prepared in high purity with no cross-contamination.
Organogelation properties
Gelation strength.
Traditionally, the minimum gelation concentration (MGC) and gel-to-sol transition temperature (Tgs) are the two commonly used parameters to assess the gel strength of a series of structurally related organogelators. However, these two methods possess some inherent limitations which could lead to misleading conclusions. First, MGC data are rather crude and may not be able to differentiate gel strength of similar magnitude. Second, at the Tgs point, the gel and sol states are in equilibrium, so the Gibbs free energy change is zero (i.e. ΔGgs = ΔHgs − TgsΔSgs = 0, or Tm = ΔHgs/ΔSgs). Hence, using the Tgs values to compare the relative stability (i.e. ΔHgs) of two gels is only valid when the entropy of changes (ΔSgs) of the two systems are equal. While the latter proposition is likely to be valid for compounds of similar structure and molecular weight, this is certainly not the case among oligomers of different lengths as ΔSgs should be size dependent. As a result, the gel-to-sol enthalpy changes (ΔHgs) will be used to assess the wet gel strength. Incidentally, the ΔSgs data could also provide additional information regarding the structural orderliness of the gel assembly.
For the OAT series, stable organogels in aromatic solvents could be formed when the minimum number of CONH units was 6, while this number was 7 for the OATe series. The enthalpy and entropy data (shown in a per gram basis) obtained from differential scanning calorimetry (DSC) studies provided more revealing insights (Table 1). For these oligomers, the number of amide units is directly proportional to its molecular weight, therefore the presented data roughly reflected the relevant values on a per amide unit basis. First, within the same series and with the same end group, the ΔHgs value increased monotonically as the oligomer length became longer, indicating that each CONH unit in the longer oligomer contributes stronger to the stability of the gel network than that in a shorter oligomer. This synergistic multiple functional group binding effect had been demonstrated earlier by Hunter37 and also by us.27,38 However, the increment began to level off when the CONH units became very large (OAT-COPrg-9vs.OAT-COPrg-11), implying the presence of a plateauing effect. Second, the strength of the OAT gels was always greater than that of corresponding OATe gels having the same number of CONH (OAT-COPrg-7vs.OATe-COPrg-7, OAT-CO2H-10vs.OATe-CO2H-10), which proved that a decrease in the density of CONH unit could weaken the gel network. Third, the end group played a very important role in determining the gel strength. The gels formed from oligomers bearing the N-propagaryl amide end group were more stable than the ones with one less amide unit but bearing the CO2H end group (OAT-COPrg-7 > OAT-CO2H-8, OAT-COPrg-9 > OAT-CO2H-10). Fourth, from the ΔSgs data, it was found that the longer oligomers always possessed a higher degree of structural order in the self-assembled gel state than the shorter oligomers for both the OAT and OATe series. Incidentally, it was also noted that the OAT oligomers formed a more order supramolecular assembly than the corresponding OATe oligomers, and that the N-propargyl amide end group also conferred higher packing order than the carboxylic acid end group. As the entropic contribution to the gelation was very different even for compounds of very similar structure, this confirmed that the simple use of Tgs or MGC values to compare gel strength is not reliable.
Table 1 Enthalpy (ΔHgs) and entropy (ΔSgs) changes of gel-to-sol transition of p-xylene gels (conc. = 25 mg mL−1) of OAT and OATe oligomersa
| Compound |
ΔHgs (J g−1) |
ΔSgs × 103 (J g−1 K−1) |
Compound |
ΔHgs (J g−1) |
ΔSgs × 103 (J g−1 K−1) |
|
Gels were aged in the sealed crucible at 25 °C for 20 min prior to measurements. Heating profile: 25 °C to 100 °C at a rate of 5 °C min−1. p-Xylene was used as the reference in the measurements.
|
|
Acids
|
|
OAT-CO2H-6
|
12.0 |
38.7 |
— |
|
|
|
OAT-CO2H-8
|
23.2 |
67.5 |
OATe-CO2H-8
|
2.6 |
8.1 |
|
OAT-CO2H-10
|
24.9 |
69.3 |
OATe-CO2H-10
|
5.8 |
17.7 |
|
OAT-CO2H-12
|
28.5 |
78.2 |
— |
|
|
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
Amides
|
|
OAT-COPrg-7
|
26.8 |
83 |
OATe-COPrg-7
|
16.1 |
52.8 |
|
OAT-COPrg-9
|
39.5 |
114 |
OATe-COPrg-9
|
26.0 |
69.6 |
|
OAT-COPrg-11
|
39.9 |
112 |
— |
|
|
Gelation mechanism.
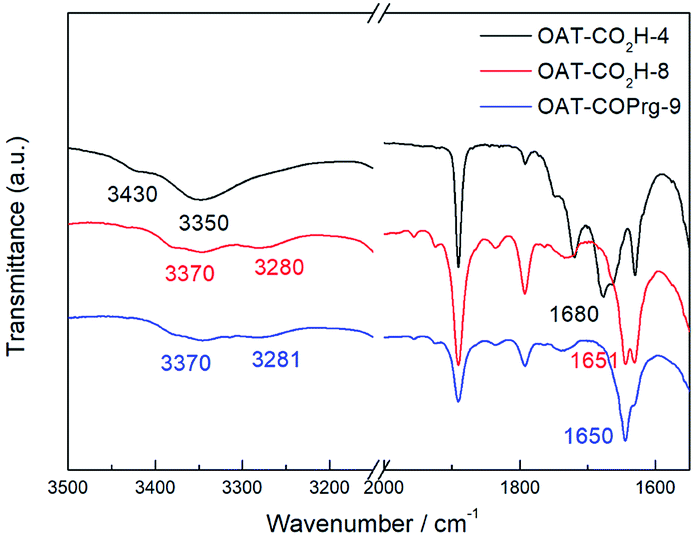
Having established the number and density of the CONH units and the end group effects on the gel strength, the gelation mechanism was then examined first by Fourier-transformed infrared (FTIR) spectroscopy. A comparison of the FTIR spectra of the non-gelator OAT-CO2H-4 (2.5% w/v solution in p-xylene) showed the adsorptions at around 3430 and 3350 cm−1 due to the non-hydrogen-bonded amide N–H stretching frequencies (Fig. 4). They were found to shift to 3370 cm−1 and 3280 cm−1 in the spectra of the gelators OAT-CO2H-8 and OAT-COPrg-9 (2.5% w/v gel in p-xylene). Similarly, the stretching frequencies of the primary amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O also shifted from 1680 cm−1 to 1650 cm−1. The FTIR results strongly proved that the amide groups were involved in the hydrogen-bonding process which was responsible for the gelation property of these oligomers.
O also shifted from 1680 cm−1 to 1650 cm−1. The FTIR results strongly proved that the amide groups were involved in the hydrogen-bonding process which was responsible for the gelation property of these oligomers.
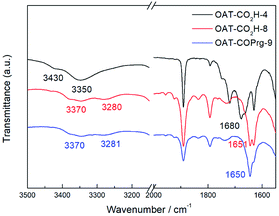 |
| | Fig. 4 Stacked FTIR spectra of OAT-CO2H-4 (2.5% solution in p-xylene), OAT-CO2H-8 (2.5% gel in p-xylene) and OAT-COPrg-9 (2.5% gel in p-xylene). | |
Gelation model.
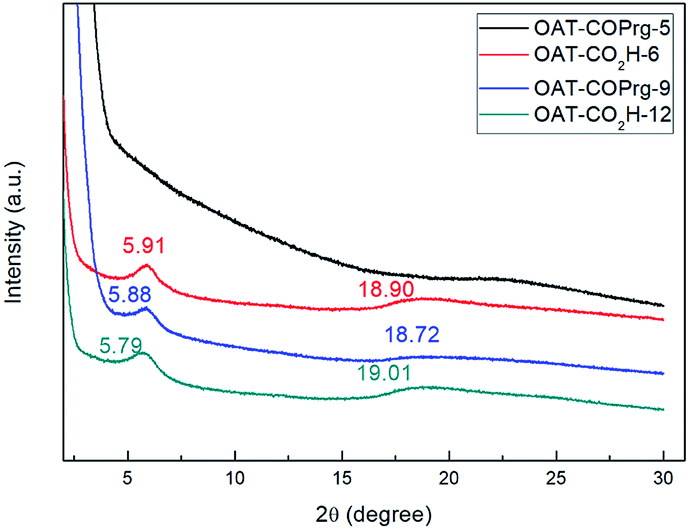
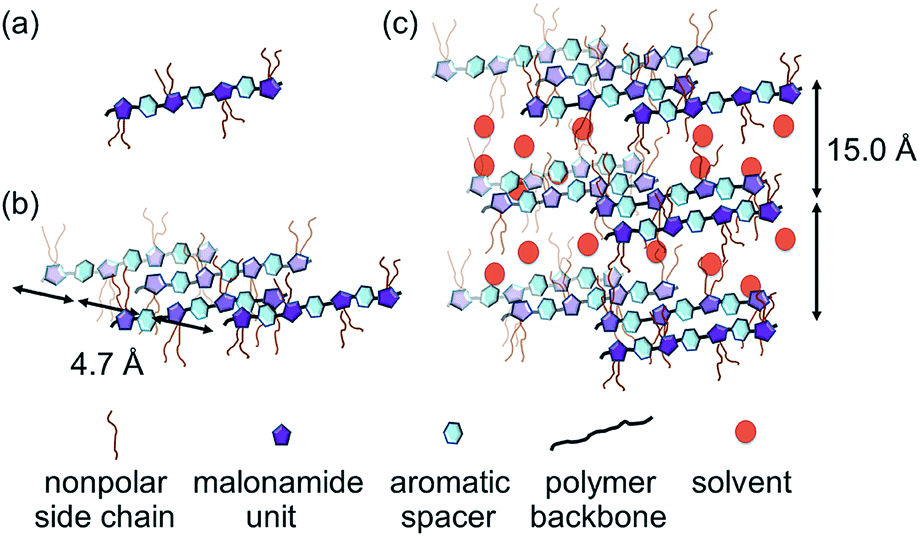
To explore the gel packing arrangement of the crystallization zone in the molecular scale, OAT-COPrg-5, OAT-CO2H-6, OAT-COPrg-9 and OAT-CO2H-12 were subjected to small angle X-ray diffraction (SAXRD) analysis. For OAT-COPrg-5 which could not form gel in p-xylene, no obvious peaks were found (Fig. 5). For the three remaining gelating oligomers, they all showed a similar pattern with two peaks at 2θ = 5.8° and 18.8°. The sharper peak at 5.8° was originated from a d-spacing of 15.0 Å, which was shorter than twice the span of the 4-butyldecyl side chain (2 × 13.5 Å). Another broad peak at 18.8° with a d-spacing of 4.7 Å fell into the range of the interatomic distance of π–π stacking. These peaks were not well resolved, indicating that the ordering of individual molecules inside the xerogel was low. This is most likely due to the backbone flexibility of the long oligomer chain and also each of the oligomers is actually a heterogeneous mixture of diastereomers, which prevent their orderly packing. Nonetheless, the result suggested that gel formation was due to the interdigitation of the hydrophobic side chains and π–π stacking interactions of the backbone aromatic moieties. The packing structures of OAT-CO2H-2n and OAT-COPrg-(2n+1) were very similar which indicated that neither the nature of the end group nor the oligomer length would lead to obviously different nanostructures. Similar results were also obtained in the patterns of the elongated series OATe-COPrg-7 and OATe-CO2H-10 (ESI Fig. S3, pS34†).
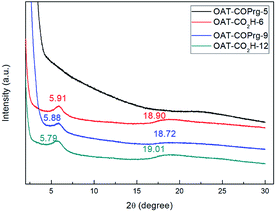 |
| | Fig. 5 SAXRD patterns of 2.5% freeze dry p-xylene solution sample of OAT-COPrg-5, and 2.5% xerogel samples from p-xylene of OAT-CO2H-6, OAT-COPrg-9 and OAT-CO2H-12. | |
To further investigate the self-assembly model of OAT and OATe compounds at the nanoscale, we would like to learn whether our oligoamides could also form duplexes similar to those reported by Hunter.31–34 Hence, 1H NMR dilution experiment was carried out using OAT-CO2H-6 in CDCl3 in order to determine the binding constant. As it turned out, the data fitted very well (r2 = 0.999) to a dimerization model with a Kdim = 2 M−1 (ESI Fig. S5 and Table S1, p S35†), indicating the binding was very weak. Besides OAT-CO2H-6, we also carried out the NMR dilution experiments on OAT-COPrg-7, OAT-COPrg-9 and OAT-CO2H-10 in CDCl3, but the results did not fit the dimerization model. It was rationalized that our oligomers did not simply bind in a dimeric fashion, as a gel network could only be formed via the supramolecular crosslinking involving many oligomer molecules.
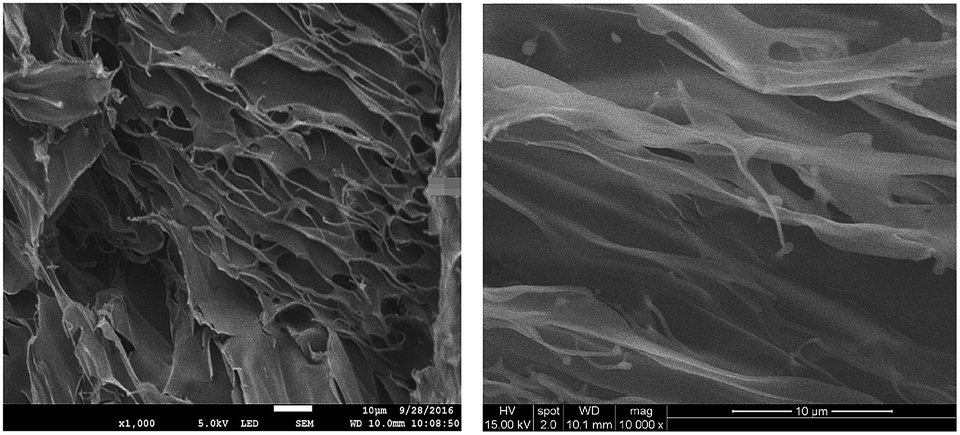
Xerogels of OAT-CO2H-12 and OATe-CO2H-10 were prepared and subjected to scanning electron microscopy (SEM) examinations to reveal the morphology of the network structure (Fig. 6). All the images presented lamellar assemblies which were further linked by entangled fibers or bundles. A gel model was proposed based on the SAXRD and SEM results (Fig. 7). First, the individual oligomers self-associated via intermolecular hydrogen bonding and π–π stacking interactions to form bundles. The bundles then further interacted via side chain interdigitation to form lamellar structures that were capable of trapping solvent molecules. It should be noted that the oligomer chains are not bundled up in a regular fashion (i.e. some oligomer chain ends protrude out from the pack while some ends bury inside the interior).
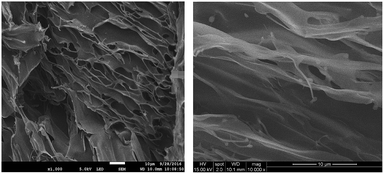 |
| | Fig. 6 SEM images (20k magnification) of freeze-dried 1.5% gels in p-xylene (left) OAT-CO2H-12 and (right) OATe-CO2H-10. | |
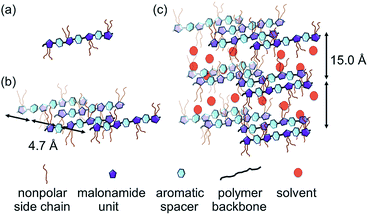 |
| | Fig. 7 Self-assembling model of oligomer gel. (a) Free oligomer chain. (b) Oligomer bundle formation via π–π stacking interaction. (c) Gel network formation via side chain interdigitation and solvent entrapment. | |
End group effect on the kinetics and stability of the self-assembled organogels
While most research works on gels focus on their gelation strength and their associated applications, the kinetics of gel formation is less explored. In our earlier DSC studies, it had already been shown that the end group made significant influence on the packing arrangement and also the strength of the gel network. In order to gain more information of the gel assembly, the xerogel samples prepared by freeze drying of wet p-xylene gels (hereafter named as initial samples), were firstly heated at a rate of 10 °C min−1 from 40 °C to 200 °C to obtain the first heating profile. The samples were then subjected to annealing at 200 °C for 5 min, then cooling down at a rate of 5 °C min−1 back to 40 °C, and the annealed samples were reheated to obtain the second heating curve. Rather unexpectedly, it was found that end group, the method of xerogel preparation (initial sample vs. annealed sample) and the cooling rate all had significant impact on the thermodynamic stability of the xerogels.
N-Propargyl amide end group.
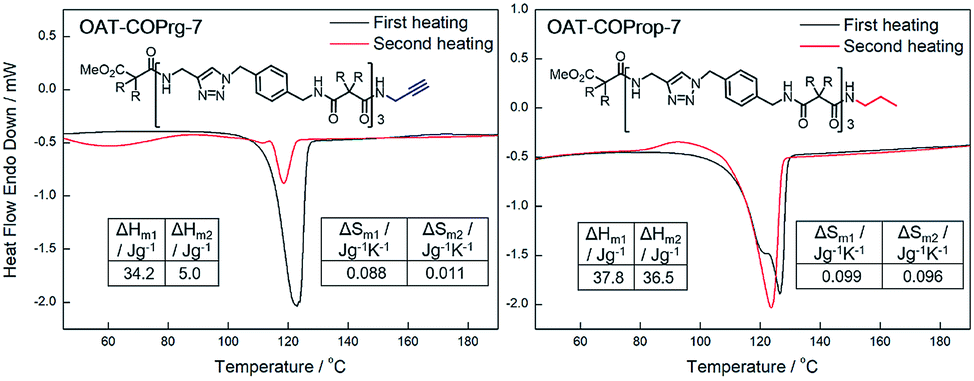
The solid-to-liquid transition temperature (Tm) of the xerogel did not change much for the first and second heatings for all the OAT-COPrg-(2n+1) samples. However, both the enthalpy (ΔHm2 = 5.0 J g−1) and entropy of melting (ΔSm2 = 11.3 × 10−3 J g−1 K−1) values of the annealed samples decreased sharply as compared to those (ΔHm1 = 34.2 J g−1 and ΔSm1 = 87.9 × 10−3 J g−1 K−1) of the initial samples (Fig. 8 and ESI Fig. S8–S9, pS37†). This was not due to sample decomposition as the 1H NMR spectra of the initial and annealed samples were the same (ESI Fig. S18, pS45†). When the annealed OAT-COPrg-7 sample was subjected to wet gel formation followed by freeze drying, the DSC profile of the initial sample re-emerged, which indicated that such a change of the DSC profile was due to a difference of the self-assembly structures prepared from freeze drying of a wet gel vs. from cooling of a melted gel. The diminishment of ΔHm and ΔSm values implied a weakening of the stability of the self-assembling structure and also a reduction of ordered crystalline zones of the annealed samples. This suggested that in the melted state, the oligomers were entangled and were unable to pack into the most stable self-assembly structure due to kinetic entrapment. More interestingly, lowering the cooling rate from 5 °C min−1 to 1 °C min−1, the drop of ΔHm2 (5.0 → 10.4 J g−1) became less, while increasing the cooling rate from 5 °C min−1 to 10 °C min−1 led to further dropping of ΔHm2 (5.0 → 2.9 J g−1) (ESI Table S3, pS38†). The results proved that the diminution of the endothermic peak in the second cycle was because that the self-assembly ability of OAT-COPrg-(2n+1) in melted state was worse than that in the diluted solution state. In the case of the length extended OATe-COPrg-(2n+1) series, no Tm could be identified in the annealed samples, although a solid-to-liquid transition could be found in the initial samples (ESI Fig. S9, pS37†). This finding implied that chain entanglement in the melted state was very severe in the elongated OATe oligomers such that no-crystalline zone could be formed during cooling of the melted state.
N-Propyl amide end group.
To further understand the end group effect on the self-assembling process, the saturated analog of OAT-COPrg-7 (i.e. N-propyl amide, OAT-COProp-7) was prepared.35 To our surprise, the ΔHm and ΔSm values of OAT-COProp-7 before and after annealing were very similar (Fig. 8), which proved that the terminal C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C unit of OAT-COPrg-7 was responsible for its inferior self-assembly ability in the melted state. This finding revealed that even a tiny structural difference could change the kinetic as well as the thermodynamic aspects of the molecular self-assembly process. In fact, Bouteiller had similarly shown that changing an alkyl to alkenyl substituent in bisurea derivatives could strongly affect the self-assembly structure, and had suggested that either steric repulsion between the alkenyl and alkyl groups or favourable solvation of the alkenyl group could be the reasons for the observed phenomenon.39
C unit of OAT-COPrg-7 was responsible for its inferior self-assembly ability in the melted state. This finding revealed that even a tiny structural difference could change the kinetic as well as the thermodynamic aspects of the molecular self-assembly process. In fact, Bouteiller had similarly shown that changing an alkyl to alkenyl substituent in bisurea derivatives could strongly affect the self-assembly structure, and had suggested that either steric repulsion between the alkenyl and alkyl groups or favourable solvation of the alkenyl group could be the reasons for the observed phenomenon.39
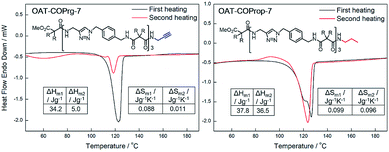 |
| | Fig. 8 DSC thermograms for xerogels of OAT-COPrg-7 and OAT-COProp-7 prepared from p-xylene (conc. = 2.5%). Black: initial samples, red: annealed samples. | |
Carboxylic acid end group.
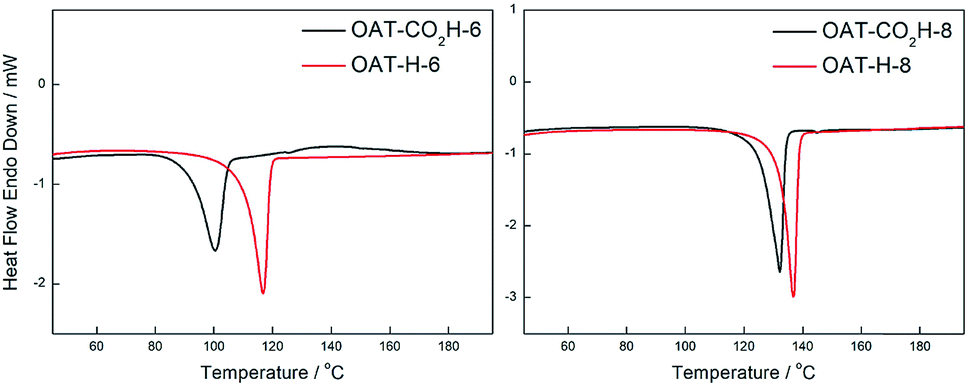
Comparing the DSC profiles of the initial and annealed samples bearing the carboxylic acid end group (OAT-CO2H-6 and OAT-CO2H-8), it was found that the Tm were different for the initial and annealed samples, while the ΔHm values remained essentially the same (Fig. 9 and Table 2). In order to figure out what happened during annealing, the annealed sample was subjected to 1H NMR analysis (ESI Fig. S17, pS45†). The 1H NMR solution spectra (CDCl3) of all the annealed OAT-CO2H-2n showed that one of the amide NHs was upfield shifted to ∼5.8 ppm as compared to that of the corresponding initial samples, which indicated that this NH was positioned in a weaker hydrogen-bonding environment after annealed once. Additionally, a new small peak showed up at ∼2.1 ppm. Based on this evidence, it was concluded that a decarboxylation had taken place during the annealing process, which resulted in the disappearance of intramolecular hydrogen-bonding interaction between the NH and carbonyl moiety of carboxylic acid end group. The new peak at 2.1 ppm should therefore be the signal of the newly generated CH. The chemical identity of the decarboxylated samples was further confirmed by mass spectral analysis (ESI Section 10†). Hence, all annealed OAT-CO2H-2n compounds were actually the corresponding OAT-H-2n derivatives.
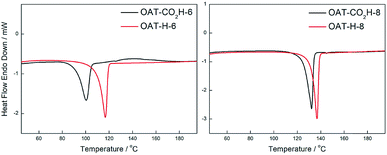 |
| | Fig. 9 DSC thermograms for xerogels of initial and annealed decarboxylated samples of OAT-CO2H-6 and OAT-CO2H-8 prepared from p-xylene (conc. = 2.5%). Black: initial samples, red: annealed decarboxylated samples. | |
Table 2 Melting temperature (Tm) and enthalpy change of melting (ΔHm) of xerogels of initiala and annealed decarboxylatedb samples of OAT-CO2H-2n
| First heating initial sample |
T
m (°C) |
ΔHm (J g−1) |
Second heating decarboxylated sample |
T
m (°C) |
ΔHm (J g−1) |
|
Xerogels were prepared from freeze drying of a p-xylene gel sample of gelator conc. = 25 mg mL−1.
Annealing was conducted at 200 °C for 5 min.
|
|
OAT-CO2H-6
|
100 |
21.0 |
OAT-H-6
|
117 |
20.3 |
|
OAT-CO2H-8
|
132 |
25.0 |
OAT-H-8
|
137 |
24.8 |
|
OAT-CO2H-10
|
149 |
29.9 |
OAT-H-10
|
149 |
27.2 |
|
OAT-CO2H-12
|
155 |
30.7 |
OAT-H-12
|
154 |
24.8 |
The fact that a decarboxylation reaction took place during annealing of the OAT-CO2H-2n derivatives prompted us to re-investigate the minimum temperature required for such a transformation. It was later determined that partial decarboxylation reaction happened only when a solution of OAT-CO2H-2n was heated to about 138 °C. Hence, all wet gel samples of OAT-CO2H-2n were prepared at below 120 °C in order to preserve their structural integrity.
Molecular self-sorting
The self-sorting studies were performed by DSC examinations since it is a simple and straight-forward method to reveal the thermodynamic properties of the pure and mixed samples. For two miscible compounds, only one broad endothermic peak will appear upon heating and the melting temperature will locate between those of the two pure samples. However, for a self-sorted binary system, two endothermic peaks will be observed.
Self-sorting between OATs with different change length.
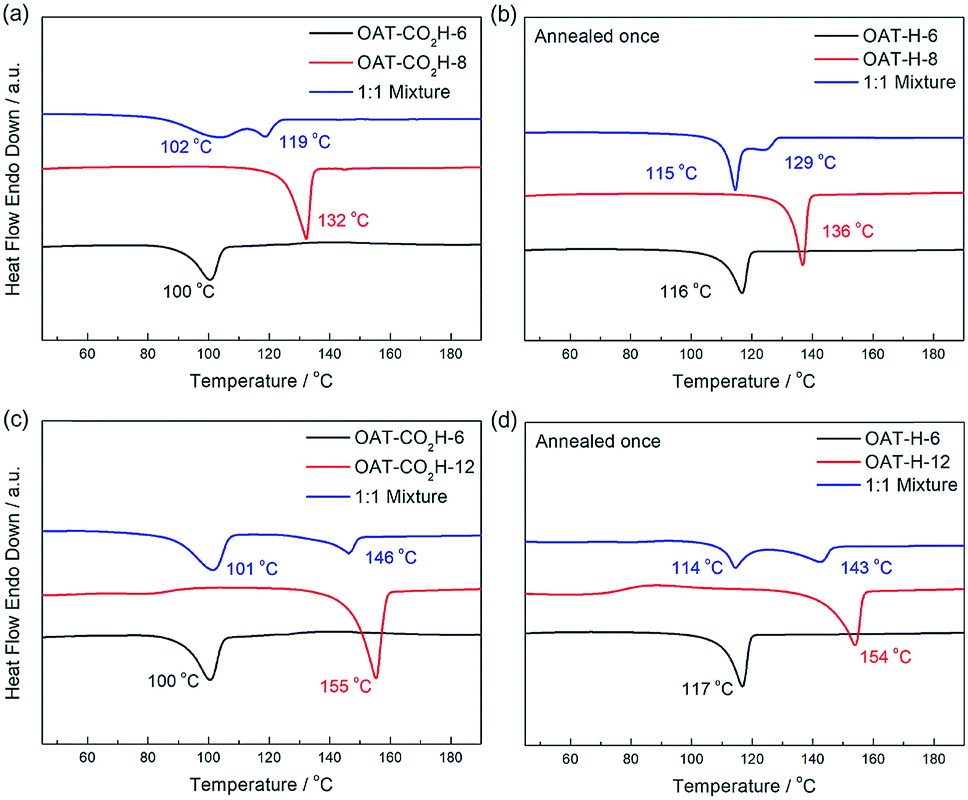
We first conducted self-sorting of organogels involving two-component binary system of oligomers of different change length (i.e. different number of CONH units). The DSC thermogram of a 1:1 mixed xerogel of OAT-CO2H-6 (1.25% w/v) and OAT-CO2H-8 (1.25% w/v) was compared to that of pure xerogel of OAT-CO2H-6 (2.5% w/v) and pure xerogel of OAT-CO2H-8 (2.5% w/v) (Fig. 10a). Two slightly overlapping peaks (102 °C and 119 °C) were identified in the mixed sample and each of them was in a similar position to that of the pure sample respectively. This indicated that some degree of self-sorting could be observed even when the difference of CONH units between the two OAT-CO2H-2n was just two. The slightly decrease of the two Tm values as compared to those of pure samples was probably due to the existence of eutectic component (i.e. not complete self-sorting). After annealing once to trigger the decarboxylation reaction, the resulting mixed sample of OAT-H-6 and OAT-H-8 also exhibited phase-separated self-sorting phenomenon in the melted state (Fig. 10b). Similar self-sortings were also observed for OAT-CO2H-6/OAT-CO2H-10 (ESI Fig. S12, pS40†), and OAT-CO2H-6/OAT-CO2H-12 (Fig. 10c). In these cases where the difference in the oligomer length becomes larger, peak separation in the mixed sample was more apparent, and the Tm values also get much closer to those of the pure components. The results indicate that when the difference in chain length becomes larger, the degree of self-sorting is also higher.
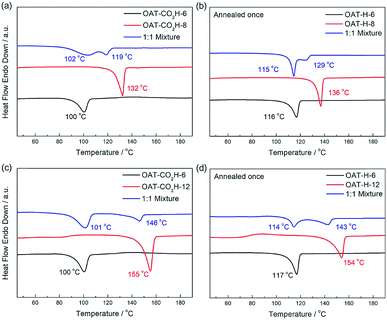 |
| | Fig. 10 (a) DSC thermograms of xerogels (2.5% w/v in p-xylene) of a 1:1 mixture of OAT-CO2H-6/OAT-CO2H-8 (blue), pure OAT-CO2H-8 (red), and pure OAT-CO2H-6 (black); (b) DSC thermograms of xerogels of a 1:1 mixture of OAT-H-6/OAT-H-8 (blue), pure OAT-H-8 (red), and pure OAT-H-6 (black); (c) DSC thermograms of xerogels (2.5% w/v in p-xylene) of a 1:1 mixture of OAT-CO2H-6/OAT-CO2H-12 (blue), pure OAT-CO2H-12 (red), and pure OAT-CO2H-6 (black); (d) DSC thermograms of xerogels of a 1:1 mixture of OAT-H-6/OAT-H-12 (blue), pure OAT-H-12 (red), and pure OAT-H-6 (black). | |
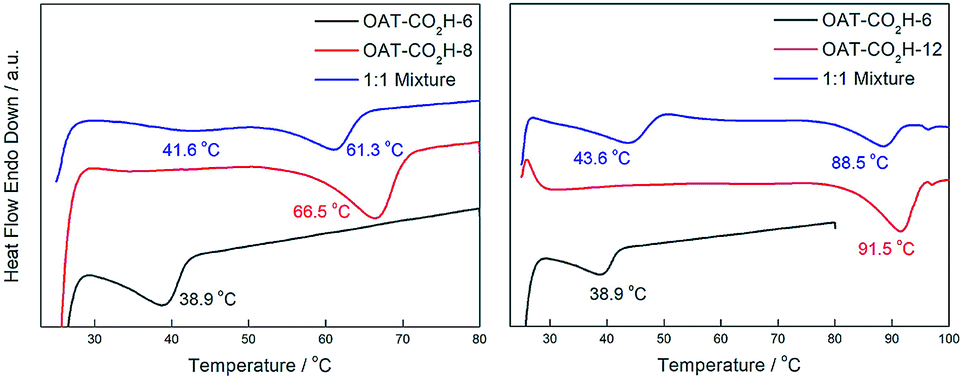
To confirm that the self-sorting actually happened during the gelation process but not due to the freeze-drying procedure, DSC experiments were also performed on the wet gels. We tested mixed wet gels (2.5% w/v in p-xylene) of OAT-CO2H-6/OAT-CO2H-8 and OAT-CO2H-6/OAT-CO2H-12 (Fig. 11). Due to a much lower gelator concentration, the signals were relatively weak. Nonetheless, two endothermic peaks in the mixed gels could still be observed, which proved that narcissistic self-sorting happened during the formation of nano-scale assemblies in wet gels. Again, the degree of self-sorting in the mixed gel OAT-CO2H-6/OAT-CO2H-12 was higher than that in OAT-CO2H-6/OAT-CO2H-8. Hence, the extent of self-sorting of the xerogels could actually reflect that of the wet gel systems.
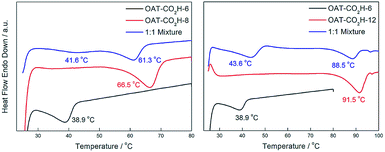 |
| | Fig. 11 Left: DSC thermograms of wet gels (2.5% w/v in p-xylene) of a 1:1 mixture of OAT-CO2H-6/OAT-CO2H-8 (blue), pure OAT-CO2H-8 (red) and pure OAT-CO2H-6 (black); right: DSC thermograms of wet gels (2.5% w/v in p-xylene) of a 1:1 mixture of OAT-CO2H-6/OAT-CO2H-12 (blue), pure OAT-CO2H-12 (red) and pure OAT-CO2H-6 (black). | |
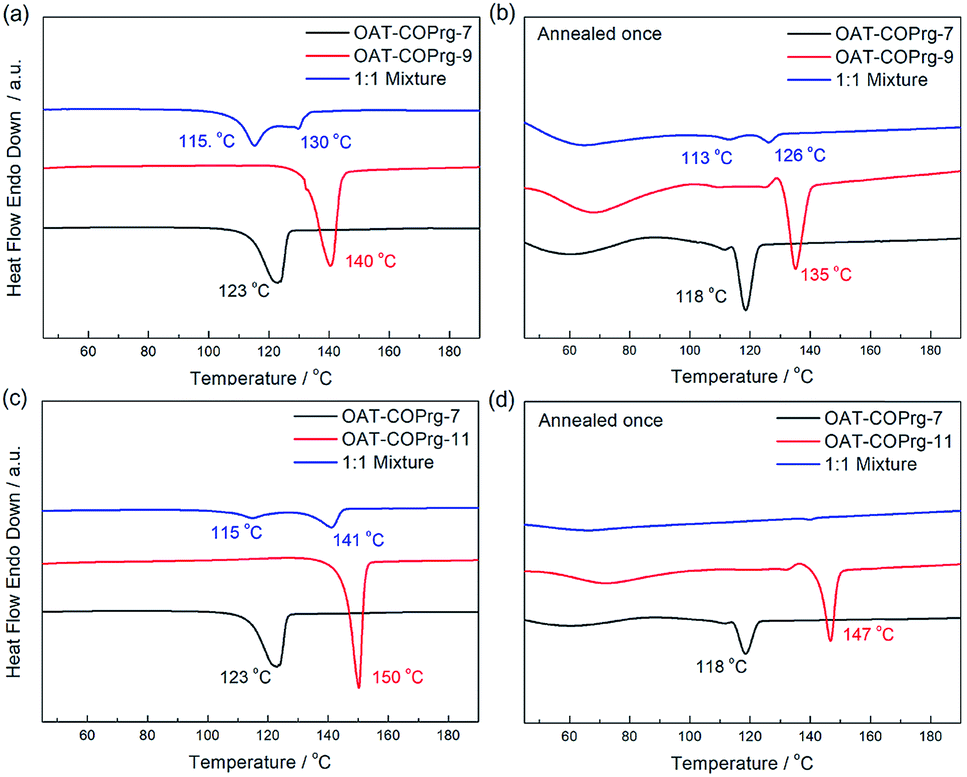
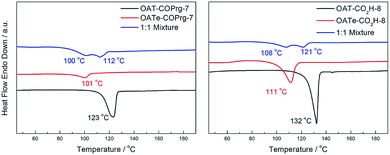
Self-sorting between oligomers of the OAT-COPrg-(2n+1) series was also investigated (Fig. 12). In the recorded DSC thermogram of a mixture of OAT-COPrg-7/OAT-COPrg-9, two peaks showed up which proved that when the difference of the number of CONH units between two OAT molecules was larger than one, self-sorting would be exhibited in the gelation process. Slightly phase separated phenomenon could also be observed in the annealed blends even though the peaks shrunk badly after the annealing procedure, implying that kinetic trapping in the melted state prevented the majority of the oligomers from self-assembling into a structurally order structure. Similarly, self-sorting was clearly observed with the mixed xerogel of OAT-COPrg-7/OAT-COPrg-11. However, no endotherm could be found in the annealed sample, suggesting that the two oligomers were kinetically trapped and could not form any crystalline zone upon cooling. It was believed that the higher oligomer OAT-COPrg-11 was completely devoid of kinetic freedom due to the chain entanglement in the melted state as a result of its longer chain structure.
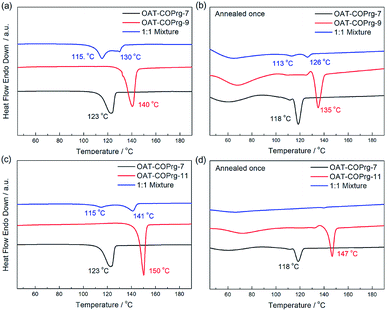 |
| | Fig. 12 DSC thermograms of xerogels (2.5% w/v in p-xylene): (a) 1:1 mixture of OAT-COPrg-7/OAT-COPrg-9 (blue), pure OAT-COPrg-9 (red), and pure OAT-COPrg-7 (black) with (b) annealed samples; (c) 1:1 mixture of OAT-COPrg-7/OAT-COPrg-11 (blue), pure OAT-COPrg-11 (red), and pure OAT-COPrg-7 (black) with (d) annealed samples. | |
On the other hand, when OAT-CO2H-6 and OAT-COPrg-7 were mixed together, only one peak appeared and the Tm was almost in the middle of those of two pure samples (ESI Fig. S13a, pS41†). This result indicated that when the difference of the number of CONH was only one, no self-sorting could be observed. Similar non-sorting was also observed for OAT-H-6 and OAT-COPrg-7 (ESI Fig. S13b, pS41†).
XRD measurement was also conducted on the self-sorted 1:1 mixed gel of OAT-CO2H-6/OAT-CO2H-12 (ESI Fig. S4, pS34†). The peak pattern of the mixture was very similar to that of OAT-CO2H-6 or that of OAT-CO2H-12, i.e. both the side chain interdigitation and π–π stacking peaks could be observed. However, it should be noted that XRD experiment could not differentiate a self-sorted or a randomly mixed system because both systems will give the same diffraction pattern. Since the oligomer chains are not bundled up in a regular fashion, hence the diffraction pattern due to a difference of oligomer chain length could not be detected. SEM analysis of the self-sorted 1:1 mixture of OAT-CO2H-6/OAT-CO2H-12 also could not identify any region showing microscopic self-sorting (ESI Fig. S16, pS44†).
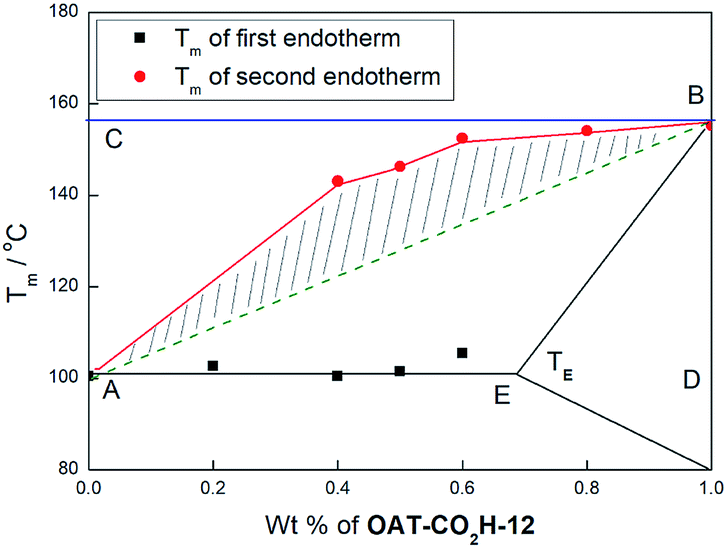
Semi-quantitative assessment of the degree of self-sorting.
To semi-quantitatively assess the degree of self-sorting in a binary mixed gel system, the Tm values at various ratios of the two components were recorded and the corresponding phase diagrams29 were constructed. Taking the phase diagram of OAT-CO2H-6/OAT-CO2H-12 xerogel as an example, the plotted eutectic phase diagram was consistent with the fact that the two oligomers exhibited limited solid solubility with each other (Fig. 13). The terminal point E was assigned to be the midpoint of the two compositions where the first endothermic peak last showed up and where it first disappeared. It was noticed that the eutectic temperature was very close to the Tm of OAT-CO2H-6 such that the terminal solid solution zone was too narrow to be represented on the phase diagram. As it turned out, at 0–20 weight% of OAT-CO2H-12, only one Tm value, corresponding to that of pure OAT-CO2H-6 was observed. This suggested well mixing of the two oligomers. On the other hand, two Tm values, the first corresponded to that of OAT-CO2H-6 and the second to OAT-CO2H-12, were found when the weight% of OAT-CO2H-12 was around 40–60%, indicating effective self-sorting in these compositions. Once the weight% of OAT-CO2H-12 reached more than 80%, again only one Tm value, corresponding to that of pure OAT-CO2H-12 was observed. This again indicated well mixing of the two oligomers. In theory, the green dash line (AB) represented the theoretical Tm at various compositions of a totally miscible mixture (i.e. no self-sorting) and the blue horizontal line (CB) represented the theoretical Tm of OAT-CO2H-12 in a completely immiscible mixture. Hence, the ratio (β) of the area of the shaded polygon to the area of the triangle defined by ABC can be used to represent the degree of self-sorting in the binary mixture (the rationale is given in ESI pS42†). In the case of the binary system OAT-CO2H-6/OAT-CO2H-12, the β value was 43%. The β values of other binary systems were similarly calculated from their corresponding phase diagrams (Table 3 and ESI Fig. S15, p43†). From the calculation results, it can be concluded that (i) when the difference of CONH number between two oligomers was larger, the self-sorting degree would be larger too, (ii) the carboxylic acid terminated oligomers exhibited slightly better self-sorting capability than the hydride terminated series, and (iii) the extent of self-sorting diminished as the lengths of the two oligomers became very large. Hence, clear self-sorting was observed in the case of OAT-CO2H-6/OAT-CO2H-8 (β = 26%), but no self-sorting was found in the case of OAT-CO2H-10/OAT-CO2H-12 (β = 0%), indicating that a levelling off effect was in operation once the number of amide units became very large. In other words, a difference of two amide units could induce good degree of self-sorting in the mixture involving two lower oligomers, but not quite enough for that of two longer ones.
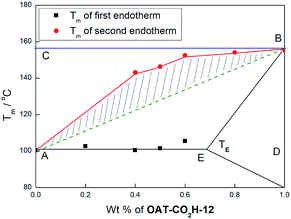 |
| | Fig. 13 Phase diagram of mixed xerogels of OAT-CO2H-6/OAT-CO2H-12 at different weight% of OAT-CO2H-12. | |
Table 3 Calculated β values of various oligomer blends
| Blend |
β
|
Blend |
β
|
|
OAT-CO2H-6/OAT-CO2H-8 |
26% |
OAT-H-6/OAT-H-8 |
6% |
|
OAT-CO2H-6/OAT-CO2H-12 |
43% |
OAT-H-6/OAT-H-12 |
30% |
|
OAT-CO2H-8/OAT-CO2H-12 |
33% |
OAT-H-8/OAT-H-12 |
8% |
|
OAT-CO2H-10/OAT-CO2H-12 |
0% |
OAT-H-10/OAT-H-12 |
0% |
Three component self-sorting system.
In order to test whether the length difference would still lead to a self-sorted system containing three components, a mixed xerogel comprising of a 1:1:1 weight ratio of OAT-CO2H-6, OAT-CO2H-8 and OAT-CO2H-12 was subjected to DSC analysis. Three slightly overlapped endothermic peaks appeared in the DSC trace (Fig. 14). This result indicated that a certain degree of self-sorting even in a three-component mixture. As expected, the ternary system involving OAT-H-6/OAT-H-8/OAT-H-12 showed slightly inferior self-sorting ability, although three peaks could be identified, but they overlapped to a higher extent.
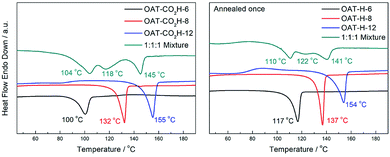 |
| | Fig. 14 Left: DSC thermograms of xerogels (2.5% w/v in p-xylene) prepared from a 1:1:1 mixture of OAT-CO2H-6/OAT-CO2H-8/OAT-CO2H-12 (green), pure OAT-CO2H-12 (blue), pure OAT-CO2H-8 (red), and pure OAT-CO2H-6 (black); right: DSC thermograms of a 1:1:1 mixture of OAT-H-6/OAT-H-8/OAT-H-12 (green), pure OAT-H-12 (blue), pure OAT-H-8 (red), and pure OAT-H-6 (black). | |
Self-sorting between OAT and OATe oligomers.
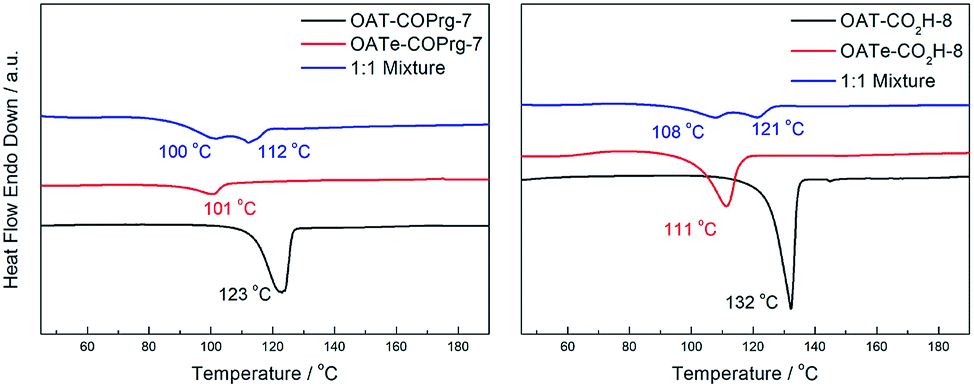
Having investigated how a difference in the number of CONH motifs would lead to and influence the self-sorting behaviour using OATs with different lengths, we continued to explore whether a difference in the density of interaction units would also contribute to self-sorting by comparing the OAT and OATe oligomers. It has already been shown that the OATe series of compounds possessed a slightly inferior gelation strength than that of the corresponding OAT oligomers. It was therefore envisaged that this stability difference could be the driving force for self-sorting. Indeed, a mixed xerogel of 1:1 OAT-COPrg-7/OATe-COPrg-7 revealed the presence of two slightly overlapping endothermic peaks (Fig. 15), indicating partial self-sorting of the system. Similarly, partial self-sorting between OAT-CO2H-8 and OATe-CO2H-8 was also noted in the mixed xerogel sample. Based on these results, it can be concluded that a difference in the density of interacting amide motifs could also lead to phase-separated assemblies during the gelation process.
 |
| | Fig. 15 Left: DSC thermograms of xerogels (2.5% w/v in p-xylene) prepared from a 1:1 mixture of OAT-COPrg-7/OAT-COPrg-7 (blue), pure OAT-COPrg-7 (red), and pure OATe-COPrg-7 (black); right: DSC thermograms of xerogels (2.5% w/v in p-xylene) prepared from a 1:1 mixture of OAT-CO2H-8/OATe-CO2H-8 (blue), pure OATe-CO2H-8 (red), and pure OAT-CO2H-8 (black). | |
Conclusions
Several homologous series of oligo(amide-triazole)s bearing various end groups, different number and density of CONH units were successfully prepared and characterized. They were able to form strong physical gels in various aromatic solvents. According to FTIR, SAXRD and SEM experiments, the gelation network was formed by hydrogen bonding, π–π stacking interactions and side chain interdigitation.
Both the number and density of CONH units were found to influence the gelation strength of the oligomers. In general, a larger number and higher density of CONH units could promote the formation of a stronger gelation network, but a levelling off effect was observed once the number became very large. Rather surprisingly, the end group was found to have a strong effect on the self-assembly in the melted state. Oligomers bearing a N-propargyl amide end group were found to be kinetically trapped and showed inferior kinetic mobility to self-assembly into order crystalline zones, while this was not seen in the saturated N-propyl amide terminated analogue.
The various oligomers were also found to exhibit different extent of self-sorting in both the wet gel and xerogel states depending on the nature of the end group, the difference in the number and density of the amide units of the constituted components. A difference of CONH number of two or higher was enough to show self-sorting behaviour both in two-component and three-component mixed systems. The plot of binary phase diagrams revealed that a larger difference in the CONH number would lead to a higher degree of self-sorting behaviour. At the same time, a difference in the density of interacting amide units could also lead to partial self-sorting behaviour.
To conclude, we demonstrated here that for structurally flexible, non-preorganized molecules, a slight change of the molecular structure would seriously affect their self-assembly structure and self-sorting phenomenon. Contrary to our initial presumption, narcissistic self-sorting does not just only prevail in mixtures making up of structurally very different molecules, but actually appears to be fairly general even in mixtures of structurally closely related homologs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Research Grants Council, HKSAR (Ref No. 14301114) and the Faculty of Science, CUHK (Account no. 3132670) for the financial support.
Notes and references
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- K. Osowska and O. Š. Miljanić, Synlett, 2011, 1643–1648 CAS.
- M. L. Saha and M. Schmittel, Org. Biomol. Chem., 2012, 10, 4651–4684 RSC.
- Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779–789 RSC.
- K. A. Jolliffe, P. Timmerman and D. N. Reinhoudt, Angew. Chem., Int. Ed., 1999, 38, 933–937 CrossRef CAS.
- Y. Ma, S. V. Kolotuchin and S. C. Zimmerman, J. Am. Chem. Soc., 2002, 124, 13757–13769 CrossRef CAS.
- A. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS PubMed.
- R. Krämer, J.-M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394–5398 CrossRef.
- M. Yoshizawa, M. Nagao, K. Kumazawa and M. Fujita, J. Organomet. Chem., 2005, 690, 5383–5388 CrossRef CAS.
- R. J. Sarma and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 377–380 CrossRef CAS.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- L. R. Holloway, P. M. Bogie and R. J. Hooley, Dalton Trans., 2017, 46, 14719–14723 RSC.
-
M. Schmittel and S. Saha, From Self-Sorting of Dynamic Metal-Ligand Motifs to (Supra)Molecular Machinery in Action, in Advances in Inorganic Chemistry, ed. R. van Eldik and R. Puchta, Elsevier Inc., London, UK, 2018, vol. 71, Supramolecular Chemistry, pp. 135–175 Search PubMed.
- B. Bilgiçer, X. Xing and K. Kumar, J. Am. Chem. Soc., 2001, 123, 11815–11816 CrossRef.
- L. Isaacs, Acc. Chem. Res., 2014, 47, 2052–2062 CrossRef CAS PubMed.
- X.-Q. Wang, W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Lett., 2015, 44, 1040–1046 CrossRef CAS.
- Q. Ji, R. C. Lirag and O. Š. Miljanić, Chem. Soc. Rev., 2014, 43, 1873–1884 RSC.
- A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC.
- A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC.
- F. Beuerle, S. Klotzbach and A. Dhara, Synlett, 2016, 27, 1133–1138 CrossRef CAS.
-
G. ten Brinke, Polym. Sci. A Compr. Ref., 2012, vol. 1, pp. 287–313 Search PubMed.
- For an example of social self-sorting of preorganized oligomers, see M. Li, K. Yamato, J. S. Ferguson and B. Gong, J. Am. Chem. Soc., 2006, 128, 12628–12629 CrossRef CAS PubMed.
- A. P. Bisson, F. J. Carver, D. S. Eggleston, R. C. Haltiwanger, C. A. Hunter, D. L. Livingstone, J. F. McCabe, C. Rotger and A. E. Rowan, J. Am. Chem. Soc., 2000, 122, 8856–8868 CrossRef CAS.
- For a review of self-sorting in polymers, see C. R. South, C. Burd and M. Weck, Acc. Chem. Res., 2007, 40, 63–74 CrossRef CAS PubMed.
- E. Wisse, L. E. Govaert, H. E. H. Meijer and E. W. Meijer, Macromolecules, 2006, 39, 7425–7432 CrossRef CAS.
- J. Kumaki, T. Kawauchi, K. Ute, T. Kitayama and E. Yashima, J. Am. Chem. Soc., 2008, 130, 6373–6380 CrossRef CAS PubMed.
- J. Zhang, H.-F. Chow, M.-C. Chan, G. K. W. Chow and D. Kuck, Chem.–Eur. J., 2013, 19, 15019–15025 CrossRef CAS PubMed.
- C.-F. Leung and H.-F. Chow, Chem.–Eur. J., 2017, 23, 4827–4836 CrossRef CAS PubMed.
- A. S. W. Chan and P. R. Sundararajan, J. Phys. Chem. B, 2016, 120, 9253–9263 CrossRef CAS PubMed.
- For a review on the self-sorting of low molecular weight multi-component gels, see L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- A. E. Stross, G. Iadevaia and C. A. Hunter, Chem. Sci., 2016, 7, 94–101 RSC.
- G. Iadevaia, A. E. Stross, A. Neumann and C. A. Hunter, Chem. Sci., 2016, 7, 1760–1767 RSC.
- A. E. Stross, G. Iadevaia and C. A. Hunter, Chem. Sci., 2016, 7, 5686–5691 RSC.
- D. Núñez-Villanueva and C. A. Hunter, Chem. Sci., 2017, 8, 206–213 RSC.
- See ESI for details.†.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- A. P. Bisson, F. J. Carver, D. S. Eggleston, R. C. Haltiwanger, C. A. Hunter, D. L. Livingstoe, J. F. McCabe, C. Rotger and A. E. Rowan, J. Am. Chem. Soc., 2000, 122, 8856–8868 CrossRef CAS.
- K.-N. Lau, H.-F. Chow, M.-C. Chan and K.-W. Wong, Angew. Chem., Int. Ed., 2008, 47, 6912–6916 CrossRef CAS PubMed.
- M. Roman, C. Cannizzo, T. Pinault, B. Isare, B. Andrioletti, P. van der Schoot and L. Bouteiller, J. Am. Chem. Soc., 2010, 132, 16818–16824 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, spectral data, SAXRD, and DSC thermograms. See DOI: 10.1039/c9sc00861f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Hak-Fun
Chow
and
Hak-Fun
Chow