
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly-chemoselective step-down reduction of carboxylic acids to aromatic hydrocarbons via palladium catalysis†
Chengwei
Liu
b,
Zhi-Xin
Qin
c,
Chong-Lei
Ji
c,
Xin
Hong
 *c and
Michal
Szostak
*ab
*c and
Michal
Szostak
*ab
aCollege of Chemistry and Chemical Engineering and Key Laboratory of Auxiliary Chemistry and Technology for Chemical Industry, Ministry of Education, Shaanxi University of Science and Technology, Xi'an 710021, China
bDepartment of Chemistry, Rutgers University, 73 Warren Street, Newark, NJ 07102, USA. E-mail: michal.szostak@rutgers.edu
cDepartment of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: hxchem@zju.edu.cn
First published on 29th April 2019
Abstract
Aryl carboxylic acids are among the most abundant substrates in chemical synthesis and represent a perfect example of a traceless directing group that is central to many processes in the preparation of pharmaceuticals, natural products and polymers. Herein, we describe a highly selective method for the direct step-down reduction of carboxylic acids to arenes, proceeding via well-defined Pd(0)/(II) catalytic cycle. The method shows a remarkably broad substrate scope, enabling to direct the classical acyl reduction towards selective decarbonylation by a redox-neutral mechanism. The utility of this reaction is highlighted in the direct defunctionalization of pharmaceuticals and natural products, and further emphasized in a range of traceless processes using removable carboxylic acids under mild, redox-neutral conditions orthogonal to protodecarboxylation. Extensive DFT computations were conducted to demonstrate preferred selectivity for the reversible oxidative addition and indicated that a versatile hydrogen atom transfer (HAT) pathway is operable.
Introduction
The reduction of carboxylic acid derivatives represents one of the most fundamental transformations in synthetic chemistry and catalysis.1 Reduction reactions of carboxylic acids and derivatives are traditionally performed using stoichiometric metal hydrides.2 However, these reagents suffer from major scope limitations and are inherently less safe than milder silane-based hydrides due to their pyrophoric nature.3 The reduction of carboxylic acid chlorides to aldehydes (acyl pathway) has been achieved by Rosenmund using Pd-catalysis4via an oxidative insertion/transmetallation/reductive elimination mechanism,5 thus establishing the classic cross-coupling tactics for the synthesis of aldehydes from carboxylic acids (Fig. 1A). More recently, a Ni-catalyzed step-down reduction of N-chelating amides directly to hydrocarbons has been achieved by Maiti and co-workers,6 while the Rueping group developed a selective methodology for the Ni-catalyzed step-down reduction of phenolic esters and N-acyl-glutarimides (Fig. 1B),7 developed earlier by our group.8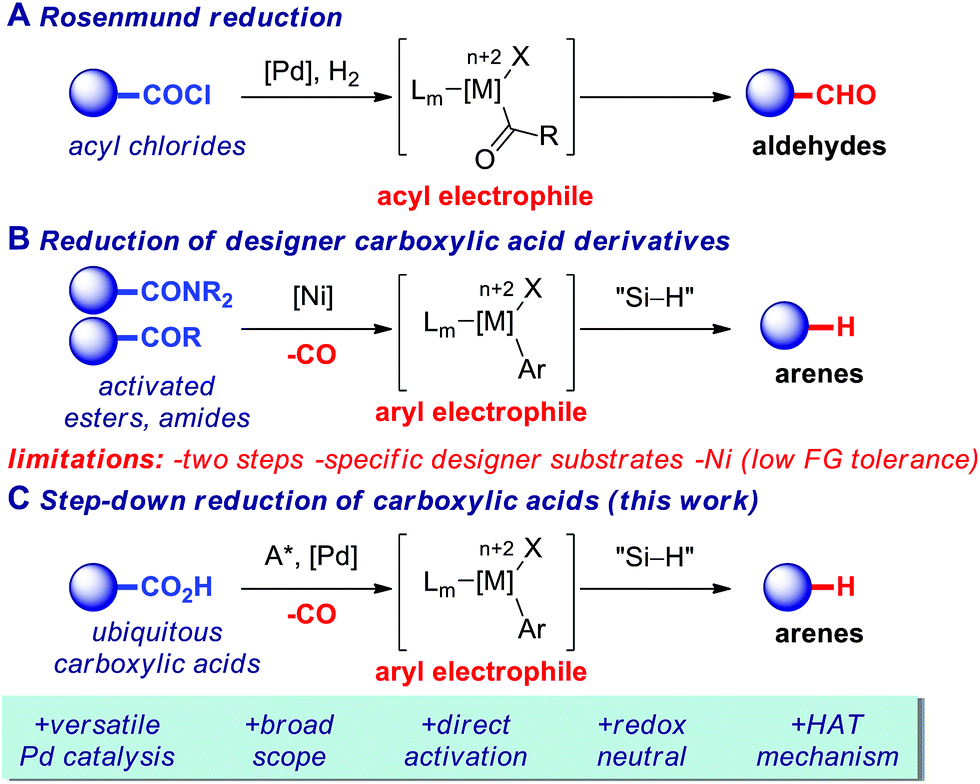 | ||
| Fig. 1 Transition-metal-catalyzed reduction of carboxylic acids. | ||
Herein, we report the first highly selective method for the direct step-down reduction of ubiquitous carboxylic acids to arenes (decarbonylative pathway), proceeding via well-defined Pd(0)/(II) cycle (Fig. 1C). The method supersedes the two-step methods using less general substrates and shows much broader reaction scope owing to the versatility of Pd-catalysis.5
The use of preformed carboxylic acid derivatives has been of choice to effect the direct reduction to hydrocarbons (Fig. 1B).6,7 Recognizing that these specifically-designed and less general N- and O-derivatives (pyrazoles, glutarimides, phenolic esters) are prepared from carboxylic acids in a separate step, we recently questioned whether a more straightforward approach engaging directly simple aromatic carboxylic acids could be realized in this important reaction class using versatile Pd-catalysis (Fig. 1C).
Specifically, we proposed that the direct reduction of carboxylic acids9,10 to hydrocarbons via a heretofore unknown redox-neutral, decarbonylative11–15 Pd(0)-catalyzed pathway could enable to use ubiquitous carboxylic acids as traceless activating groups, offering high level of predictability5a,b and functional group tolerance5e–g under redox-neutral conditions orthogonal to protodecarboxylation.16 Furthermore, the method would allow us for a more convenient approach to hydrocarbons that the reduction of designer N- and O-carboxylic acid derivatives, which are further limited by substrate scope.6,7 Finally, since aromatic carboxylic acids are commercially synthesized from the corresponding and cheaper toluenes, the method would establish Pd-promoted access to benzenes from feedstock toluenes, thus enabling to valorize crude oil as a mild alternative to toluene hydrodealkylation.17
Results and discussion
We initiated our studies by probing the direct reduction of electronically- and sterically-unbiased 4-phenyl benzoic acid as a model substrate. After extensive optimization (see ESI†), we found that the reduction of 4-Ph-benzoic acid in the presence of Pd(OAc)2 (1 mol%), dppb (2 mol%, dppb = 1,4-bis(diphenylphosphino)butane), piv2O (1.5 equiv., piv = 2,2-dimethylpropanoyl), Et3N (1.5 equiv.) and Et3SiH (1.5 equiv.) as a hydride source afforded the desired reduction product in 97% yield on gram scale, attesting to the scalability of the method. Under the optimized conditions reduction to the aldehyde or aldehyde derived products was not observed, consistent with high facility of catalytic system to trigger the reduction (cf. acyl pathway) under redox-neutral conditions.With optimal conditions in hand, we next focused on examining the scope of the decarbonylative reduction of carboxylic acids (Fig. 2A). We were delighted to find that the scope of the reaction is very broad and compatible with a variety of functional groups. As shown, unbiased as well as sterically-hindered aryl (2a–a′, 2c–f) and alkenyl benzoic (2b–b′) acid underwent smooth reduction. Substitution with electron-donating (2c–d) or electron-withdrawing (2e–f) groups was readily tolerated. Simple (2g–g′) and substituted naphthalenes (2h–i) were found to be competent substrates. Notably the reduction is not limited to conjugated arenes6 and can be applied to a broad array of simple benzoic acids bearing a plethora of functional groups poised for further manipulation, including unprotected hydroxy (2j–j′), ethers (2k–k′), amines (2l–l′), nitriles (2m–m′), esters (2n–n′), ketones (2o–o′), aldehydes (2p), sulfonyl (2q), acyl groups (2r–s), amides (2t–u), amines (2v–w), and halides (2x). It is noteworthy that a range of heterocycles, including quinolines (2y), indoles (2z–z′), pyridines (2aa), thiophenes (2ab), benzofurans (2ac) and benzothiophenes (2ad), as well as extremely sterically-hindered carboxylic acids (2af) proceeded in high yields and with exquisite selectivity for decarbonylation. Overall, the scope of the reaction shows a number of clear advantages over other methods.6,7,9–11,16
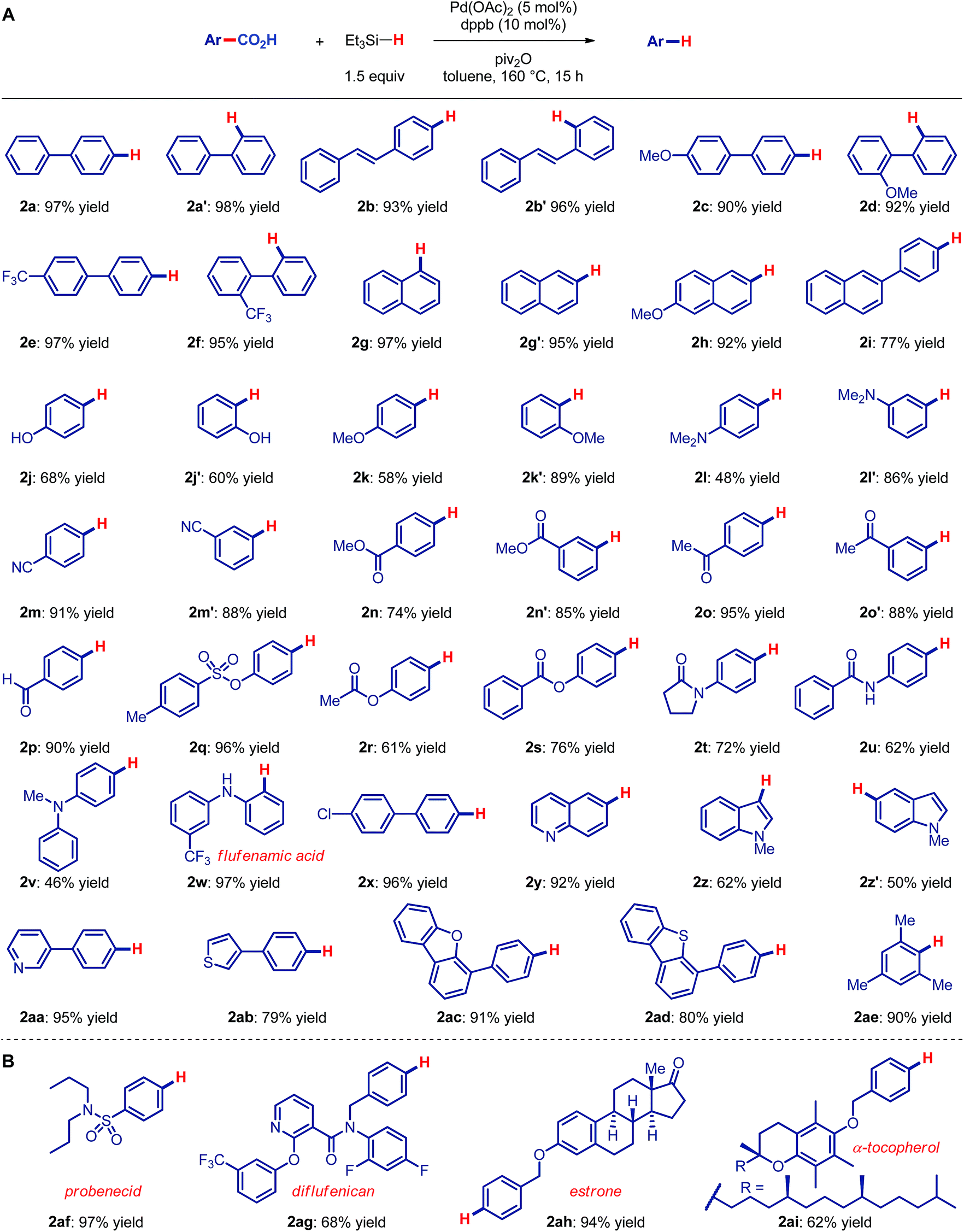 | ||
| Fig. 2 Scope of decarbonylative reduction of carboxylic acids. Conditions: carboxylic acid (1.0 equiv.), Pd(OAc)2 (5 mol%), dppb (10 mol%), Et3SiH (1.5 equiv.), piv2O (1.5 equiv.), toluene, 160 °C, 15 h. Dppb = 1,4-bis(diphenylphosphino)butane; piv = pivaloyl. See ESI for details.† | ||
To demonstrate the generality and potential impact of this new reduction method, we applied this protocol to late-stage derivatization of bioactive natural products and pharmaceuticals (Fig. 2B). We were delighted to find that decarboxylation of probenecid (2af) as well as of carboxylic acids derived from a fluorine-containing18 diflufenican (2ag), estrone (2ah) and tocopherol (2ai) afforded the decarbonylation products in high yields, underscoring the mild conditions and high potential impact of the present protocol.
As a further illustration of the synthetic utility we conducted a series of metal-catalyzed and metal-free reactions using carboxylic acid as a traceless directing group (Fig. 3A–D). We were pleased to find that Ru-catalyzed ortho-arylation directed by a carboxylic acid,19 electrophilic meta-iodination/Suzuki cross-coupling20 and electrophilic meta-iodination/Heck cross-coupling20 provide rapid access to a range of valuable products in high yields, thus signifying a clear appeal of this novel method to organic synthesis. Furthermore, the decarbonylative reduction of carboxylic acids establishes valuable access to benzenes from feedstock toluenes (Fig. 3D). New valorization methods of oil processing products are of high interest from the industrial and sustainability standpoints.17a,b
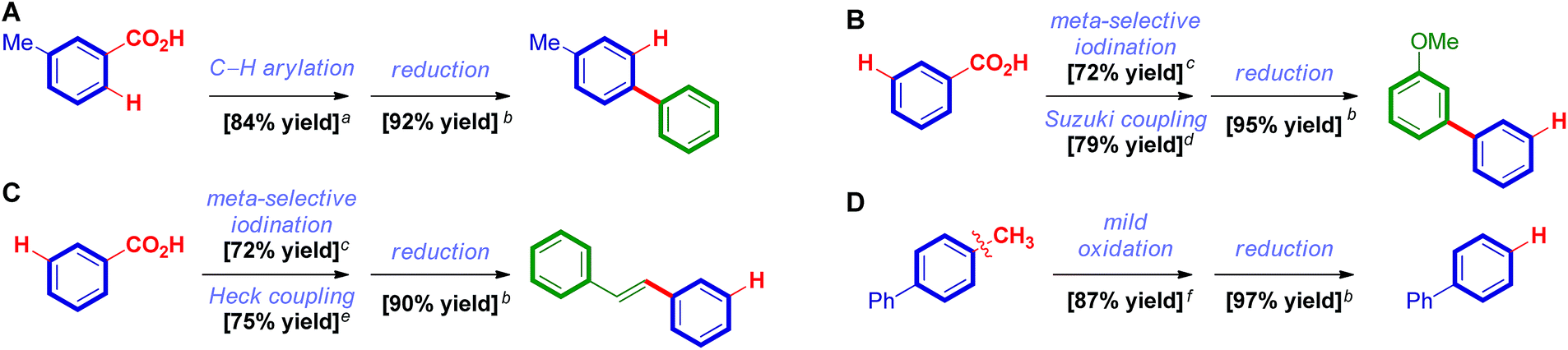 | ||
Fig. 3 Synthetic applications. Conditions: aPhI, [Ru(p-cym)Cl2]2, PCy3HBF4, K2CO3, NMP, 100 °C. bStandard conditions. cI2, NaIO4, H2SO4, 23 °C. d3-MeO-C6H4-B(OH)2, Pd2(dba)3, SPhos, K3PO4, PhMe, 100 °C. ePhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, Pd2(dba)3, Pt-Bu3HBF4, Et3N, dioxane, 23 °C. fKMnO4, Na2CO3, H2O, 120 °C. CH2, Pd2(dba)3, Pt-Bu3HBF4, Et3N, dioxane, 23 °C. fKMnO4, Na2CO3, H2O, 120 °C. | ||
Altogether, the broad scope of reactivity, tolerance to various sensitive functional groups and the potential to predictably use in functionalization of complex acids provide distinct advantages from other processes for removing carboxylic acid group6,7,9–11,16 and Bode well for future applications.
Although the scope of the reaction is very broad, several points should be noted: (1) typically, the yield can be improved by using small excess of Et3SiH (3.0 equiv.) and/or triethylamine as a base. For example, the yield of 2l can be improved to 67% under these conditions. (2) In general, the reduction of electron-rich aromatics is less efficient than that of electron-deficient counterparts (vide infra). (3) Direct reduction of electro-rich five-membered heterocycles is feasible; for example, the reduction of 5-chlorothiophene-2-carboxylic acid proceeds in 91% yield. (4) Multiple reductions are feasible; for example, reduction of [1,1′-biphenyl]-3,3′,4,4′-tetracarboxylic acid (bis-phthalic acid) proceeds in 81% yield. (5) Halides on the carboxylic acid containing ring are possible. (6) We typically did not observe side reactions (e.g. aldehyde formation, reduction to the alcohol, deoxygenation). Studies on further expansion of the substrate scope are underway.
Extensive computational studies were conducted to gain insight into the reaction mechanism and elucidate the controlling factors of selectivity. Recent advances in computational organometallic catalysis make this approach appealing to design more efficient catalytic systems and predict selectivity of bond activation events.21–24 The computed free energy profile of the catalytic cycle of decarbonylative reduction is shown in Fig. 4A. The C–O bond activation of benzoic pivalic anhydride proceeds viaTS4, generating the acylpalladium intermediate 5. Subsequent decarbonylation through TS6 leads to the penta-coordinated arylpalladium intermediate 7, and CO then dissociates to produce the LPd(aryl)(OPiv) intermediate 8. From 8, the model silane (TMSH) coordinates, and a subsequent hydrogen atom transfer (HAT) occurs viaTS10 to produce the reduced arene and the LPd(silyl)(OPiv) intermediate 12. 12 undergoes a Si–O reductive elimination to regenerate the active Pd(0) catalyst for the next catalytic cycle. We were not able to locate the transition states for palladium-hydride formation despite extensive efforts. Based on the free energy changes of the overall catalytic cycle, the acylpalladium intermediate 5 is the on-cycle resting state, and the HAT step viaTS10 is the rate-limiting step with an overall barrier of 31.8 kcal mol−1.
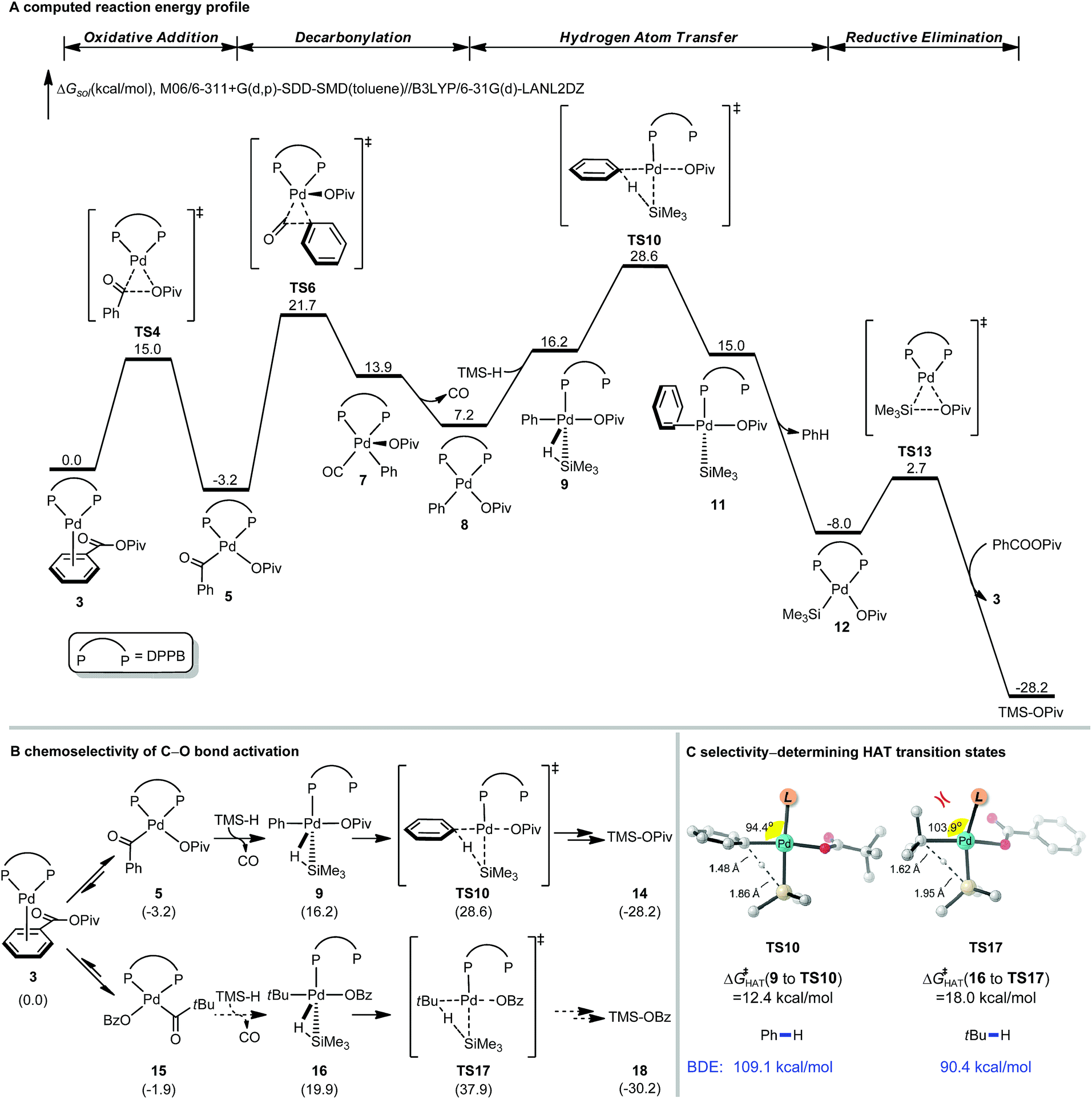 | ||
| Fig. 4 DFT-calculated reaction energy profile and chemoselectivity of C–O bond activation of [Pd(dppb)]-catalyzed decarbonylative reduction of benzoic pivalic anhydride. See ESI for computational details.† | ||
The chemoselectivity of C–O bond activation is determined by the substituent of anhydride. Fig. 4B shows the free energies of the key intermediates and HAT transition states of the competing C–O bond activation pathways. The HAT step determines the overall catalytic efficiency and differentiates the competing pathways by 9.3 kcal mol−1 (TS10vs.TS17). This computed selectivity is consistent with the experimental observations that the C–O bond activation only occurs on the benzoic acid. Detailed free energy changes of the reduction pathway involving C–O bond activation of pivalic acid are included in the ESI (Fig. S1†). Two factors contribute to this chemoselectivity. First, the steric repulsions between the bulky tBu group and dppb ligand disfavor the pivalic acid C–O bond activation pathway. The highlighted angles in the HAT transition states reflect these steric effects; the phosphine ligand in TS17 is significantly bent away from tBu group (Fig. 4C). In addition, the phenyl group is intrinsically a better hydrogen atom acceptor comparing with tBu group based on the bond dissociation energies (Ph–H: 109.1 kcal mol−1, tBu–H: 90.4 kcal mol−1, Fig. 4C). This leads to the differences of intrinsic HAT barriers (12.4 kcal mol−1viaTS10, 18.0 kcal mol−1viaTS17, Fig. 4C), which further increases the chemoselectivity.
Additional studies were conducted to gain insight into the reaction mechanism (see ESI, Fig. S2–S7†). (1) To investigate whether benzoic pivalic anhydride was a possible reaction intermediate, 4-Ph-benzoic pivalic anhydride was prepared and subjected to the reaction conditions. Formation of product 2a was observed (87% yield). Moreover, 4-Ph-benzoic acetic anhydride served as a competent intermediate (83% yield). (2) To investigate electronic effect on the decarbonylative step-down reduction, a Hammett correlation study employing differently substituted 4-Ar-benzoic acids was conducted. The study showed a large positive ρ-value of 1.57 (R2 = 0.99), which can be compared with the ρ+-value of 0.94 (R2 = 0.92) using Hammett–Brown σ+-constants, suggesting that electron-deficient arenes are inherently more reactive substrates, consistent with facility of metal insertion and decarbonylation. (3) To investigate steric effect on the decarbonylative step-down reduction, intermolecular competition experiments between differently substituted carboxylic acids were conducted, revealing that sterically-hindered carboxylic acids react preferentially, consistent with decarbonylation favored by steric demand of acylpalladium complexes. (4) To gain additional insight, relative reactivity studies regarding the use of biaryls, conjugated arenes and conjugated vinyl-arenes were conducted. The experiments revealed the following order of reactivity: biaryl = Np > vinyl-Ar. (5) To investigate the reduction selectivity, experiments at lower temperatures were conducted. The formation of aldehyde reduction products was not detected. (6) To investigate the effect of low catalytic loading, the reduction was conducted at 0.10 mol% of Pd(OAc)2. Formation of product 2a from 1a was observed in high yield (82% yield), consistent with the high efficiency of the reduction. Note that the reduction at low loading is also possible using Ac2O (51% yield), albeit with a decreased reaction efficiency. The beneficial effect of piv2O vs. Ac2O is consistent with the role of steric repulsion of the t-Bu group shutting down the alternative C–O cleavage pathway.12h Studies are currently in progress to investigate the mechanistic details of the decarbonylative cross-coupling manifold of carboxylic acids.
Finally, additional points regarding the impact and utility are in order. The facile reduction of carboxylic acids via a redox-neutral pathway should be benchmarked against the known methods for the reduction of carboxylic acid derivatives using Ni6,7 and the known methods via protodecarboxylation mechanism.16 (1) The benefits of the direct use of carboxylic acids cf. designer analogues are clear. (2) Furthermore, the broad scope of the reaction and the orthogonal mechanism for removing the carboxylic acid group expand the utility of carboxylic acids as directing groups in organic synthesis. (3) Perhaps most importantly, the transformation encompasses a general manifold for decarbonylative redox-neutral cross-coupling of ubiquitous carboxylic acids via a unified mechanism that provides a range of new compelling methods for manipulation of this privileged functional group.25
Conclusions
In conclusion, we have reported the first method for a direct reduction of carboxylic acids to arenes via well-defined redox-neutral decarbonylative Pd(0)/(II) catalytic cycle. The reaction provides a number of practical advantages for the construction of arenes over recently established two-step methods using designer derivatives of ubiquitous carboxylic acids. The reaction conditions are mild and tolerate a remarkably broad range of functional groups. The practical value of this transformation is evident from the potential to manipulate densely-functionalized substrates, including bioactive natural products and pharmaceuticals as well as in traceless reaction sequences. Detailed DFT study of the reaction mechanism has provided insight into the selectivity of bond activation events and elucidated steric requirements for a direct activation of carboxylic acids via a decarbonylative pathway. Studies towards expanding the scope of decarbonylative transformations of carboxylic acids and related substrates are underway and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Rutgers University (M. S.), the NSF (CAREER CHE-1650766, M. S.), NSFC (21702182 and 21873081, X. H.), the Chinese “Thousand Youth Talents Plan” (X. H.), and Zhejiang University (X. H.) for generous financial support. The Bruker 500 MHz spectrometer used in this study was supported by the NSF-MRI grant (CHE-1229030). Calculations were performed on the high-performance computing system at the Department of Chemistry, Zhejiang University.Notes and references
- For reviews, see: (a) D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004 CrossRef CAS PubMed; (b) S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289 CrossRef CAS; (c) P. G. Andersson and I. J. Munslow, Modern Reduction Methods, Wiley, 2008 CrossRef; (d) M. Hudlicky, Reductions in Organic Chemistry, Ellis Horwood, 1984 Search PubMed.
- (a) M. B. Smith and J. March, Advanced Organic Chemistry, Wiley, 2007 Search PubMed; (b) B. M. Trost and I. Fleming, Comprehensive Organic Synthesis, Pergamon Press, 1991 Search PubMed.
- A. F. Abdel-Magid, Reductions in Orgnaic Synthesis, ACS Symposium Series, 1996 Search PubMed.
- (a) K. W. Rosenmund, Chem. Ber., 1918, 51, 585 CrossRef CAS; (b) E. Mosettig and R. Mozingo, The Rosenmund Reduction of Acid Chlorides to Aldehydes, in Organic Reactions, R. Adams, John Wiley and Sons, 1948 Search PubMed; (c) J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156 CrossRef CAS; (d) A. V. Iosub, C. J. Wallentin and J. Bergman, Nat. Catal., 2018, 1, 645 CrossRef.
- (a) R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley, 2005 Search PubMed; (b) J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, 2010 Search PubMed; (c) Metal-Catalyzed Cross-Coupling Reactions and More, A. de Meijere, S. Bräse and M. Oestreich, Wiley, 2014 Search PubMed; (d) Science of Synthesis: Cross-Coupling and Heck-Type Reactions, G. A. Molander, J. P. Wolfe and M. Larhed, Thieme, 2013 Search PubMed; (e) T. Colacot, New Trends in Cross-Coupling: Theory and Applications, RSC, 2015 Search PubMed; (f) X. F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047 CrossRef CAS PubMed; (g) C. C. C. Johansson-Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS PubMed.
- A. Dey, S. Sasmai, K. Seth, G. K. Lahiri and D. Maiti, ACS Catal., 2017, 7, 433 CrossRef CAS.
- H. Yue, L. Guo, S. C. Lee, X. Liu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 3972 CrossRef CAS PubMed.
- For reviews on N-acyl-glutarimides, see: (a) S. Shi, S. P. Nolan and M. Szostak, Acc. Chem. Res., 2018, 51, 2589 CrossRef CAS PubMed; (b) G. Meng and M. Szostak, Eur. J. Org. Chem., 2018, 20–21, 2352 CrossRef.
- For a leading review on the use of carboxylic acids as substrates in catalysis, see: L. J. Gooßen, N. Rodriguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef PubMed.
- For additional reviews, see: (a) N. Rodriguez and L. J. Gooßen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (b) W. Dzik, P. Lange and L. Gooßen, Chem. Sci., 2012, 3, 2671 RSC.
- For recent reviews on decarbonylative cross-couplings, see: (a) C. Liu and M. Szostak, Org. Biomol. Chem., 2018, 16, 7998 RSC; (b) L. Guo and M. Rueping, Acc. Chem. Res., 2018, 51, 1185 CrossRef CAS PubMed; (c) A. Dermenci and G. Dong, Sci. China: Chem., 2013, 56, 685 CrossRef CAS; (d) J. B. Johnson and T. Rovis, Acc. Chem. Res., 2008, 41, 327 CrossRef CAS PubMed; for selected mechanistic studies on decarbonylation, see: (e) P. Fristrup, M. Kreis, A. Palmelund, P. O. Norrby and R. Madsen, J. Am. Chem. Soc., 2008, 130, 5206 CrossRef CAS PubMed; (f) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed; (g) K. J. Cavell, Coord. Chem. Rev., 1996, 155, 209 CrossRef CAS.
- For selected decarbonylative cross-couplings from our groups, see: (a) G. Meng and M. Szostak, Angew. Chem., Int. Ed., 2015, 54, 14518 CrossRef CAS PubMed; (b) S. Shi, G. Meng and M. Szostak, Angew. Chem., Int. Ed., 2016, 55, 6959 CrossRef CAS PubMed; (c) G. Meng and M. Szostak, Org. Lett., 2016, 18, 796 CrossRef CAS PubMed; (d) C. Liu, G. Meng and M. Szostak, J. Org. Chem., 2016, 81, 12023 CrossRef CAS PubMed; (e) S. Shi and M. Szostak, Org. Lett., 2017, 19, 3095 CrossRef CAS PubMed; (f) G. Meng and M. Szostak, ACS Catal., 2017, 7, 7251 CrossRef CAS; (g) C. L. Ji and X. Hong, J. Am. Chem. Soc., 2017, 139, 15522 CrossRef CAS PubMed; (h) C. Liu, C. L. Ji, X. Hong and M. Szostak, Angew. Chem., Int. Ed., 2018, 57, 16721 CrossRef CAS PubMed.
- For selected recent examples of decarbonylative and decarboxylative cross-coupling reactions, see: (a) S. T. Keaveney and F. Schoenebeck, Angew. Chem., Int. Ed., 2018, 57, 4073 CrossRef CAS PubMed; (b) A. Chatupheeraphat, H. H. Liao, W. Srimontree, L. Guo, Y. Minenkov, A. Poater, L. Cavallo and M. Rueping, J. Am. Chem. Soc., 2018, 140, 3724 CrossRef CAS PubMed; (c) A. N. Desnoyer, F. W. Friese, W. Chiu, M. W. Drover, P. O. Patrick and J. A. Love, Chem.–Eur. J., 2016, 22, 4070 CrossRef CAS PubMed; (d) A. N. Desnoyer and J. A. Love, Chem. Soc. Rev., 2017, 46, 197 RSC; (e) K. Muto, J. Yamaguchi, D. G. Musaev and K. Itami, Nat. Commun., 2015, 6, 7508 CrossRef PubMed; (f) C. Liu and M. Szostak, Chem. Commun., 2018, 54, 2130 RSC; (g) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed; (h) D. C. Behenna, Y. Liu, T. Yurino, J. Kim, D. E. White, S. C. Virgil and B. M. Stoltz, Nat. Chem., 2012, 4, 130 CrossRef CAS PubMed; (i) D. V. Gribkov, S. J. Pastine, M. Schnürch and D. Sames, J. Am. Chem. Soc., 2007, 129, 11750 CrossRef CAS PubMed.
- For further recent examples demonstrating the utility of decarbonylative manifold, see: (a) S. K. Murphy, J. W. Park, F. A. Cruz and V. M. Dong, Science, 2015, 347, 56 CrossRef CAS PubMed; (b) J. Hu, Y. Zhao, J. Liu, Y. Zhang and Z. Shi, Angew. Chem., Int. Ed., 2016, 55, 8718 CrossRef CAS PubMed; (c) X. Fang, B. Cacherat and B. Morandi, Nat. Chem., 2017, 9, 1105 CrossRef CAS PubMed; (d) M. De La Higuera Macias and B. A. Arndtsen, J. Am. Chem. Soc., 2018, 140, 10140 CrossRef CAS PubMed; (e) Y. H. Lee and B. Morandi, Nat. Chem., 2018, 10, 1016 CrossRef CAS PubMed; (f) C. A. Malapit, J. R. Bour, C. E. Brigham and M. S. Sanford, Nature, 2018, 563, 100 CrossRef CAS PubMed.
- (a) For a recent review on reactions of thioesters, including decarbonylations, see: V. Hirschbeck, P. H. Gehrtz and I. Fleischer, Chem.–Eur. J., 2018, 24, 7092 CrossRef CAS PubMed; for a review on Fukuyama reduction, see: (b) T. Fukuyama and H. Tokuyama, Aldrichimica Acta, 2004, 37, 87 CAS.
- (a) For a review, see: J. Schwarz and B. König, Green Chem., 2018, 20, 323 RSC and references cited therein; (b) For additional examples, see: ref. 9 and 10 and references cited therein; For reviews on acyl-metal intermediates, see: ; (c) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef; (d) A. Zapf, Angew. Chem., Int. Ed., 2003, 42, 5394 CrossRef CAS PubMed; (e) See, ref. 9, 8a and references cited therein.
- (a) H. A. Wittcoff, B. G. Reuben and J. S. Plotkin, Chemicals from Toluene, in Industrial Organic Chemicals, H. A. Wittcoff, B. G. Reuben and J. S. Plotkin, Wiley, 2013 Search PubMed; (b) A. Ouattara, C. Pibouleau, C. Azzaro-Pantel and S. Domenech, Energy Convers. Manage., 2013, 74, 129 CrossRef CAS; (c) M. Beller and H. U. Blaser, Organometallics as Catalysts in the Fine Chemical Industry, Springer, 2012 CrossRef.
- For selected recent reviews, see: (a) C. N. Neumann and T. Ritter, Acc. Chem. Res., 2017, 50, 2822 CrossRef CAS; (b) K. N. Lee, J. W. Lee and M. Y. Ngai, Tetrahedron, 2018, 74, 7127 CrossRef CAS PubMed.
- L. Huang and D. J. Weix, Org. Lett., 2016, 18, 5432 CrossRef CAS PubMed.
- J. Mortier, Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, Wiley, 2016 Search PubMed.
- (a) M. Garcia-Melchor, A. A. C. Braga, A. Lledos, G. Ujaque and F. Maseras, Acc. Chem. Res., 2013, 46, 2626 CrossRef CAS PubMed; (b) A. Fromm, C. van Wüllen, D. Hackenberger and L. J. Gooßen, J. Am. Chem. Soc., 2014, 136, 10007 CrossRef CAS PubMed.
- For selected computational studies of oxidative addition with palladium catalysis, see: (a) L. J. Gooßen, D. Koley, H. Hermann and W. Thiel, Organometallics, 2005, 24, 2398 CrossRef; (b) M. Ahlquist, P. Fristrup, D. Tanner and P. O. Norrby, Organometallics, 2006, 25, 2066 CrossRef CAS; (c) M. Ahlquist and P. O. Norrby, Organometallics, 2007, 26, 550 CrossRef CAS; (d) K. C. Lam, T. B. Marder and Z. Lin, Organometallics, 2007, 26, 758 CrossRef CAS; (e) T. E. Barder, M. R. Biscoe and S. L. Buchwald, Organometallics, 2007, 26, 2183 CrossRef CAS; (f) F. Schoenebeck and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 2496 CrossRef CAS PubMed.
- For a computational study of decarbonylation with palladium catalysis, see: M. Lesslie, Y. Yang, A. J. Canty, E. Piacentino, F. Berthias, P. Maitre, V. Ryzhov and R. A. J. O′Hair, Chem. Commun., 2018, 54, 346 RSC.
- For a computational study of reductive elimination with palladium catalysis, see: M. Pérez-Rodríguez, A. A. C. Braga, M. Garcia-Melchor, M. H. Pérez-Temprano, J. A. Casares, G. Ujaque, A. R. de Lera, R. Álvarez, F. Maseras and P. Espinet, J. Am. Chem. Soc., 2009, 131, 3650 CrossRef PubMed.
- Q. Zhao and M. Szostak, ChemSusChem, 2019, 12 DOI:10.1002/cssc.201900408.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. See DOI: 10.1039/c9sc00892f |
| This journal is © The Royal Society of Chemistry 2019 |