
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNucleophile-intercepted Beckmann fragmentation reactions†
Samuel J.
Touchette
,
Evan M.
Dunkley
,
Leah L.
Lowder
and
Jimmy
Wu
 *
*
Department of Chemistry, Dartmouth College, Hanover, New Hampshire 03755, USA. E-mail: jimmy.wu@dartmouth.edu
First published on 4th July 2019
Abstract
We describe the first examples of nucleophile-intercepted Beckmann fragmentations of indoline oximes. This reaction uses MsCl as a promoter to give cyano chlorides and is believed to proceed through an aziridinium intermediate via a double stereoinvertive process. Mechanistic insights have led to the further discovery that oxygen, nitrogen, and bromide nucleophiles can be employed for this fragmentation by the use of other promoters. We envision that these products may be useful in the syntheses of members of the akuammiline and koumine families of indoline alkaloids.
Introduction
First reported in 1886, the classic Beckmann rearrangement converts ketoximes to the corresponding amides (Fig. 1).1 In the intervening time, numerous variants of this reaction have been reported and can be broadly classified into two categories: (1) trapping of the intermediate nitrilium ions and/or imidates with nucleophiles other than water, and (2) fragmentation to give nitriles.2 These fragmentation reactions nearly always involve Grob-like elimination processes to give an unsaturated component, such as an alkene or carbonyl, concomitant with scission of a C–C bond. Furthermore, aldoximes routinely undergo direct E2 elimination at the oxime to give nitriles.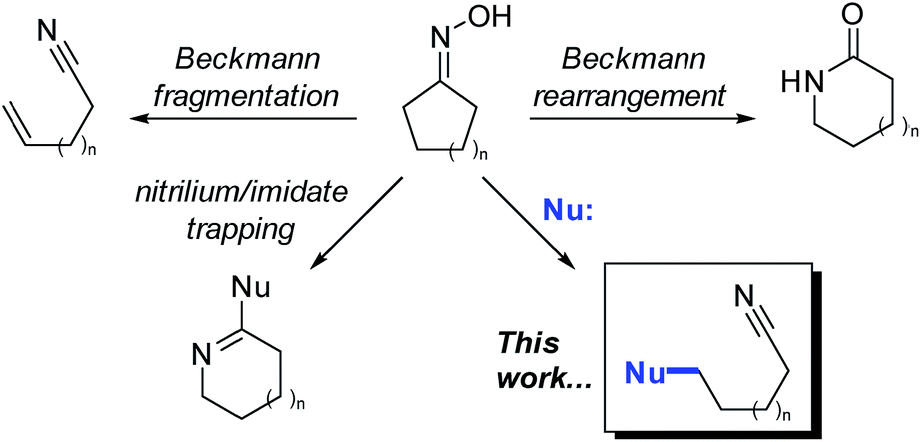 | ||
| Fig. 1 Major classes of variations on the Beckmann rearrangement. | ||
In contrast, Beckmann fragmentation reactions of ketoximes that circumvent the elimination step, and instead proceed by nucleophilic attack, are exceedingly rare (Fig. 1; this work). The few reported instances of nucleophile-intercepted Beckmann fragmentations are typically minor components of undesired side-reactions and constrained to highly specialized substrates.3–9 Despite the scarcity of precedents, we envisioned that a broadly general variant of such a transformation could potentially serve as a powerful tool in the preparation of densely-functionalized indoline systems. Herein, we report the first instance of nucleophile-intercepted Beckmann fragmentations of indolinyl oximes using a diverse set of nucleophiles to generate cyclohexa[b]indolines featuring four contiguous stereocenters (eqn (1)). We anticipate that rapid and efficient access to these compounds may be useful in the synthesis of members of the akuammiline10–12 and koumine13–16 families of natural products (Fig. 2). These are important classes of bioactive indoline alkaloids of which there are several dozen members.17–20
 | ||
| Fig. 2 Nucleophile-intercepted Beckmann fragmentation reactions of indolinyl oximes and representative members of the akuammiline and koumine alkaloids. | ||
Our research group has been interested in the development of novel reactions involving indoles and related heterocycles for some time.21–28 In this context, we have previously reported the (3 + 2) annulation between 3-substituted indoles and various oxyallyl cations generated from α-haloketones to give bicycloketone 3 (Scheme 1).23 In the following work, we converted 3 to the corresponding oxime with NH2OH·HCl. Upon treatment with MsCl, none of the bicyclic lactam regioisomers resulting from prototypical Beckmann rearrangement was obtained. Instead, we were surprised to isolate cyano chloride 4a in 52% yield along with 17% of its regioisomer 5a, both of whose gross structures and relative stereochemical relationships were assigned based on X-ray crystallography and extensive NMR spectroscopy. No epimeric compounds were observed.
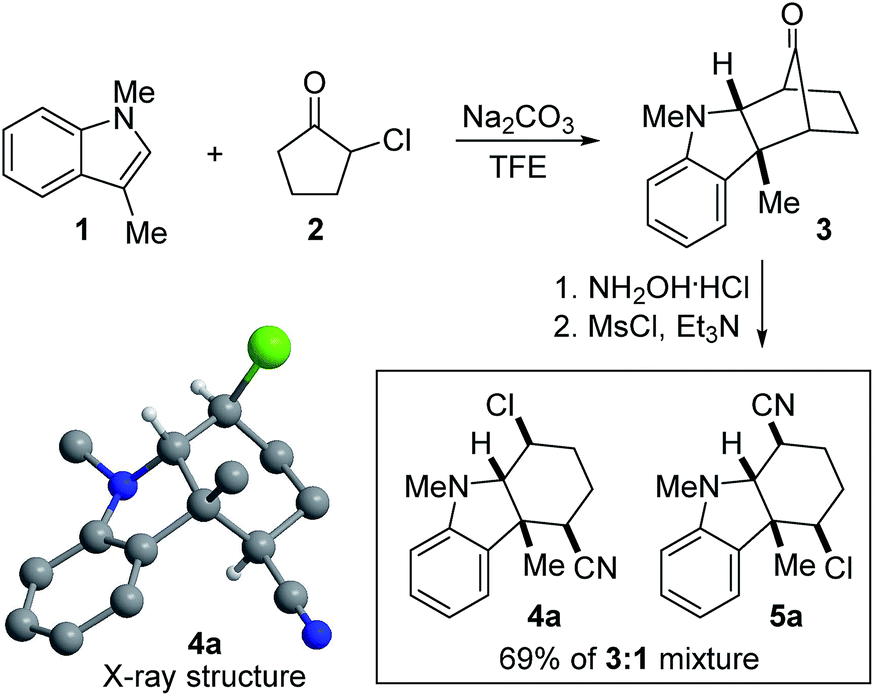 | ||
| Scheme 1 Discovery of the nucleophile-intercepted Beckmann fragmentation. | ||
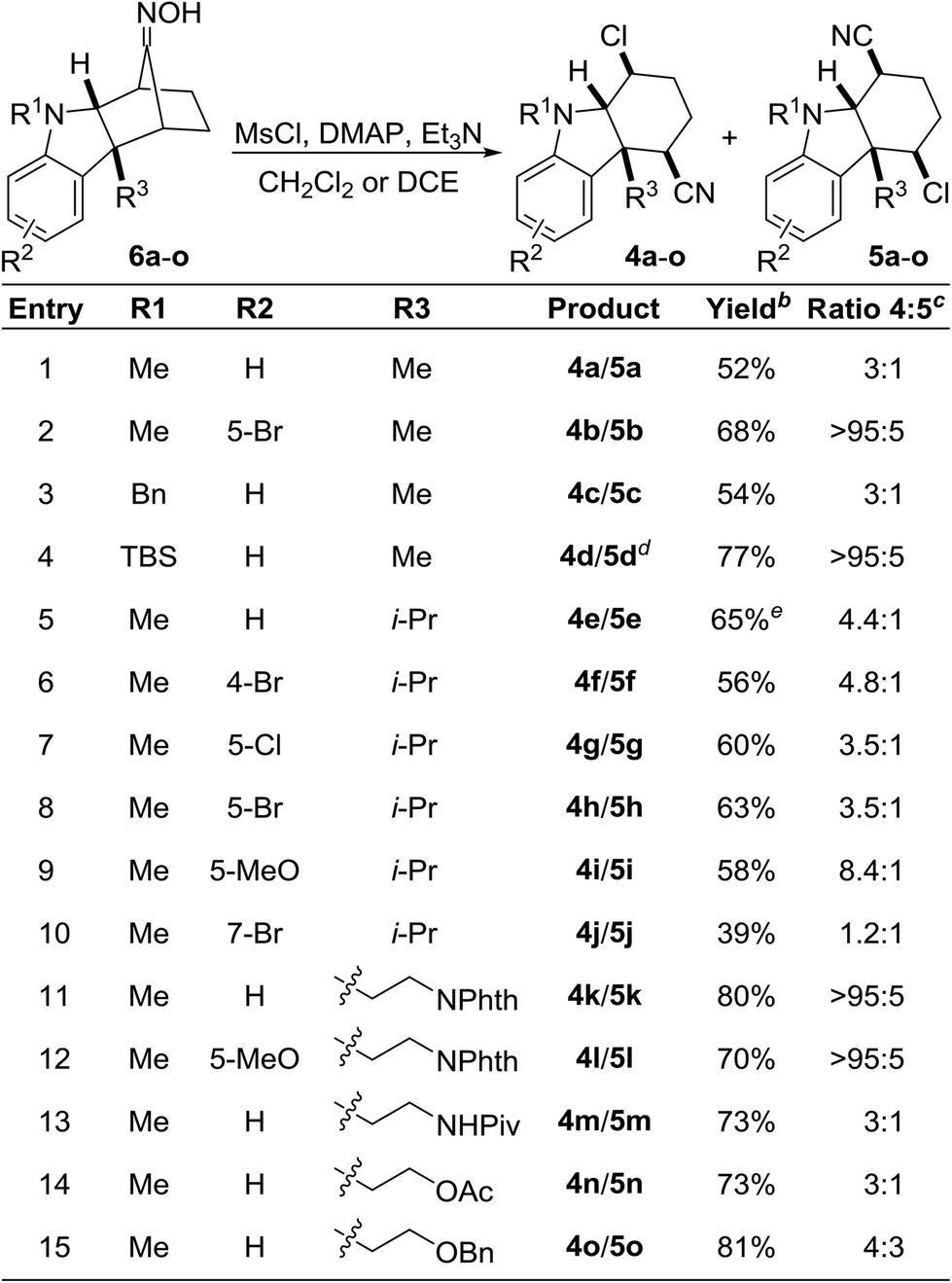
Intrigued by this unexpected finding, we wondered whether the transformation was more broadly general. Indeed, the nucleophile-intercepted Beckmann fragmentation was compatible with a wide range of substituents at R1–R3 and functional groups (Table 1). The indoline nitrogen may be functionalized with methyl, benzyl, and TBS (entries 1–4); although for the silylated substrate, the product was isolated as the desilylated N–H compound (entry 4). Furthermore, the aromatic nucleus of the indoline may be substituted with various halides, methyl, or the electron-rich methoxy group (entries 2, 6–10 and 12). Reactions involving both tryptamine- and tryptophol-derived oximes 6k–o also proceeded smoothly to provide the desired products (entries 11–15). The use of oximes with a free N–H indoline nitrogen resulted in decomposition. The range of regioselectivity between 4 of 5 was broad and varied from 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to greater than 95:5. In nearly all instances, the regioisomers were separable by silica gel flash chromatography. We did not detect any Grob-like fragmentation products in any of the reactions.
:1 to greater than 95:5. In nearly all instances, the regioisomers were separable by silica gel flash chromatography. We did not detect any Grob-like fragmentation products in any of the reactions.
Based on what is known about the Beckmann rearrangement and the stereochemical outcome of the product,29 we propose the mechanism depicted in Scheme 2. It is known that the functionalization of compounds containing carbon–nitrogen double bonds with electron-withdrawing groups at nitrogen facilitates its interconversion between E and Z isomers.30 We therefore hypothesize that the equilibration between mesylates (E)-7 and (Z)-7 is rapid and propose that the ensuing fragmentation is both rate-limiting and irreversible. This may proceed via a stereospecific process enabled in part by neighboring group participation of the non-bonding electrons of the indoline nitrogen to give aziridinium intermediate 8 with inversion of configuration. There is little steric bias between (E)-7 and (Z)-7. However, due to the stereoelectronic requirement of the mesylate leaving group to be anti to the migrating carbon, we believe that the geometry of the oxime mesylate is the primary determinant of the regiochemical outcome of the reaction. Thus, this sets up a classic Curtin–Hammett scenario in which the product distribution is governed by the relative difference in the transition state energies (ΔΔG‡) of (E)-7 → 8versus (Z)-7 → 9. This proposal is supported, in part, by the observation that the product distribution between 4 and 5 is consistently greater than that of the E/Z diastereomeric ratio of the corresponding oxime starting materials (typically 1:1 dr). Subsequent attack of aziridinium 8 by chloride also occurs with inversion to give the major product 4a with the stereochemical outcome shown. Formation of the minor regioisomeric product 5a may occur analogously through phenonium intermediate 9.
 | ||
| Scheme 2 Proposed mechanism of the nucleophile-intercepted Beckmann rearrangement. | ||
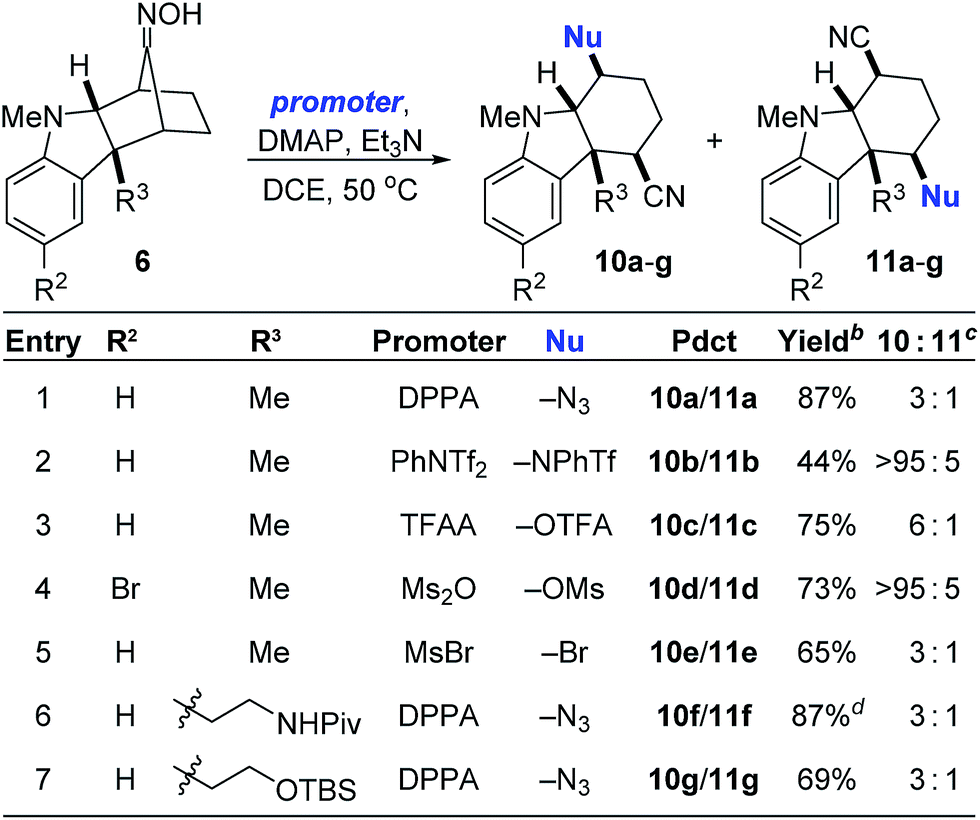
Guided by these mechanistic insights, we next sought to expand the scope of the nucleophile-intercepted Beckmann fragmentation to include nucleophiles besides chloride. We reasoned that the use of promoters other than MsCl would activate the oxime for fragmentation yet provide different nucleophiles for opening the putative intermediates 8 and 9. Table 2 summarizes these results. Thus, treating various oximes 6 with diphenylphosphoryl azide (DPPA) resulted in the formation of the expected cyano azides (entries 1, 6 and 7). Surprisingly, relatively weak nucleophiles generated by promoters such as PhNTf2, TFAA, and Ms2O were also effective in giving the corresponding triflamide, trifluoroacetate, and mesylate products (entries 2–4). Bromides can be obtained when using MsBr (entry 5). The use of Tf2NBz resulted in benzoylation of the oxime oxygen without further reaction (see ESI†). As was also the case in Table 1, the ratio of major to minor products fluctuates greatly in response to subtle changes in substrate structure. We do not yet fully understand the factors that dictate these product distributions.
|
a Reaction conditions: 6 (1 equiv.), promoter (3 equiv.), DMAP (5 mol%), Et3N (5 equiv.), [0.2] M.
b Isolated yield of only the major isomer 10.
c As determined by 1H NMR spectroscopy of the unpurified residue.
d Isolated as a 3:1 mixture of 10f/11f. DPPA = diphenylphosphoryl azide; TFAA = trifluoroacetic anhydride; OTFA = trifluoroacetate.
|
|---|
|
To test the validity of the proposed aziridinium intermediate 8, we elected to study the stereochemical outcome of the conversion of 10c and 12 to the corresponding bromide (Scheme 3). Our working hypothesis was that the activation of alcohol 12 may lead to the regeneration of the putative aziridinium intermediate 8, which may then be opened by bromide. This process would proceed by double stereoinversion to give 10e with the stereochemical relationship shown. However, if the reaction were to progress through the free carbocation 14, then we might reasonably expect to observe the formation of diastereomeric or alkene by-products. Thus, trifluoroacetate 10c was hydrolyzed to 12 (94% yield) and then treated with PBr3 to furnish 10e (88% yield) as the only observable product. That we did not detect the presence of either olefinic or stereoisomeric compounds is consistent with the intermediacy of aziridinium 8, although stereoselective trapping of 14 by the nucleophile cannot be definitively ruled out.
 | ||
| Scheme 3 Preliminary data in support of aziridinium 8. | ||
Conclusions
In summary, we have reported the first instance of a general nucleophile-intercepted Beckmann rearrangements of indolinyl oximes. The reaction is amenable to a wide range of functional groups on the starting material. Furthermore, various promoters can be utilized to introduce oxygen, nitrogen, and halide substituents into the final products. The available evidence supports a stereospecific fragmentation of the activated oxime to give an aziridinium intermediate as the likely rate-limiting step. We believe that this reaction may be useful in future synthetic endeavors towards indoline alkaloids such as the akuammiline family and koumine-type of natural products. Efforts are ongoing in our laboratories towards these targets.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. W. acknowledges the generous support of the NIH NIGMS (R01GM111638).Notes and references
- E. Beckmann, Ber. Dtsch. Chem. Ges., 1886, 19, 988–993 CrossRef.
- R. E. Gawley, in Organic Reactions, ed. A. S. Kende, John Wiley & Sons, Inc., 1988, vol. 35 Search PubMed.
- A. Hassner and E. G. Nash, Tetrahedron Lett., 1965, 6, 525–529 CrossRef.
- K. Hirao, H. Miura and O. Yonemitsu, Heterocycles, 1977, 7, 857 CrossRef CAS.
- P. N. Confalone, G. Pizzolato, D. Lollar-Confalone and M. R. Uskokovic, J. Am. Chem. Soc., 1980, 102, 1954–1960 CrossRef CAS.
- R. J. Hunadi and G. K. Helmkamp, J. Org. Chem., 1981, 46, 2880–2884 CrossRef CAS.
- M. Kirihara, K. Niimi, M. Okumura and T. Momose, Chem. Pharm. Bull., 2000, 48, 220–222 CrossRef CAS.
- W. Ai, Y. Liu, Q. Wang, Z. Lu and Q. Liu, Org. Lett., 2018, 20, 409–412 CrossRef CAS.
- E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744–748 CrossRef CAS.
- A. Ramirez and S. García-Rubio, Curr. Med. Chem., 2003, 10, 1891–1915 CrossRef CAS.
- R. Eckermann and T. Gaich, Synthesis, 2013, 45, 2813–2823 CrossRef CAS.
- J. M. Smith, J. Moreno, B. W. Boal and N. K. Garg, Angew. Chem., Int. Ed., 2015, 54, 400–412 CAS.
- P. Magnus, B. Mugrage, M. DeLuca and G. A. Cain, J. Am. Chem. Soc., 1989, 111, 786–789 CrossRef CAS.
- P. Magnus, B. Mugrage, M. R. DeLuca and G. A. Cain, J. Am. Chem. Soc., 1990, 112, 5220–5230 CrossRef CAS.
- Y.-K. Xu, L. Yang, S.-G. Liao, P. Cao, B. Wu, H.-B. Hu, J. Guo and P. Zhang, J. Nat. Prod., 2015, 78, 1511–1517 CrossRef CAS.
- J. K. Kerkovius and M. A. Kerr, J. Am. Chem. Soc., 2018, 140, 8415–8419 CrossRef CAS.
- H. Arai, Y. Hirasawa, A. Rahman, I. Kusumawati, N. C. Zaini, S. Sato, C. Aoyama, J. Takeo and H. Morita, Bioorg. Med. Chem., 2010, 18, 2152–2158 CrossRef CAS.
- Y. Xu, H.-Q. Qiu, H. Liu, M. Liu, Z.-Y. Huang, J. Yang, Y.-P. Su and C.-X. Yu, Pharmacol., Biochem. Behav., 2012, 101, 504–514 CrossRef CAS.
- B.-J. Xiong, Y. Xu, G.-L. Jin, M. Liu, J. Yang and C.-X. Yu, Sci. Rep., 2017, 7, 14269 CrossRef.
- S. E. Kearney, G. Zahoránszky-Kőhalmi, K. R. Brimacombe, M. J. Henderson, C. Lynch, T. Zhao, K. K. Wan, Z. Itkin, C. Dillon, M. Shen, D. M. Cheff, T. D. Lee, D. Bougie, K. Cheng, N. P. Coussens, D. Dorjsuren, R. T. Eastman, R. Huang, M. J. Iannotti, S. Karavadhi, C. Klumpp-Thomas, J. S. Roth, S. Sakamuru, W. Sun, S. A. Titus, A. Yasgar, Y.-Q. Zhang, J. Zhao, R. B. Andrade, M. K. Brown, N. Z. Burns, J. K. Cha, E. E. Mevers, J. Clardy, J. A. Clement, P. A. Crooks, G. D. Cuny, J. Ganor, J. Moreno, L. A. Morrill, E. Picazo, R. B. Susick, N. K. Garg, B. C. Goess, R. B. Grossman, C. C. Hughes, J. N. Johnston, M. M. Joullie, A. D. Kinghorn, D. G. I. Kingston, M. J. Krische, O. Kwon, T. J. Maimone, S. Majumdar, K. N. Maloney, E. Mohamed, B. T. Murphy, P. Nagorny, D. E. Olson, L. E. Overman, L. E. Brown, J. K. Snyder, J. A. Porco, F. Rivas, S. A. Ross, R. Sarpong, I. Sharma, J. T. Shaw, Z. Xu, B. Shen, W. Shi, C. R. J. Stephenson, A. L. Verano, D. S. Tan, Y. Tang, R. E. Taylor, R. J. Thomson, D. A. Vosburg, J. Wu, W. M. Wuest, A. Zakarian, Y. Zhang, T. Ren, Z. Zuo, J. Inglese, S. Michael, A. Simeonov, W. Zheng, P. Shinn, A. Jadhav, M. B. Boxer, M. D. Hall, M. Xia, R. Guha and J. M. Rohde, ACS Cent. Sci., 2018, 4, 1727–1741 CrossRef CAS.
- X. Han, H. Li, R. P. Hughes and J. Wu, Angew. Chem., Int. Ed., 2012, 51, 10390–10393 CrossRef CAS PubMed.
- X. Han and J. Wu, Angew. Chem., Int. Ed., 2013, 52, 4637–4640 CrossRef CAS.
- H. Li, R. P. Hughes and J. Wu, J. Am. Chem. Soc., 2014, 136, 6288–6296 CrossRef CAS.
- M. C. DiPoto, R. P. Hughes and J. Wu, J. Am. Chem. Soc., 2015, 137, 14861–14864 CrossRef CAS.
- O. G. Chepurny, C. A. Leech, M. Tomanik, M. C. DiPoto, H. Li, X. Han, Q. Meng, R. N. Cooney, J. Wu and G. G. Holz, Sci. Rep., 2016, 6, 28934 CrossRef.
- J. Chen and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 3951–3955 CrossRef CAS.
- M. C. DiPoto and J. Wu, Org. Lett., 2018, 20, 499–501 CrossRef CAS.
- J. Chen and J. Wu, Chem. Sci., 2018, 9, 2489–2492 RSC.
- S. Yamabe, N. Tsuchida and S. Yamazaki, J. Org. Chem., 2005, 70, 10638–10644 CrossRef CAS.
- J. Gálvez and A. Guirado, J. Comput. Chem., 2009, 520–531 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all new compounds, selected 2D-NOESY, COSY, HMBC, HSQC data, and X-ray data for 4a and 5a. CCDC 1898982 and 1898983. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00926d |
| This journal is © The Royal Society of Chemistry 2019 |