
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssembly of 1H-isoindole derivatives by selective carbon–nitrogen triple bond activation: access to aggregation-induced emission fluorophores for lipid droplet imaging†
Dandan
He
a,
Zeyan
Zhuang
b,
Xu
Wang
a,
Jiawei
Li
 a,
Jianxiao
Li
a,
Wanqing
Wu
*ab,
Zujin
Zhao
*b,
Huanfeng
Jiang
*a and
Ben Zhong
Tang
bc
a,
Jianxiao
Li
a,
Wanqing
Wu
*ab,
Zujin
Zhao
*b,
Huanfeng
Jiang
*a and
Ben Zhong
Tang
bc
aKey Laboratory of Functional Molecular Engineering of Guang Dong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510641, P. R. China. E-mail: cewuwq@scut.edu.cn; jianghf@scut.edu.cn
bState Key Laboratory of Luminescent Materials and Devices, Center for Aggregation-Induced Emission, South China University of Technology, Guangzhou 510640, China. E-mail: mszjzhao@scut.edu.cn
cDepartment of Chemistry, Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, The Hong Kong University of Science & Technology, Kowloon, Hong Kong, China
First published on 12th June 2019
Abstract
A novel strategy has been established to assemble a series of single (Z)- or (E)-1H-isoindole derivatives through selectively and sequentially activating carbon–nitrogen triple bonds in a multicomponent system containing various nucleophilic and electrophilic sites. The reaction provides efficient access to structurally unique fluorophores with aggregation-induced emission characteristics. These new fluorophores show fluorescence wavelengths and efficiencies that can be modulated and have excellent potential to specifically light up lipid droplets (LDs) in living cells with bright fluorescence, low cytotoxicity and better photostability than commercially available LD-specific dyes.
Introduction
Organic fluorophores are of significant interest in a wide range of disciplines on account of their promising applications in organic light-emitting diodes, chemical sensing and biological imaging.1 Particularly, organic fluorophores containing N-heterocycles are highly desirable for organic chemistry research due to their ubiquity in biologically active natural products, organic functional materials and pharmaceuticals.2 Over the past few decades, N-heterocycle-based fluorophores have been developed rapidly for biological imaging owing to their good biological activity.3,4 Recently, some interesting fluorophores based on nitrogen-containing heterocycles which exhibit aggregation-induced emission (AIE) properties have been reported by the groups of Huang, Tian, Liu and us.5,6 Thanks to the AIE effect, these fluorophores are free of aggregation-caused emission quenching, and can fluoresce strongly in the aggregated state, such as nanoparticles, which enables them to perform efficiently in tracking various organelles in living cells via fluorescence imaging techniques. To facilitate the advancement in this field, novel design and direct synthesis of new fluorophores containing N-heterocycles are highly demanded.It is known that the molecular structure determines the properties, and the control and modification of the structure would endow materials with different properties.7 Developing efficient and selective synthetic methods is key to construct various new skeletons. For research concerning precise creation and controlled synthesis of functional molecules, traditional chemistry faces challenges of increasing systematicness and comprehensiveness. Consequently, the design, synthesis and discovery of properties of functional molecules are undoubtedly a tendency of organic chemistry. Given the importance of elemental nitrogen in functional materials, it can be expected that when introducing multiple nitrogen atoms or various nitrogen-containing groups into the target molecules, novel properties should be obtained. Nitriles are common platform chemicals in the chemical commodity industry, and the low cost coupled with versatile transformation of these molecules underlies their great use in industrial and academic communities. For instance, nitriles may serve as a valuable precursor toward structurally diverse compounds such as amines,8 amides,9 ketones,10 and heterocycles.11 In most cases, however, nitriles are used as solvents or ligands in organometallic reactions,12 presumably due to the inherently inert nature of carbon–nitrogen triple bonds. To address this issue, many methods for activating nitriles have been reported recently. Generally, three strategies for nitrile transformation are typically employed: (i) nucleophilic addition reaction,13 (ii) electrophilic addition reaction,14 and (iii) radical addition reaction.15 Despite recent advances in carbon–nitrogen triple bond transformation,16 a selective and sequential activation of carbon–nitrogen triple bonds is yet to be realized.
Based on our previous work, we anticiapte that a convenient and concise approach to assemble a series of carbon–nitrogen triple bonds with different activities could be provided by rational design. As illustrated in the proposed synthetic route (Scheme 1), isocyanide 3 is initially activated via an insertion reaction to give intermediate I. Subsequently, the nucleophilic addition of 2 to the nitrile group of I forms intermediate II, which is selectively driven by different reactivities of electrophilic sites. Finally, the desired 1H-isoindole product 4 would be obtained through the reductive elimination and isomerization of II. This protocol, a palladium-catalyzed multicomponent cross-coupling reaction, provides direct access to novel fluorescent scaffolds with desirable potential properties. Moreover, the photophysical properties and potential bioimaging application of the synthesized 1H-isoindole derivatives can be further investigated.
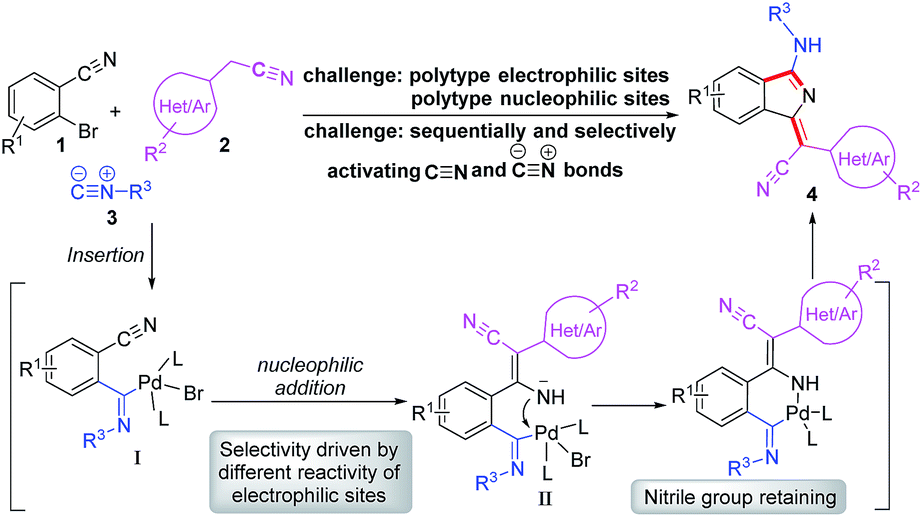 | ||
| Scheme 1 Proposed synthetic route for 1H-isoindole derivatives. | ||
Results and discussion
We commenced our study by optimizing reaction conditions with 2-bromobenzonitrile (1a), 2-phenyacetonitrile (2a) and tert-butyl isocyanide (3a) as model substrates. Initially, the feasibility of the hypothesis was confirmed by product 4a obtained in 51% yield when the reaction was treated with 10 mol% Pd(OAc)2, 20 mol% PPh3 and 3 equivalents of t-BuOK in 1 mL DMSO at 120 °C for 12 h under air (Table 1, entry 1). Encouraged by this observation, further study of other reaction parameters was carried out. The examination of different bases revealed that t-BuOK was the most appropriate choice (Table 1, entries 2 and 3). Subsequent screening of catalysts showed that Pd(OAc)2 still gave the best result (Table 1, entries 4 and 5). In addition, a series of ligands and solvents were tested (Table 1, entries 6–10) and the highest yield (79%) of 4a was attributed to the increased equivalents of 2a and 3a in dry DMSO (Table 1, entry 12). The structure and configuration of 1H-isoindole derivatives were confirmed by X-ray crystal analysis (Fig. 1).17 The single E configuration of the carbon–carbon double bond in compound 4a could be explained by intramolecular C–H⋯N hydrogen bonding, which will effectively rigidify the molecular structure and increase the stability of the molecule (Fig. 1A).|
|
|||||
|---|---|---|---|---|---|
| Entrya | [Pd] | Ligand | Base | Solvent | Yieldb (%) |
| a All reactions were performed with 1a (0.2 mmol), 2a (1 equiv.), 3a (1 equiv.), palladium catalyst (10 mol%), ligand (20 mol%), base (3 equiv.), and solvent (1 mL), at 120 °C under air for 12 h. b The yield was determined by GC with n-dodecane as the internal standard based on 1a. n.d. = not determined. c 2a (2 equiv.) and 3a (2 equiv.). d 2a (2 equiv.), 3a (2 equiv.), and dry DMSO. L1: tri-o-tolyphosphine. L2: thiosemicarbazide. | |||||
| 1 | Pd(OAc)2 | PPh3 | t-BuOK | DMSO | 51 |
| 2 | Pd(OAc)2 | PPh3 | t-BuONa | DMSO | n.d. |
| 3 | Pd(OAc)2 | PPh3 | Cs2CO3 | DMSO | n.d. |
| 4 | PdCl2 | PPh3 | t-BuOK | DMSO | 28 |
| 5 | Pd(TFA)2 | PPh3 | t-BuOK | DMSO | Trace |
| 6 | Pd(OAc)2 | PPh3 | t-BuOK | Dioxane | 48 |
| 7 | Pd(OAc)2 | PPh3 | t-BuOK | DMF | Trace |
| 8 | Pd(OAc)2 | L1 | t-BuOK | DMSO | Trace |
| 9 | Pd(OAc)2 | P(t-Bu)3 | t-BuOK | DMSO | Trace |
| 10 | Pd(OAc)2 | L2 | t-BuOK | DMSO | 38 |
| 11c | Pd(OAc)2 | PPh3 | t-BuOK | DMSO | 67 |
| 12d | Pd(OAc)2 | PPh3 | t-BuOK | DMSO | 81 (79) |
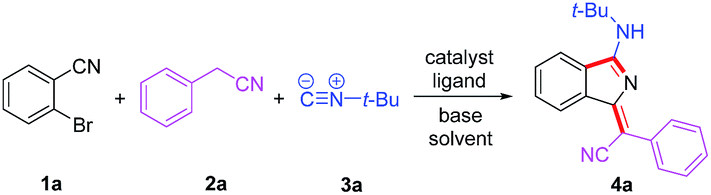
 | ||
| Fig. 1 (A–D) Single crystal structures of 4a, 4p, 4m, and 4m-M. (E and F) Molecular packing of 4a (A) and 4p (B) in crystals. | ||
Under the optimized conditions, the substrate scope was then explored (Table 2). Diversely substituted acetonitriles were employed as the substrates. Both electron-donating (OMe, t-Bu and Me) and electron-withdrawing (F, Cl and Br) groups in 2-phenylacetonitriles were compatible and the corresponding products were afforded in 53–73% yields. Notably, when using 2-thiopheneacetonitrile as the substrate, the corresponding products 4m and 4y were isolated in good yields of 74% and 76%, respectively. In addition, other tested alkyl isocyanides such as n-butyl isocyanide, 1,1,3,3-tetramethylbutyl isocyanide, adamantyl isocyanide, cyclopentyl isocyanide and cyclohexyl isocyanide were found to be suitable for this transformation, converting to the corresponding products 4p–4y in 44–84% yields. Additionally, the reactions of 2-bromobenzonitriles with different functional groups also proceeded smoothly under the standard conditions to deliver products 4j, 4o and 4x in acceptable yields. The good functional group compatibility, especially for the thiophene group, could be conveniently modified for the purpose of fluorescence–structure relationship study. Compound 4m-M containing an extended conjugation system could be constructed easily from 4m in 85% yield via bromination and Suzuki coupling reaction.
| a Reaction conditions: all reactions were performed with 1 (0.2 mmol), 2 (2 equiv.), 3 (2 equiv.), Pd(OAc)2 (10 mol%), PPh3 (20 mol%), t-BuOK (3 equiv.), and dry DMSO (1 mL), at 120 °C under air for 12 h. |
|---|
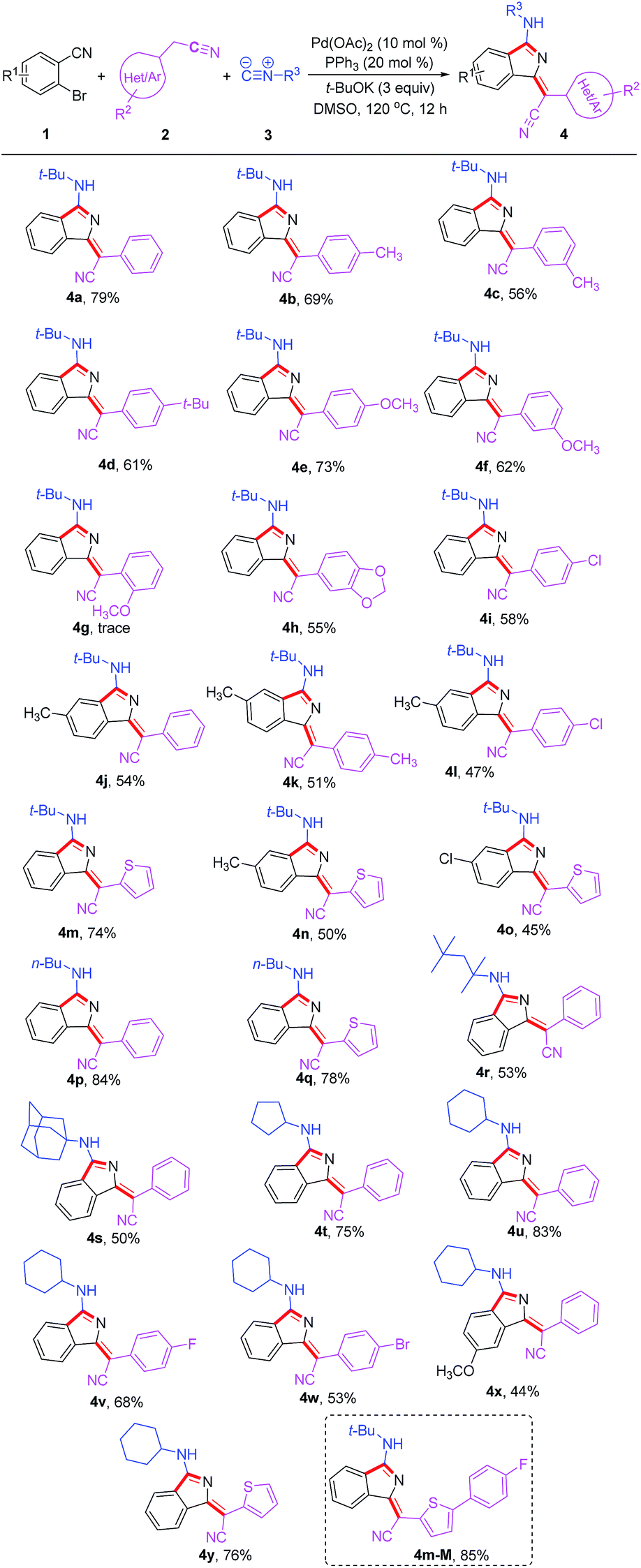
|
To demonstrate the potential application of this method, the photophysical properties of these synthetic fluorophores were carefully investigated (Tables 3 and S1†). 1H-isoindole derivatives, which were converted from 2-phenylacetonitriles with different functional groups, show similar absorption maxima in the range of 392–415 nm in CH2Cl2 solution. They emit very weak fluorescence in solutions with extremely low fluorescence quantum yields (ΦFs) of 0.2–0.9%, but exhibit intense fluorescence in the range of 408–438 nm with a much higher ΦFs of 1.9–26.3% in solid films, demonstrating AIE characteristics, which was further confirmed from the emission spectra of 4a in water/methanol with different water fractions (Fig. S1†). For these new AIE fluorophores, the intramolecular motion of flexible rotors (e.g. the aromatic rings linked with R2) accounts for the quenching of fluorescence in the solution state, while the restriction of such a kind of motion results in emission enhancement in the aggregated state.18 Besides, the efficient fluorescence in the aggregated state is also attributed to the bulky R3 fragment, which suppresses close packing and thus strong intermolecular interactions.19 In other words, the solid-state fluorescence behaviours can be well modulated by changing R3. As shown in Fig. 1, compared to 4p with a normal butyl group, 4a bearing a bulkier tertiary butyl group adopts a looser packing mode in crystals to avoid close π–π stacking, leading to a higher ΦFs for 4a than 4p in both film and powder. Moreover, the fluorescence wavelength could be further tuned by utilizing substrates 2 with different functional groups. For instance, 4m shows a yellow fluorescence peak at 553 nm in film, which is apparently red-shifted relative to the green fluorescence of 4a (518 nm) and 4p (526 nm). This can be ascribed to the strengthened electron donor–acceptor interaction when electron-donating thiophene is introduced.20 In addition, containing a more extended conjugation system, 4m-M can emit orange light at 589 nm.
| Compound | λ abs (nm)a | λ em (nm)e | Stokes shift (cm−1)f | Φ F (%) | |||
|---|---|---|---|---|---|---|---|
| In CH2Cl2b (ε (104 M−1 cm−1)c) | In filmd | In film | In CH2Cl2 | In film | In powder | ||
| a Maximum absorption wavelength. b Measured in CH2Cl2 at 10.0 μM. c Molar absorption coefficient. d Measured in a drop-cast film on a quartz plate. e Emission peak (excited using the maximum absorption wavelength in CH2Cl2 as the excitation wavelength). f Stokes shift = 1/λabs (in film) − 1/λem (in film). g Absolute fluorescence quantum yield measured by calibrated integration. | |||||||
| 4a | 394 (1.76) | 411 | 518 | 50 | 0.2 | 25.0 | 33.8 |
| 4m | 423 (3.44) | 431 | 553 | 51 | 0.5 | 2.7 | 16.4 |
| 4p | 396 (2.27) | 423 | 526 | 46 | 0.6 | 13.5 | 21.0 |
| 4m-M | 453 (3.17) | 456 | 589 | 50 | 0.9 | 2.6 | 8.0 |
With an AIE fluorophore library in hand, compound 4a and modified 4m-M were selected as fluorescent probes for live cell imaging. Lipid droplets (LDs) are uniquely encapsulated by a phospholipid monolayer, which segregates their hydrophobic neutral lipid core from the aqueous cytosol.21 Considering the lipophilic properties of the fluorophores used, we tried to perform hydrophobic LD localization experiments with HeLa cells. Cell image data were obtained via irradiation at 405 nm on HeLa cells co-stained with 4a or 4m-M and HCS LipidTOX™ Deep Red neutral lipid stain, a commercial probe enabling the differentiation of LDs from other organelles. As shown in Fig. 2, the merged images, with high overlap ratios (88% and 84% respectively), indicate the excellent specific targeting ability of 4a and 4m-M toward LDs in living cells.
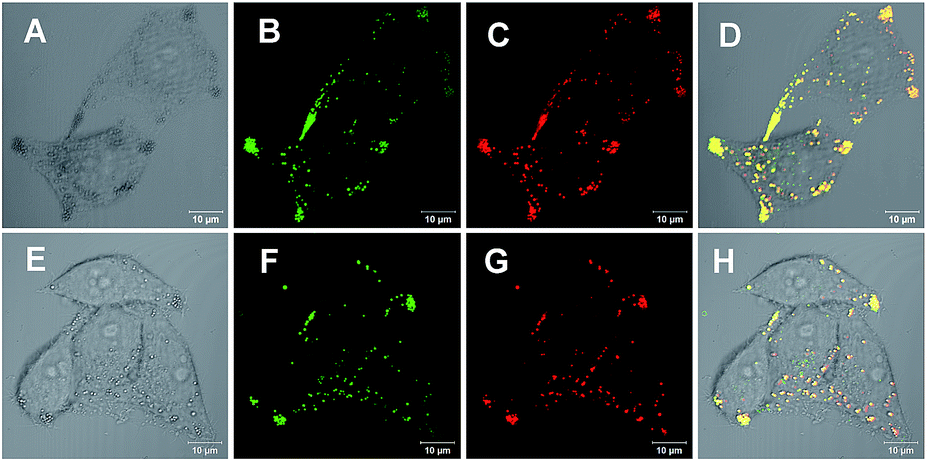 | ||
Fig. 2 Co-localization experiments of HeLa cells pretreated with oleic acid and stained with 5 μM 4a or 4m-M for 15 min and then co-stained with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 dilution of HCS LipidTOX™ Deep Red neutral lipid stain: (A and E) bright-field images, (B and F) images from 4a and 4m-M on channel 1 (4a: λex = 405, λem = 450–570; 4m-M: λex = 405, λem = 480–600), (C and G) images from HCS LipidTOX™ Deep Red neutral lipid stain on channel 2 (λex = 633, λem = 640–740), and (D and H) merged images from A–C and E–G, respectively. :1000 dilution of HCS LipidTOX™ Deep Red neutral lipid stain: (A and E) bright-field images, (B and F) images from 4a and 4m-M on channel 1 (4a: λex = 405, λem = 450–570; 4m-M: λex = 405, λem = 480–600), (C and G) images from HCS LipidTOX™ Deep Red neutral lipid stain on channel 2 (λex = 633, λem = 640–740), and (D and H) merged images from A–C and E–G, respectively. | ||
The photostability, as an important evaluation criterion for fluorescence probes, was measured quantitatively together with three commercially available LD-specific dyes, BODIPY, Nile red and HCS LipidTOX™ Deep Red neutral lipid stain. After exposure to a 405 nm laser with a power of 10% (3 mW) for 10 s between every scan interval for 30 scans, the fluorescence signal intensity of three commercial trackers was drastically lost, even Nile red was reduced to less than 20% of its initial value, while approximately 80% of the signal intensity of 4a and 4m-M was retained under the same conditions (Fig. 3A). The result suggests that the two fluorophores have excellent photostability, while BODIPY, Nile red and HCS LipidTOX™ Deep Red neutral lipid stain suffer from serious photobleaching. In addition, based on 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay, we also evaluated the cytotoxicity of the two fluorophores by using them to incubate HeLa cells. As presented in Fig. 3B, a negligible change in HeLa cell viability was observed even when 20 μM 4a or 4m-M was added to the culture medium for 24 h, which indicated that the two fluorophores have good biocompatibility to living cells.
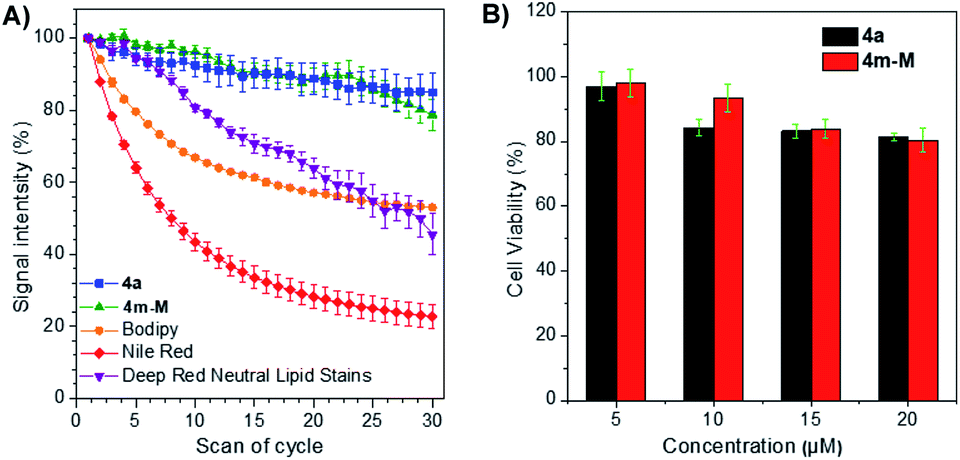 | ||
| Fig. 3 (A) Photobleaching experiment: the signal intensity changed in HeLa cells stained with 5 μM 4a, 4m-M, BODIPY or Nile red or a 1:1000 dilution of HCS LipidTOX™ Deep Red neutral lipid stain upon continuous scanning (the cells were exposed to 405 nm with 10% powder (3 mW) for 10 s in every interval). (B) Cell viability of HeLa cells after incubation with different concentrations of 4a or 4m-M (5, 10, 15 and 20 μM of 4a or 4m-M) for 24 h. | ||
Conclusions
In conclusion, we have developed a highly stereoselective Pd-catalyzed cross-coupling reaction to access a library of AIE fluorophores, 1H-isoindole derivatives, by selective carbon–nitrogen triple bond activation. The system exhibits several impressive characteristics including single Z or E selectivity, simple and diverse structures, and tunable and bright fluorescence. These AIE fluorophores have been proven to be considerably efficient reagents for cell imaging, which show excellent LD-targeting specificity and much higher photostability than commercial LD-staining dyes. The protocol should provide a new strategy for the systematic study of the design, synthesis and discovering new specific properties of functional molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Key Research and Development Program of China (2016YFA0602900), the National Natural Science Foundation of China (21490572 and 21672072), the Guangdong Natural Science Foundation (2018B030308007), and the Pearl River S&T Nova Program of Guangzhou (201610010160) for financial support.Notes and references
- (a) L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622 RSC; (b) X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590 CrossRef CAS PubMed; (c) S. W. Yun, N. Y. Kang, S. J. Park, H. H. Ha, Y. K. Kim, J. S. Lee and Y.-T. Chang, Acc. Chem. Res., 2014, 47, 1277 CrossRef CAS PubMed; (d) Z. Yang, A. Sharma, J. Qi, X. Peng, D. Y. Lee, R. Hu, D. Lin, J. Qu and J. S. Kim, Chem. Soc. Rev., 2016, 45, 4651 RSC; (e) V. Fernández-Luna, P. D. Coto and R. D. Costa, Angew. Chem., Int. Ed., 2018, 57, 8826 CrossRef PubMed; (f) J. Tagare and S. Vaidyanathan, J. Mater. Chem. C, 2018, 6, 10138 RSC.
- Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485 CrossRef CAS PubMed.
- (a) K. Tikhomirova, A. Anisimov and A. Khoroshutin, Eur. J. Org. Chem., 2012, 2201 CrossRef CAS; (b) B. Liu, Z. Wang, N. Wu, M. Li, J. You and J. Lan, Chem.–Eur. J., 2012, 18, 1599 CrossRef CAS PubMed; (c) S. Takahashi, Y. Kagami, K. Hanaoka, T. Terai, T. Komatsu, T. Ueno, M. Uchiyama, I. Koyama-Honda, N. Mizushima, T. Taguchi, H. Arai, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2018, 140, 5925 CrossRef CAS PubMed; (d) Y. Cheng, G. Li, Y. Liu, Y. Shi, G. Gao, D. Wu, J. Lan and J. You, J. Am. Chem. Soc., 2016, 138, 4730 CrossRef CAS PubMed; (e) J. A. González-Vera, F. Fueyo-González, I. Alkorta, M. Peyressatre, M. C. Morris and R. Herranz, Chem. Commun., 2016, 52, 9652 RSC; (f) F. D. Moliner, N. Kielland, R. Lavilla and M. Vendrell, Angew. Chem., Int. Ed., 2017, 56, 3758 CrossRef PubMed; (g) A. C. Shaikh, D. S. Ranade, P. R. Rajamohanan, P. P. Kulkarni and N. T. Patil, Angew. Chem., Int. Ed., 2017, 56, 757 CrossRef CAS PubMed; (h) Y. Chen, W. Zhang, Z. Zhao, Y. Cai, J. Gong, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 5011 CrossRef CAS PubMed; (i) J. Ohata, M. K. Miller, C. M. Mountain, F. Vohidov and Z. T. Ball, Angew. Chem., Int. Ed., 2018, 57, 2827 CrossRef CAS PubMed.
- (a) A. L. Odom and T. J. McDaniel, Acc. Chem. Res., 2015, 48, 2822 CrossRef CAS PubMed; (b) J. Xuan and A. Studer, Chem. Soc. Rev., 2017, 46, 4329 RSC.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed; (b) G. Yu, D. Wu, Y. Li, Z. Zhang, L. Shao, J. Zhou, Q. Hu, G. Tang and F. Huang, Chem. Sci., 2016, 7, 3017 RSC; (c) J. Mei, Y. Huang and H. Tian, ACS Appl. Mater. Interfaces, 2018, 10, 12217 CrossRef CAS PubMed; (d) F. Hu, X. Cai, P. N. Manghnani, Kenry, W. Wu and B. Liu, Chem. Sci., 2018, 9, 2756 RSC.
- (a) J. H. Kim, T. Gensch, D. Zhao, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 10975 CrossRef CAS PubMed; (b) H. Lu, Y. Zheng, X. Zhao, L. Wang, S. Ma, X. Han, B. Xu, W. Tian and H. Gao, Angew. Chem., Int. Ed., 2016, 55, 155 CrossRef CAS PubMed; (c) S. Sasaki, S. Suzuki, W. M. C. Sameera, K. Igawa, K. Morokuma and G. Konishi, J. Am. Chem. Soc., 2016, 138, 8194 CrossRef CAS PubMed; (d) B. Tang, C. Wang, Y. Wang and H. Zhang, Angew. Chem., Int. Ed., 2017, 56, 12543 CrossRef CAS PubMed.
- (a) J.-B. Feng and X.-F. Wu, Adv. Synth. Catal., 2016, 358, 2179 CrossRef CAS; (b) S. Chakraborty, G. Leitus and D. Milstein, Chem. Commun., 2016, 52, 1812 RSC; (c) X. Zhang, X. Xie and Y. Liu, Chem. Sci., 2016, 7, 5815 RSC; (d) M. Xiong, X. Xie and Y. Liu, Org. Lett., 2017, 19, 3398 CrossRef CAS PubMed; (e) X. Yang, H. Yu, Y. Xu and L. Shao, J. Org. Chem., 2018, 83, 9682 CrossRef CAS PubMed.
- (a) G. C. Midya, A. Kapat, S. Maiti and J. Dash, J. Org. Chem., 2015, 80, 4148 CrossRef CAS PubMed; (b) P. Marcé, J. Lynch, J. Blackerb and J. M. J. Williams, Chem. Commun., 2016, 52, 1436 RSC; (c) Y. Li, H. Chen, J. Liu, X. Wan and Q. Xu, Green Chem., 2016, 18, 4865 RSC.
- (a) C. Zhou and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 2302 CrossRef CAS PubMed; (b) J. Lindh, P. J. R. Sjöberg and M. Larhed, Angew. Chem., Int. Ed., 2010, 49, 7733 CrossRef CAS PubMed; (c) T. Miao and G.-W. Wang, Chem. Commun., 2011, 47, 9501 RSC; (d) J.-C. Hsieh, Y.-C. Chen, A.-Y. Cheng and H.-C. Tseng, Org. Lett., 2012, 14, 1282 CrossRef CAS PubMed; (e) M. Meng, L. Yang, K. Cheng and C. Qi, J. Org. Chem., 2018, 83, 3275 CrossRef CAS PubMed; (f) J. Zhang, F. Zhang, L. Lai, J. Cheng, J. Sun and J. Wu, Chem. Commun., 2018, 54, 3891 RSC.
- (a) M. Ramanathan and S.-T. Liu, J. Org. Chem., 2015, 80, 5329 CrossRef CAS; (b) Y.-L. Chen, P. Sharma and R.-S. Liu, Chem. Commun., 2016, 52, 3187 RSC; (c) R.-R. Zhou, Q. Cai, D.-K. Li, S.-Y. Zhuang, Y.-W. Wu and A.-X. Wu, J. Org. Chem., 2017, 82, 6450 CrossRef CAS PubMed; (d) M. Ramanathan, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Org. Lett., 2017, 19, 5840 CrossRef CAS; (e) M. Ramanathan and S.-T. Liu, J. Org. Chem., 2017, 82, 8290 CrossRef CAS PubMed; (f) U. T. Das, L. J. W. Shimon and D. Milstein, Chem. Commun., 2017, 53, 13133 RSC; (g) S. Yao, K. Zhou, J. Wang, H. Cao, L. Yu, J. Wu, P. Qiu and Q. Xu, Green Chem., 2017, 19, 2945 RSC; (h) K. T. Ashitha, V. P. Kumar, C. T. F. Salfeena and B. S. Sasidhar, J. Org. Chem., 2018, 83, 113 CrossRef CAS PubMed; (i) X. Yu, L. Gao, L. Jia, Y. Yamamoto and M. Bao, J. Org. Chem., 2018, 83, 10352 CrossRef CAS PubMed; (j) A. Kishi, K. Moriyama and H. Togo, J. Org. Chem., 2018, 83, 11080 CrossRef CAS PubMed; (k) M. Ramanathan and S.-T. Liu, J. Org. Chem., 2018, 83, 14138 CrossRef CAS PubMed; (l) L. Su, K. Sun, N. Pan, L. Liu, M. Sun, J. Dong, Y. Zhou and S.-F. Yin, Org. Lett., 2018, 20, 3399 CrossRef CAS PubMed; (m) Y. Zhang, Y. Shao, J. Gong, K. Hu, T. Cheng and J. Chen, Adv. Synth. Catal., 2018, 360, 3260 CrossRef CAS.
- (a) F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035 CrossRef CAS PubMed; (b) S. F. Rach and F. E. Kühn, Chem. Rev., 2009, 109, 2061 CrossRef CAS PubMed.
- (a) S. Huang, Y. Shao, L. Zhang and X. Zhou, Angew. Chem., Int. Ed., 2015, 54, 14452 CrossRef CAS PubMed; (b) F. Xue and T. Hayashi, Angew. Chem., Int. Ed., 2018, 57, 10368 CrossRef CAS PubMed; (c) K. Hu, Q. Zhen, J. Gong, T. Cheng, L. Qi, Y. Shao and J. Chen, Org. Lett., 2018, 20, 3083 CrossRef CAS PubMed.
- (a) J. Sheng, Y. Wang, X. Su, R. He and C. Chen, Angew. Chem., Int. Ed., 2017, 56, 4824 CrossRef CAS PubMed; (b) M. Ramanathan, Y.-H. Wang, Y.-H. Liu, S.-M. Peng, Y.-C. Cheng and S.-T. Liu, J. Org. Chem., 2018, 83, 6133 CrossRef CAS PubMed.
- (a) Y. Yan, Z. Zhang, Y. Wan, G. Zhang, N. Ma and Q. Liu, J. Org. Chem., 2017, 82, 7957 CrossRef CAS PubMed; (b) C. Zhang, J. Pi, S. Chen, P. Liu and P. Sun, Org. Chem. Front., 2018, 5, 793 RSC; (c) J. Zheng, Z. Deng, Y. Zhang and S. Cui, Adv. Synth. Catal., 2016, 358, 746 CrossRef CAS.
- (a) X. Wang, Y. Huang, J. Xu, X. Tang, W. Wu and H. Jiang, J. Org. Chem., 2017, 82, 2211 CrossRef CAS PubMed; (b) X. Wang, W. Xiong, Y. Huang, J. Zhu, Q. Hu, W. Wu and H. Jiang, Org. Lett., 2017, 19, 5818 CrossRef CAS PubMed; (c) X. Wang, D. He, Y. Huang, Q. Fan, W. Wu and H. Jiang, J. Org. Chem., 2018, 83, 5458 CrossRef CAS PubMed; (d) X. Wang, J. Li, Y. Huang, J. Zhu, R. Hu, W. Wu and H. Jiang, J. Org. Chem., 2018, 83, 10453 CrossRef CAS PubMed.
- CCDC 1868897 (4a), CCDC 1880913 (4m), CCDC 1880930 (4p), CCDC 1869980 (4m-M) contain the supplementary crystallographic data for this paper.†.
- G. Lin, L. Chen, H. Peng, S. Chen, Z. Zhuang, Y. Li, B. Wang, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 4867 RSC.
- J. Wang, X. Gu, P. Zhang, X. Huang, X. Zheng, M. Chen, H. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16974 CrossRef CAS PubMed.
- J. Guo, S. Hu, W. Luo, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, Chem. Commun., 2017, 53, 1463 RSC.
- D. J. Murphy, Prog. Lipid Res., 2001, 40, 325 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1868897, 1880913, 1880930 and 1869980. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01035a |
| This journal is © The Royal Society of Chemistry 2019 |