
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBrønsted acid catalysis – the effect of 3,3′-substituents on the structural space and the stabilization of imine/phosphoric acid complexes†
Maxime
Melikian‡
,
Johannes
Gramüller‡
,
Johnny
Hioe
,
Julian
Greindl
and
Ruth M.
Gschwind
 *
*
Institut für Organische Chemie, Universität Regensburg, D-93053 Regensburg, Germany. E-mail: ruth.gschwind@chemie.uni-regensburg.de
First published on 8th April 2019
Abstract
BINOL derived chiral phosphoric acids (CPAs) are widely known for their high selectivity. Numerous 3,3′-substituents are used for a variety of stereoselective reactions and theoretical models of their effects are provided. However, experimental data about the structural space of CPA complexes in solution is extremely rare and so far restricted to NMR investigations of binary TRIP/imine complexes featuring two E- and two Z-imine conformations. Therefore, in this paper the structural space of 16 CPA/imine binary complexes is screened and 8 of them are investigated in detail by NMR. For the first time dimers of CPA/imine complexes in solution were experimentally identified, which show an imine position similar to the transition state in transfer hydrogenations. Furthermore, our experimental and computational data revealed an astonishing invariance of the four core structures regardless of the different steric and electronic properties of the 3,3′-substituent. However, a significant variation of E/Z-ratios is observed, demonstrating a strong influence of the 3,3′-substituents on the stabilization of the imine in the complexes. These experimental E/Z-ratios cannot be reproduced by calculations commonly applied for mechanistic studies, despite extensive conformational scans and treatment of the electronic structure at a high level of theory with various implicit solvent corrections. Thus, these first detailed experimental data about the structural space and influence of the 3,3′-substituent on the energetics of CPA/imine complexes can serve as basis to validate and improve theoretical predictive models.
Introduction
In the field of Brønsted acid catalysis1,2 a variety of different catalyst classes have emerged in recent years, one of the most famous being BINOL (1,1′-bi-2-naphthol) derived chiral phosphoric acids (CPAs).3,4 CPAs have been successfully applied in various enantioselective syntheses such as transfer hydrogenations (see Fig. 1a),5,6 reductive aminations,7,8 Mannich type reactions,9–11 Strecker reactions,12 and enantioselective additions.13,14 The main difference between these BINOL derived CPAs are the 3,3′-substituents, which significantly influence not only yields, but especially the stereoselectivity of these reactions. Typical 3,3′-substituents successfully employed are e.g. 2,4,6-triisopropylphenyl groups in the Brønsted acid catalyst TRIP, 3,5-bis(trifluoromethyl) groups in TRIFP, or triphenylsilyl groups in TiPSY (see Fig. 1b).5,6,15 When the bulkiness of the 3,3′-substituents is increased, the cited transfer hydrogenations and the reductive amination generally show an increase in enantiomeric excess, but also a sudden drop in yield and enantioselectivity when using extended aromatic systems such as naphthyl, or 4-biphenyl groups.5–7 The reason for these differences is for most of these reactions largely unexplored and vary from reaction to reaction.16 As a result many synthetic methods using BINOL derived CPAs had to be developed via trial and error or screening methods.17,18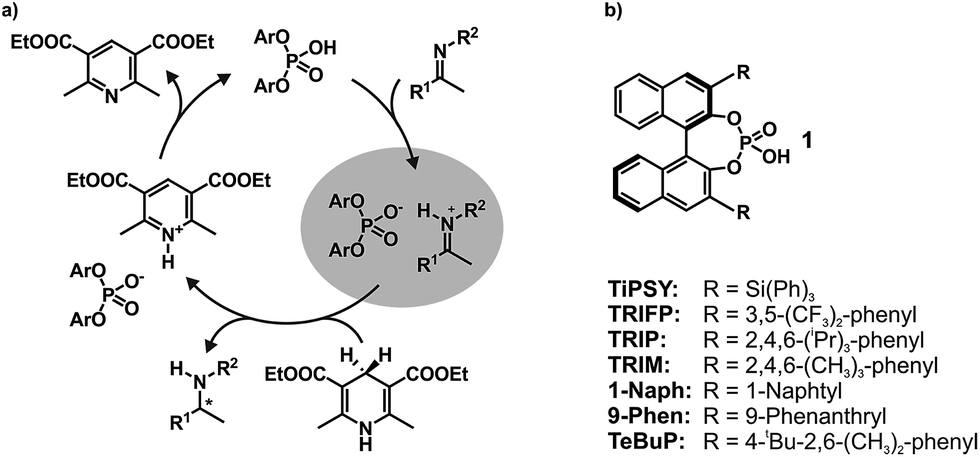 | ||
| Fig. 1 (a) Catalytic cycle of the transfer hydrogenation of imines with a chiral phosphoric acid catalyst and a Hantzsch ester as reducing agent. The binary complexed highlighted in grey is the focus of this work; (b) Brønsted acid catalysts with different 3,3′-groups. | ||
To shed light on the involved structures and to reveal the key interactions of reactivity and stereoselectivity so far mainly theoretical calculations and multivariate linear regression models have been applied.19,20 Thus, Goodman et al. linked the enantioselectivity to structural parameters of CPAs, such as the rotational barrier of the 3,3′-substituent describing the steric bulk close to the hydrogen bond and the cone angle AREA (θ) of the substrate binding pocket, reflecting remote steric bulk.21 Furthermore, combining that with a steric classification of electrophiles and nucleophiles Goodman et al. developed a webtool for predicting suitable CPA catalysts based on reactant structures.22–24 Parallel to that, Sigman et al. demonstrated how a data-driven approach can be capable of revealing nonintuitive insights about interactions involved in stereoselectivity determination by analysing the dependence of ee on steric and electronic molecular descriptors.20,25–27
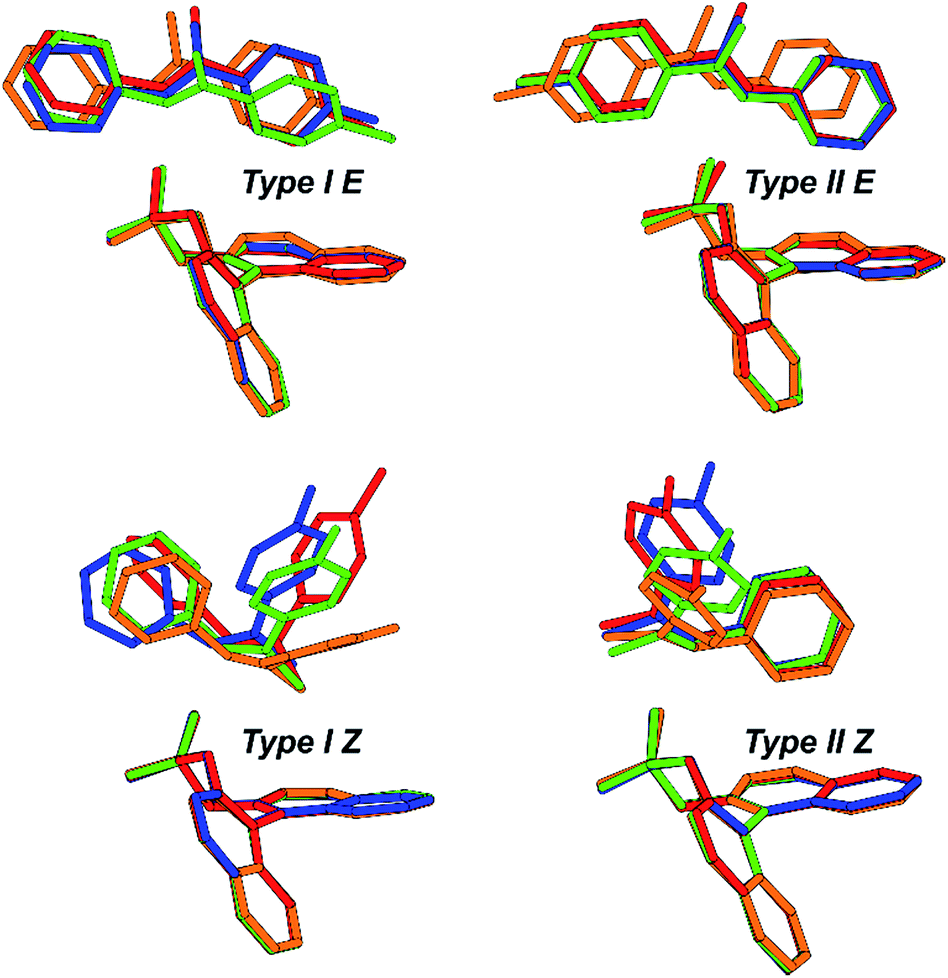
In contrast to the theoretical studies the information about the structures of CPAs/imine complexes based on experimental data is rather limited so far. To our knowledge, only two crystal structures were provided from the groups of MacMillan7 and Schneider28 with strongly deviating structural features (see Fig. 2 for structures and Fig. 1 for substituents). While a TiPSY/imine complex adopts core structure Type II E proposed by theoretical calculations (see Fig. 2a and c)29 the second crystal structure of a TeBuP/imine complex exhibits an intermediate position of structures Type I E and Type II E (see Fig. 2b and c).
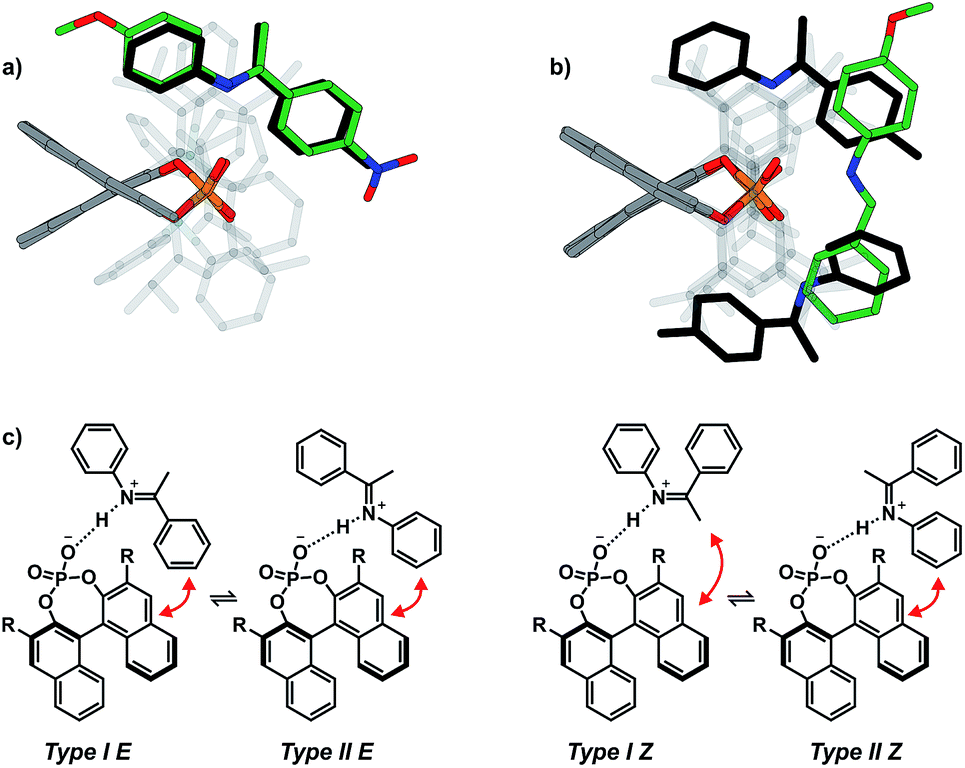 | ||
| Fig. 2 (a) Structure similarity of a crystal structure of a TiPSY/imine complex7 (green) and structure Type II E of a TRIP/imine complex identified in our previous work29 (black). (b) Structure deviation of the crystal structure of a TeBuP/imine complex28 (green) vs. both Type I E (black above) and Type II E (black below) of a TRIP/imine complex. The imine shows an intermediate position of Type I E and Type II E. (c) The four core structures of the binary complex identified in our previous work.29 The red arrows mark some of the identified NOE interactions. | ||
In solution the experimental data base is similarly sparse. While two remarkable studies provided some NMR data about a ternary CPA complex30 and a CPA reaction intermediate31 the first comprehensive insight about the structural space of CPA imine complexes and their hydrogen bond properties in solution was provided by our group on the example of TRIP/imine complexes.29,32 In depth NMR investigations revealed ionic complexes with extremely strong and highly covalent hydrogen bonds32 and a structural space covering the four core structures (Type I/II E/Z, shown in Fig. 2c),29 which were previously already predicted for ternary CPA/imine/nucleophile complexes by theoretical calculations.33 However, the effect of varying 3,3′-substituents on the structure of CPA/imine complexes as well as on their population has so far remained elusive.
Therefore, in this report we present the first experimental data about the influence of different 3,3′-substituents on the structures and E/Z populations of complexes between chiral phosphoric acids and imines. A screening covering 16 binary complexes with 5 different chiral phosphoric acids and 7 imines and in depths structural investigations on selected examples reveal an astonishing invariance of the four main structures Type I E, Type II E, Type I Z and Type II Z. Moreover, for the first time dimeric CPA/imine complexes in solution were characterised, hence extending the structural space of those complexes in solution and forming a bridge to the dimeric crystal structure. In addition, comprehensive data about the E/Z population of these complexes are provided, which deviate significantly from the calculated values (e.g. for TRIP/5: experimental ΔGrel E/Z = 2.0 kJ mol−1; theoretical ΔGrel E/Z = 9.3 kJ mol−1).29 This indicates a strong influence of refined dispersion interactions and/or solvent interactions not reflected by the commonly applied solvent models and offers the possibility to validate energetic results for theoretical prediction models in ion pairing catalysis.
Results and discussion
Investigated complexes and their NMR properties
In order to investigate the influence of different 3,3′-substituents of CPAs as well as different substituents of the imines on the structures of CPA/imine complexes and their E/Z populations, we selected several CPA catalysts and imines (see Fig. 3) used in synthesis34 and screened their NMR properties. Dichloromethane was used as solvent, since it gave small signal linewidths and the highest achievable signal dispersion at low temperatures.29 Furthermore, temperatures between 170 K and 200 K were needed to sufficiently slow down exchange processes in solution and most of the structural investigations were performed at 180 K unless otherwise noted.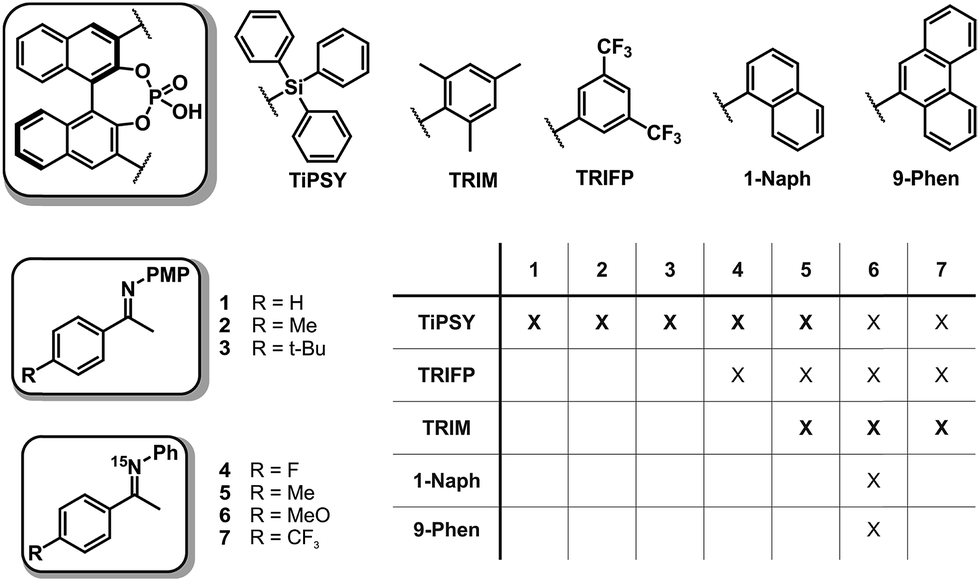 | ||
Fig. 3 Chiral phosphoric acids and imines with different functional groups that were used for the NMR-spectroscopic investigations of CPA/imine complexes (PMP = p-methoxyphenyl). All screened CPA/imine combinations are shown in the table. A bold X marks complexes, which could be investigated in detail. For all systems, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of CPA and imine was used. :1 ratio of CPA and imine was used. | ||
In principle, CPA/imine complexes with TiPSY and TRIFP produced basic signal pattern very similar to TRIP (see Fig. 4). The spectra showed highly overcrowded aromatic regions and two well separated hydrogen bond signals for the E and Z complexes. Thus, quantification of E and Z complexes is straightforward by integration of the H-bond protons. The access to a detailed structural analysis however depended individually on the overlap of key signals and their linewidths. For TiPSY complexes a structural analysis could be completed despite the significantly higher signal overlap in the aromatic region compared to TRIP. For TRIFP complexes line width factors and chemical shift overlap of key signals prevented any further structural analysis.
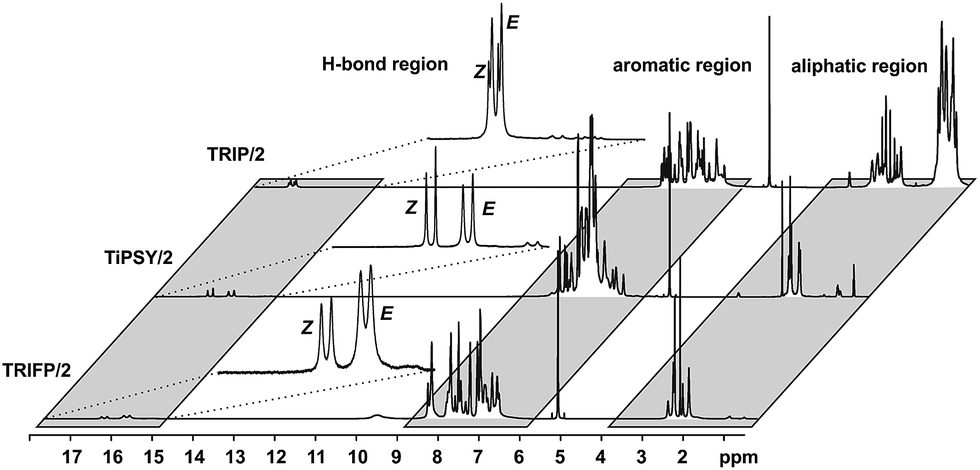 | ||
| Fig. 4 Spectral resolution of the 1H spectra of complexes between catalysts TRIP, TiPSY and TRIFP and imine 2 at 180 K in CD2Cl2; in all cases well separated signals of the hydrogen bonds indicate the E/Z populations, while deviations in linewidths and chemical shift overlap of key signals allow the structural investigations via NOE analysis only for complexes with TIPSY. | ||
For complexes with TRIM, 9-Phen, and 1-Naph as catalyst additional signals in the hydrogen bond region appeared at slightly lower chemical shifts (12.0–14.0 ppm) indicating an extended structural space for these complexes (see Fig. 5). The detailed structural investigations of TRIM complexes (see below) and dilution experiments for TRIM, 9-Phen and 1-Naph (see ESI S1–3†) identified these species as dimers, most probably enabled by attractive interactions between the 3,3′-substituents of TRIM, 9-Phen, and 1-Naph and the imines. For 9-Phen and even more pronounced for 1-Naph, a plethora of additional signals appeared in the hydrogen bond region of the 1H spectrum at 180 K. Given the asymmetry of the 3,3′-substituents of these catalysts and a rotational barrier of ≈14 kcal mol−1,21 at least two slow exchanging rotational isomers of these catalyst are expected at 180 K, causing a signal splitting of the E and Z complexes, hence revealing the whole conformational space of these binary complexes.
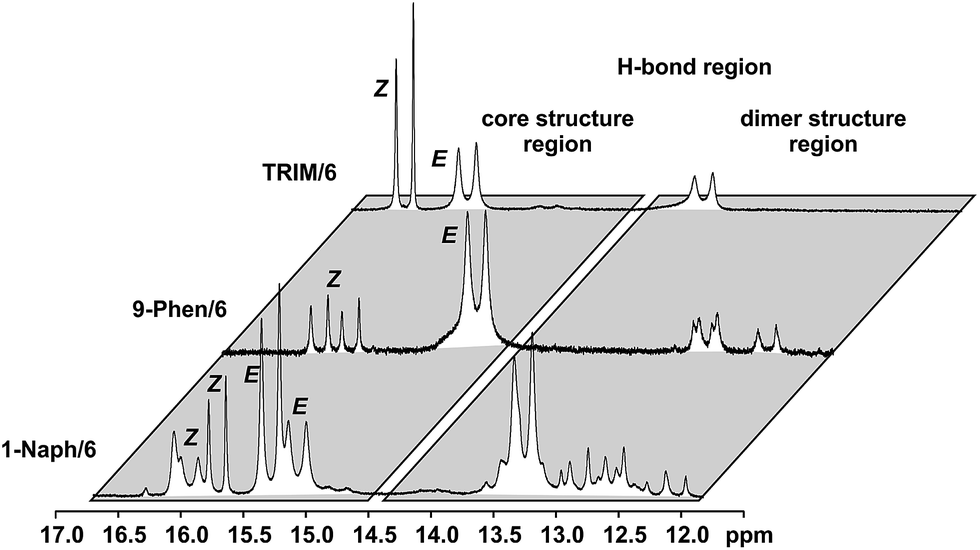 | ||
| Fig. 5 1H spectra of the H-bond region of catalysts TRIM, 9-Phen and 1-Naph at 180 K in CD2Cl2 reveal the extended structural space of these catalysts including dimeric complexes and additional conformations due to asymmetric 3,3′-substituents in 9-Phen and 1-Naph. | ||
Based on this NMR screening and initial 2D assignments, binary complexes with TiPSY and imines 1, 2, 3, 4 and 5 as well as TRIM and imines 5–7 were selected for the in detail structural investigations described in the following.
Structural space of TiPSY/imine complexes
For complexes providing such degree of signal overlap as shown in the aromatic region in Fig. 4, there are quite a few NMR spectroscopic techniques that are typically used to improve signal dispersion. These are mainly 3D experiments such as 3D-15N NOESY HSQC35 or 3D 13C NOESY HSQC36 or pure shift techniques.37–39 Especially the multidimensional approach40–42 is broadly applied in biochemistry and requires the use of specific labelling strategies.43–45 Due to the limited possibilities to include isotope labelling via different synthetic strategies into the catalyst or the imine, the chemical exchange even at low temperatures29 and the lowered solubility at the required temperatures, all of these more advanced techniques proved to be too insensitive to provide structural information in our case. Instead we focused mainly on the most sensitive one- and two-dimensional NMR methods (1H,1H/1H,13C 2D spectra for assignments and selective 1D NOESY46–48/2D NOESY/1H,19F HOESY49,50 spectra for structural investigations).In TiPSY/imine mixtures (1:1) at 180 K varying E/Z complex ratios were found with a trend to higher Z amounts compared to TRIP/imine and TRIFP/imine complexes (see Table 1). A similar trend can be observed for TRIM/imine complexes. In TiPSY/4, the E/Z ratio reached 68:32, in TiPSY/2 53:47 and in TiPSY/3 51:49. For TiPSY/1 even a 45:55 E/Z ratio was found, a rare example with higher Z-imine concentration (see ESI S4 and S5† for spectra). A complete assignment of the TiPSY/imine complexes was done for imines 1, 2, 3 and 4 at 180 K (for spectra see ESI S6–S28†). Despite a severe signal overlap in the crowded aromatic region, most of the signals were unambiguously assigned.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|
| TRIP | n.d. | 62:38 |
n.d. | n.d. | 67:33 |
77:23 |
86:14 |
| TiPSY | 45:55 |
53:47 |
51:49 |
68:32 |
56:44 |
70:30 |
71:29 |
| TRIFP | n.d. | n.d. | n.d. | 83:17 |
70:30 |
81:19 |
69:30 |
| TRIM | n.d. | n.d. | n.d. | n.d. | 47:53 |
49:51 |
55:45 |
The assignment of the E Type core structures of the TiPSY/imine complexes (Type I E, Type II E) is shown exemplarily on imine 4. With the help of 19F spectroscopy the spectral resolution can be vastly improved by reducing the complexity of the obtained spectra.51–53 Orientation Type I E was identified in a 2D 1H 19F HOESY experiment. A total of four HOE cross-peaks were found between the fluorine atom of imine 4 and the BINOL backbone of the catalyst (Fig. 6).
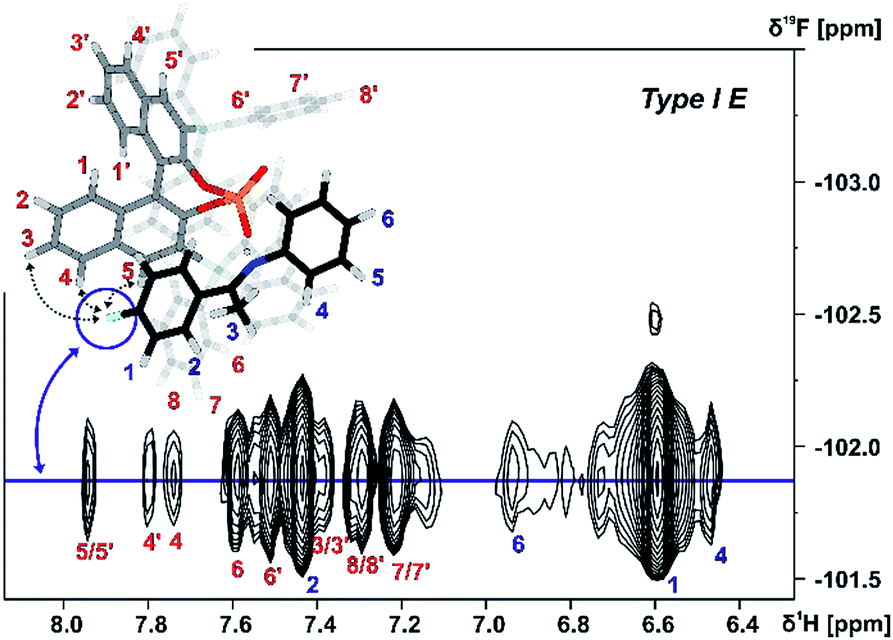 | ||
| Fig. 6 Section of a 1H, 19F 2D HOESY spectrum of TiPSY/4 at 180 K in CD2Cl2 at 600 MHz; Red dashed lines correspond to the intermolecular HOEs identifying complex structure Type I E (for detailed NMR parameters see ESI S19†). | ||
The presence of these HOE cross-peaks between imine 4 and the protons 3, 4 and 5 of the TIPSY backbone in combination with the hydrogen bond confirm the existence of the Type I E structure. Due to a dissociation, rotation and re-association of the TiPSY/imine complexes (Fig. 7), HOEs are also observed between the fluorine atom of 4 and the opposite naphthyl moiety of the catalyst (Fig. 6, HOE to proton 4′). This exchange is slow on the NMR timescale compared to the tilting mechanism that leads to an exchange between structures Type I E and Type II E (Fig. 7). A rotation of the E-imine inside the complex can be excluded, since the rotational barrier for this process would be by far too high due to the steric hindrance of the imine inside the complex. Orientation Type II E in the TiPSY/4 complex was similarly identified by using selective 1D NOESY experiments with saturation on proton 6 (see ESI S20†). Similar NOE patterns for conformations Type I E and Type II E were observed for TiPSY complexes with the imines 1, 2 and 3 (ESI S14–S18†).
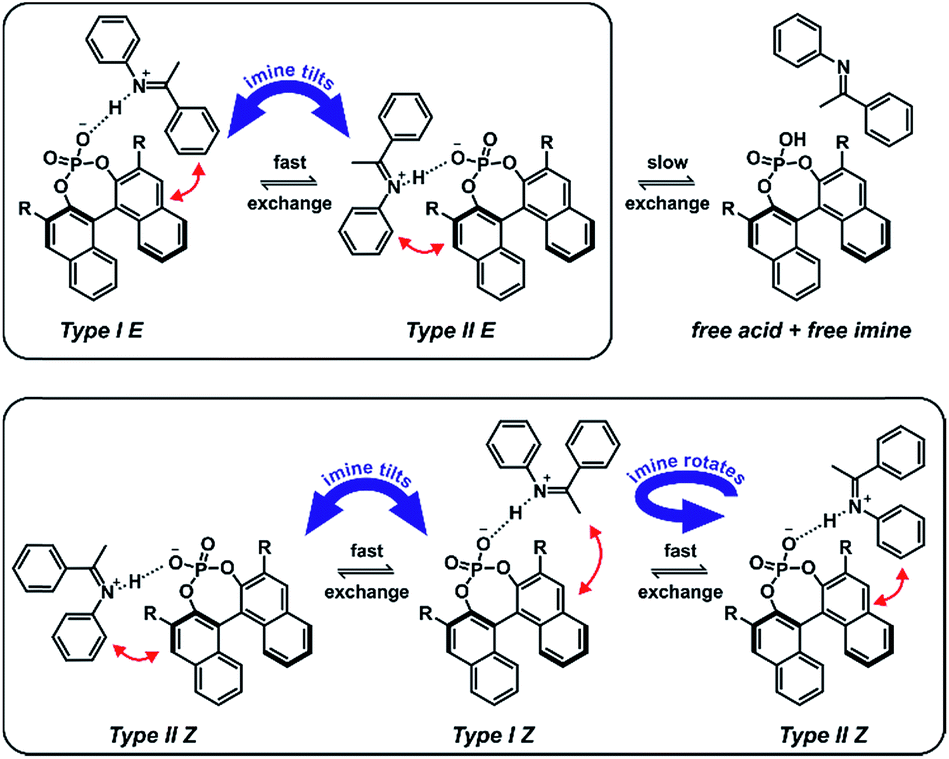 | ||
| Fig. 7 Complex exchange processes in TiPSY/imine complexes; an exchange via tilting of the imine is observed between structures Type I E and Type II E as well as a disassociation and association process of the TiPSY/E-imine complexes; the exchange via tilting is fast on the NMR time-scale and leads to a different interaction pattern for each half of the catalyst; the dissociation and association process is slow on the NMR time-scale and leads to exchange peaks between the catalyst halves; in addition to an exchange via tilting of the imine, the reduced steric hindrance of the Z-imine enables an additional fast exchange between structures Type I Z and Type II Z via rotation, leading to a different signal pattern. | ||
In contrast to the TiPSY/E-imine structures, only one set of signals is observed for the BINOL backbone of the catalyst in the TiPSY Z-imine complexes. This can be explained by the reduced steric hindrance of the Z-imine enabling an exchange between orientation Type I Z and Type II Z via rotation of the imine around the hydrogen bond (Fig. 7). This exchange pathway in addition to the exchange via tilting of the imine (see TiPSY/E-imine), results in only one average set of 1H signals for the BINOL backbone of the catalyst.29
Both orientations Type I Z and Type II Z were detected for the TiPSY/Z-imine complexes. Complex TiPSY/3 is used exemplarily for the assignment of Type I Z (Fig. 8). In the 1H NOESY spectrum, two specific intermolecular NOEs between the α-methyl group of the imine and the BINOL backbone of the catalyst were found. This interaction, in combination with the strong hydrogen bond between catalyst and imine shows the existence of orientation Type I Z in solution. Similarly, orientation Type II Z could be confirmed in selective 1D NOESY spectra (ESI S26†) and the same NOE pattern were observed for all investigated complexes TiPSY/1,2,3,4 (ESI S21–S28†).
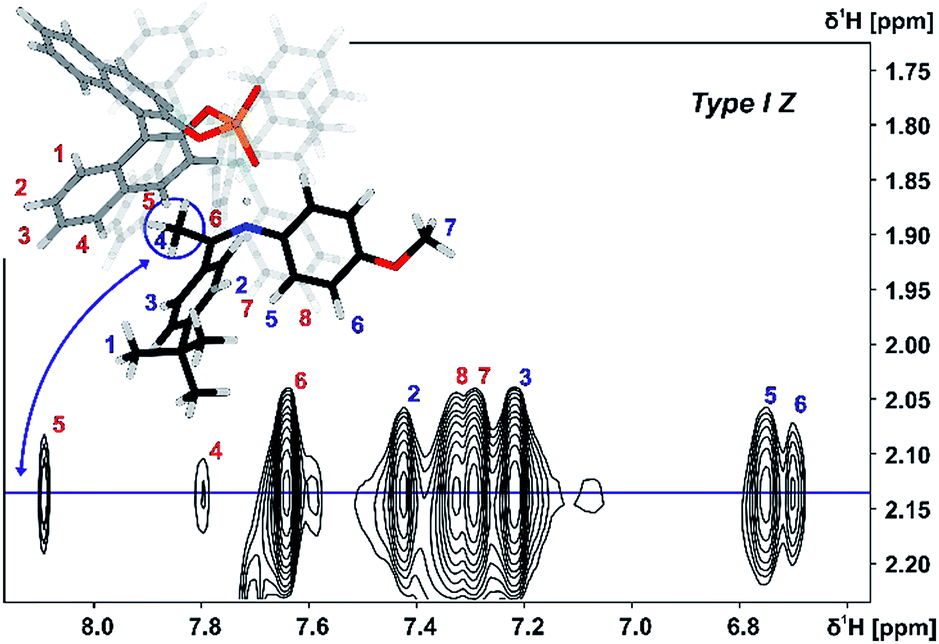 | ||
| Fig. 8 Excerpt of the 2D NOESY spectrum of TiPSY/3 at 180 K in CD2Cl2 at 600 MHz; intermolecular cross-peaks (red numbers) detail the interaction between α-methyl group of 3 (blue spin 4) and BINOL backbone of the catalyst (for detailed parameters see ESI S25†). | ||
Thus, despite significantly deviating 3,3′-substituent/imine interactions causing different E/Z ratios in TiPSY/imine complexes (see Table 1), for all TiPSY/imine complexes studied in detail the structures Type I E and Type II E as well as Type I Z and Type II Z could be elucidated by NMR.
Structural space of TRIM/imine complexes and dimerization trends of CPAs
For TRIM complexes with imines 5, 6, and 7 the same four core structures Type I/II E/Z were experimentally identified by NOE analysis (for assignment and spectra see ESI S33–42†). In contrast to TRIP and TiPSY only one set of catalyst signals for both Type I/II E structures even at 180 K is observed, which can be explained by an additional exchange pathway. This pathway is potentially enabled via facilitated rotation like it is the case for Type I/II Z and/or an enhanced dissociation-association process of the complex (for detailed explanation see ESI S43†).21Furthermore, the additional hydrogen bonded signals as discussed above (see Fig. 5) were identified as [TRIM/E-imine]2 dimers and were in detail investigated on complex TRIM/7.§ Chemical exchange between TRIM/7E and [TRIM/7E]2 was identified by EXSY signals and dilution of the sample lead to a complete shift of the monomer–dimer equilibrium towards the monomers (see ESI S1–3†). In addition, DOSY measurements corroborated the assignment as a dimer (see ESI S44†). Theoretical calculations supported by distinct changes of the measured chemical shifts (see Fig. 9) confirm the observed dimer structures to be similar to the reported crystal structure of Schneider28 (see Fig. 11 below).
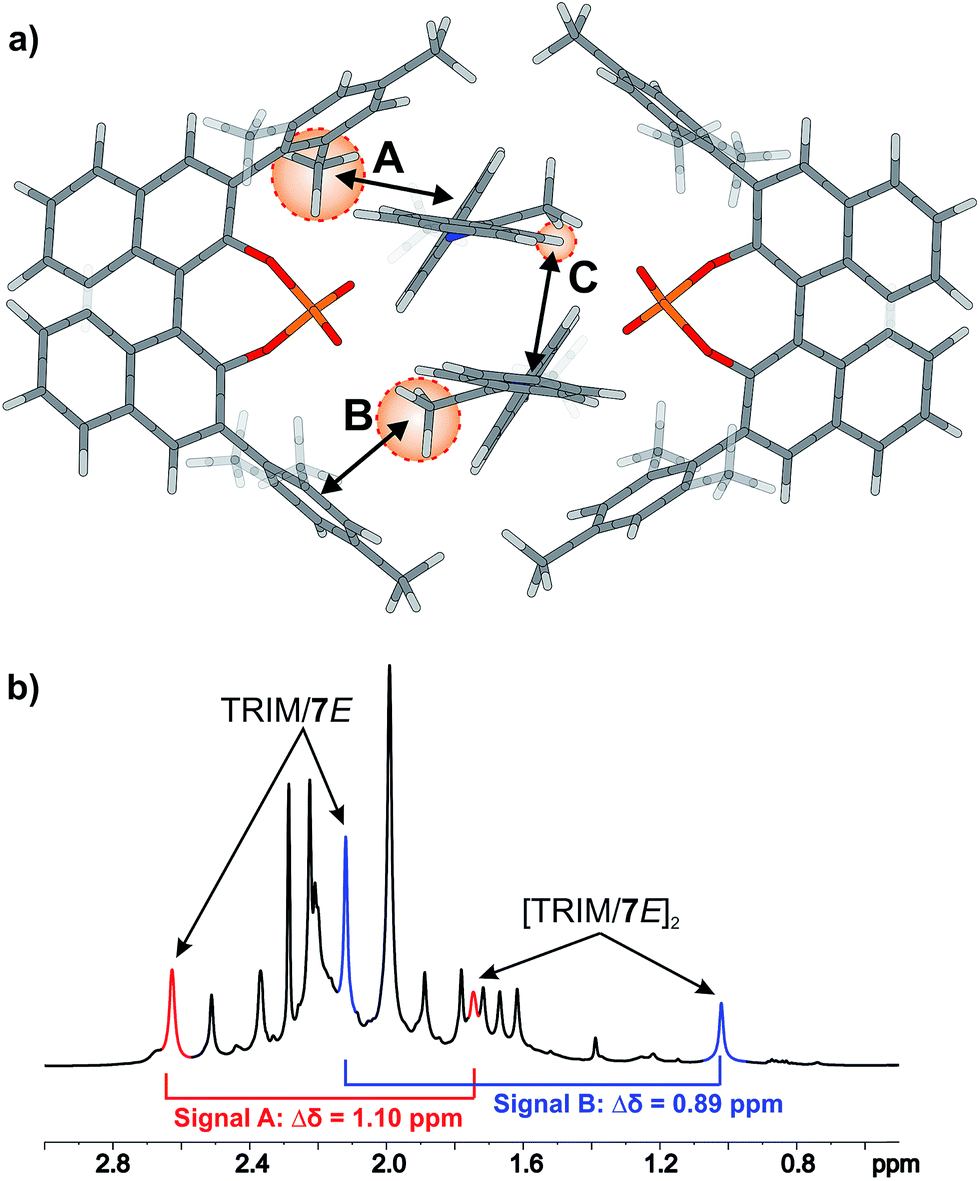 | ||
| Fig. 9 (a) Theoretically calculated dimer structure model on the example of the [TRIM/5E]2 complex reveals the steric proximity of the protons A–C to phenyl entities which causes paratropic shielding effects and induces severe highfield shifts. (b) Excerpt of the 1H spectrum of TRIM/7 at 180 K in CD2Cl2 at 600 MHz highlighting the highfield shift of proton A (red) and B (blue) of the dimer compared to the monomer. The significant highfield shifts of A–C compared to the monomeric structure (A: 1.10 ppm, B: 0.89 ppm, C: 1.56 ppm) experimentally corroborate the computed structure and indicate CH–π and π–π interactions. | ||
The 3,3′-substituent dependent formation of CPA/imine dimers gave rise to the question, which 3,3′-substituent properties enable or restrict dimer formation. Remarkably, all catalysts forming dimers possess medium rotational barriers of the 3,3′-substituent (13.63–21.58 kcal mol−1) as described by Goodman.21 This hints at the influence of putative van der Waals interactions between the catalyst 3,3′-substituent and the imine on the energetic interaction profile supporting dimer formation. The presence of such interactions is indicated for [TRIM/7E]2 by a strong highfield shift of proton A (Fig. 9), caused by steric proximity to a phenyl entity of the imine. For TRIFP and TiPSY a low rotational barrier (2.02 and 1.35 kcal mol−1 respectively)21 points out the lack of moieties capable of such interactions, while the high rotational barrier of TRIP (28.40 kcal mol−1)21 potentially indicates steric repulsion overwriting attractive interactions. Furthermore, putative CH–π interactions between the α-methyl group of the imine and the phenyl entities of the 3,3′-substituent (Interaction B in Fig. 9a) as well as π–π interactions between the two imines (Interaction C in Fig. 9a) might compensate the loss of interactions between the imine and BINOL-backbone present in the monomeric structures.29 Moreover, other 3,3′-substituent properties such as electrostatic repulsion, e.g. due to the CF3-groups in a hypothetic TRIFP dimer, or severe entropic penalties upon dimerization for catalysts with a high degree of conformational freedom of the 3,3′-substituents such as TiPSY and TRIP¶ might also affect dimerization trends.
It was reported that interlocked CPA catalysts can form P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯(HO)P hydrogen bonded pseudo-dimers which provide significantly higher stereoselectivity as their non-interlocked analogues.54 However, for synthetic applications CPA/imine dimers are not expected to have any influence as their population diminishes when approaching catalyst concentrations used in synthesis.6,7,55
O⋯(HO)P hydrogen bonded pseudo-dimers which provide significantly higher stereoselectivity as their non-interlocked analogues.54 However, for synthetic applications CPA/imine dimers are not expected to have any influence as their population diminishes when approaching catalyst concentrations used in synthesis.6,7,55
Hence, the existence of a dimer in the crystal structure of TeBuP and in the NMR spectra of TRIM, 9-Phen, and 1-Naph corroborates our previous assumption29 that attractive interactions between 3,3′-substituent and imine play a key role in the energetics of CPA complexes. Thus, the dimerization trend might be tentatively proposed as a qualitative measure for steric properties and polarizability of the 3,3′-substituent.
Effect of substituents on enantioselectivity in the transfer hydrogenation of ketimines
After ensuring the analogy of all catalytic relevant and NMR accessible structures, the influence of the 3,3′-substituents on the enantioselectivity was investigated. Therefore, the ee values for the transfer hydrogenation of imines 5, 6 and 7 with ethyl Hantzsch ester catalyzed by TRIP, TiPSY, TRIFP, TRIM, 1-Naph and 9-Phen were determined (see ESI S45†). As anticipated based on earlier work of Rueping,5 List6 and MacMillan7 and on the web tool BINOPtimal developed by Goodman,24 TiPSY (90–95% ee) gave the highest enantiomeric excess, followed by TRIP (83–86% ee) and TRIFP (74–91% ee), while TRIM (56–70% ee), 1-Naph (46–56% ee) and 9-Phen (47–59% ee) gave lower enantioselectivities. While imine 5 and 6 gave almost identical ee values for all investigated catalysts, the CF3 substituted imine 7 deviated up to ±15% for TRIFP, TRIM, 1-Naph, and 9-Phen. Since imines 5, 6 and 7 have a similar steric bulk this deviation suggests that steric interactions are not the only key factor, but that also electrostatic interactions can significantly modulate the enantioselective outcome. Next, linear correlations between the observed enantioselectivities and steric descriptors (AREA (θ) or rotational barrier) of the 3,3′-substituents as previously postulated by Goodman21 were tested. However, neither with AREA (θ) nor with the rotational barrier a linear correlation was found for our data (see ESI S46†). Moreover, the enantioselectivities also showed no correlation with the E/Z ratios of the binary complexes (see ESI S46†). This shows that a single parameter derived from catalyst alone or binary complex is not sufficient to adequately predict the stereoselective outcome at least of the investigated reaction. Rather multiple factors seem to affect the energetics of the reaction pathway, which will be subject of further investigations.Theoretical model structures and structure comparison
The above mentioned experimentally identified dimeric structures seem to be stabilized by attractive interactions, often associated with London dispersion forces. The presence of such interaction modes was already investigated and proven for TRIP/imine complexes in the previous theoretical structural studies utilizing NCI analysis and NOE analysis.29,32 The binary complex is mainly anchored by a strong hydrogen bond,32 however equally important, it is also bolstered by numerous weak attractive interactions between the imine and the catalyst's backbone as well as the bulky 3,3′-substituents.29 These interactions are also pervasive in other catalysts with different 3,3′-motives.Our theoretical calculations of TRIP, TiPSY, TRIFP, TRIM/5–7 confirm the general existence of the four core structures (Type I/II E/Z) (for computational details and data on additional CPAs, see ESI S49†) in accordance with previous results for similar complexes.29 Each of the E- and Z-imine complexes features two different orientations of the imine (Fig. 10). In the Type I orientation, the ketone moiety is located in close proximity to the BINOL backbone of the catalyst. In the Type II orientation, the imine is rotated around the hydrogen bond by ∼180° and the aniline moiety is located close to the BINOL backbone.
 | ||
| Fig. 10 Theoretical calculations of the TiPSY, TRIP, TRIFP, TRIM/5 complex show the invariance of four core structures. Despite that, the E/Z ratios and the tendency towards dimerization vary strongly within the CPA/imine complexes. Moreover, quantum chemical calculations of E/Z-ratio showed a significant offset to the experimental values. Colour code: TiPSY red, TRIP blue, TRIFP orange, TRIM green. | ||
Geometrical comparison of the four core structures from the calculation revealed a high degree of invariance in the CPA/imine complexes, which is, to our surprise, retained despite significant variation of 3,3′-substituent of the catalyst (Fig. 10). As in TRIP/imine complexes29 the two orientations of the E- and Z-imine are obtained in TiPSY, TRIFP, TRIM/5–7 binary complexes. Furthermore, similar van der Waals interaction types (π–π, CH–π) are clearly recognizable from the structural analysis. In accordance with the previous studies,32 the acidic proton is located closer to the imine in TiPSY and TRIFP binary complexes, which indicates a strong predominant zwitter ionic, but also partly covalent character (POH angle ≈ 110°–120°). Due to the high degree of invariance of the core structures, the E/Z-ratio in the binary complexes is altered due to the difference in stabilization of E- and Z-imine by the 3,3′-substituent. It is noteworthy to mention that despite a good agreement between experimentally and theoretically determined structures, the theoretical E/Z-ratio deviates significantly from the measured E/Z-ratio (Table 2).
| Experimental E:Z |
Theoretical E:Z |
|
|---|---|---|
| TRIP/5 | 79:21 |
99.80:0.20 |
| TRIFP/5 | 70:30 |
96.80:3.20 |
| TiPSY/5 | 56:44 |
99.97:0.03 |
| TRIM/5 | 47:53 |
80.41:19.59 |
Mostly, only the qualitative trend could be predicted correctly with exception of TRIM-complexes, i.e. experimentally the E-complex is less stable than the Z-complex. Despite extensive conformational search (for computational details see below), application of various dispersion corrected density functionals (GGA, hybrid GGA, meta-hybrid GGA), post-HF methods (MP2, SCS-MP2, DLPNO-CCSD(T)) and implicit solvation models (SMD, COSMO-RS), the thermodynamic stability of the considered CPA/Z-isomers is gravely underestimated. One of the most common encountered error sources in the theoretical calculation during the simulation of large flexible molecules is the initial sampling using molecular mechanics not being able to find the initial structure near to global minimum. This problem is augmented, especially in our case, by the poor parameterization of strongly hydrogen-bridged and zwitter-ionic complexes. Nevertheless, given the number of sampling conformations, experimental NOE contacts and the remarkable agreement of calculated coupling constants with the experimental values,32 the possibility of not finding the global minimum of the Z-complex is reduced significantly.
Since the weak interactions, e.g. dispersion effect in the complex, were already accounted in the post-HF methods, the underlying cause for the deviation might be either the missing explicit solvent–solute interaction favouring the Z-isomers due to its compactness, and hence a larger available solvent–solute interaction space, or the inaccurate implicit solvation of such zwitter-ionic species. For the former, tremendous increase of computational cost is expected making full ab initio calculations very restricted. An initial calculation of solvated complexes in a solvent box consisting of 300 solvent molecules has been performed and accurate calculations are planned for the future. For the latter, recent thorough investigation of solvent effect on the attenuation of inter- and intramolecular dispersive interaction showed that the current frequently employed implicit solvent models fail to describe dispersive solvent–solute interaction.56 In summary, several underlying reasons for the failure to predict the E/Z-ratio are multiple. First, considering the huge conformational space of the complex, accurate initial sampling is necessary to catch the nearest minimum for DFT refined optimization. In this case, accurate semi-empirical methods, such as tight-binding method57 and sampling using meta-dynamics may resolve the sampling issue in large electronic structures. Second, accurate description for solvation might be still underdeveloped for strongly bound zwitter-ionic species with large aromatic surfaces causing the solvent attenuation of dispersive interaction. Therefore, further improvement for implicit solvent correction is necessary.
In addition, a structure model for dimeric CPA/imine complexes was computed on the example of TRIM/5 (see Fig. 9 and 11). This dimeric structure features two imines, nested between two catalysts. An extended dispersion force between the 3,3′-substituents of the two catalysts and the two extended aromatic imines is observed. The additional imine in the dimeric complex seems to be positioned at the nucleophilic attack site, when comparing the structure to the hydride transfer transition state with Hantzsch ester as the reducing agent. An overlap of the imine substrates and the Hantzsch ester is observed which suggests similar interaction modes (Fig. 11c).
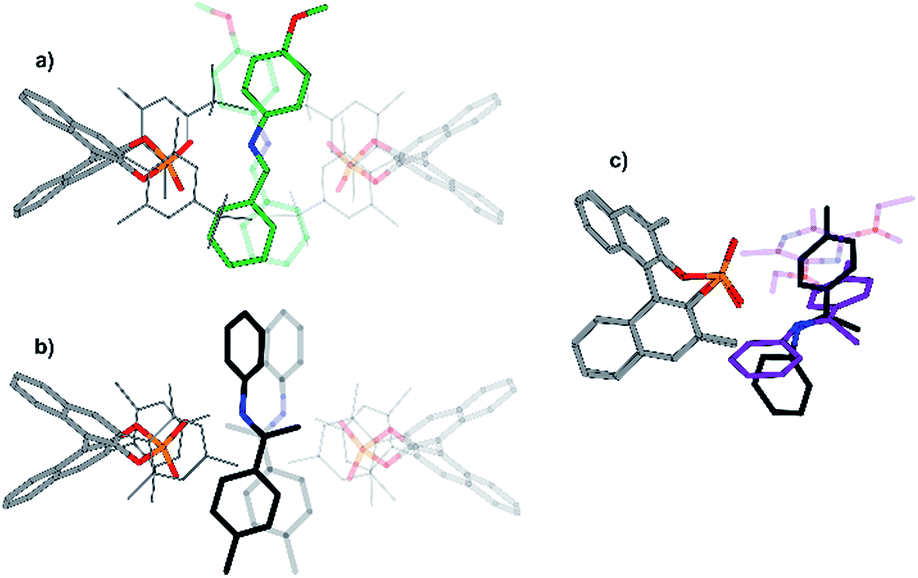 | ||
| Fig. 11 Reported crystal structure of a TeBuP/imine dimer (a) and calculated structure of a TRIM/imine dimer (b). The structures are similar despite comparing an aldimine (a) to a ketamine structure (b). (c) Comparison of the dimeric structure of a TRIM complex (black imine) with the calculated transition state consisting of a CPA (the 3,3′-substituents of the catalysts have been omitted to improve the visibility), an imine (purple) and a Hantzsch ester (purple, transparent). The steric influence of the second imine in the dimeric structure (b) is similar to the additional bulk of the Hantzsch ester in the transition state. | ||
Computational details
All binary complexes were optimized at DFT level of theory using TPSS functional with D3 correction in continuum of DCM. To mimic experimental low temperature condition the dielectric constant of DCM was increased to 16.20. Preconformational sampling was performed using force-field with MMFF parameter. Overall 1137 conformations of TRIP/imine were generated in the force-field procedure and subjected to ab initio geometry optimization. Low lying conformations were selected according to experimental constrains (NOE contacts). For other catalyst/imine conformations, core structures based on TRIP/imine complexes were taken and adapted. Post-HF single point calculations at SCS-MP2/CBS level of theory were performed on the optimized geometry (see ESI S46† for extrapolation procedure).Conclusion
A combination of detailed NMR spectroscopic studies and theoretical calculations was used to investigate the structural space of CPA/imine complexes with varying 3,3′-substituents. Our experimental data revealed the coexistence of four core structures (Type I/II E and Type I/II Z) in solution, similar to those described for binary complexes of TRIP. These core structures show a high invariance, independent of the used CPA, which is corroborated by theoretical calculations of TiPSY/imine, TRIFP/imine and TRIM/imine complexes. For the first time dimeric CPA/imine complexes were identified in solution for TRIM, 9-Phen and 1-Naph with an imine occupying the space analogous to the transition state in the transfer hydrogenation. Surprisingly, this dimer consisting of two binary complexes is stabilized exclusively by attractive non covalent interactions. Hence, not only the steric repulsion term but also the attractive term of the large 3,3′-substituents has to be considered for the energetics of CPA complexes. Despite the good agreement of experiment and theory for the identified structures, a significant disparity was found for the E/Z ratios of the complexes. All binary complexes exhibit an increased population of the Z-structures, which cannot be reproduced in the calculations. A possible reason is an insufficient description of solvent–complex interactions. This study may allow to validate and improve theoretical predictive models by providing for the first time detailed experimental data about the structural space and influence of the 3,3′-substituent on the energetics of CPA/imine complexes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the European Research Council (ERC-CoG 614182 − IonPairsAtCatalysis) and intellectual support was provided by SPP 1807 dispersion. We would like to thank Dr Nils Sorgenfrei and Thomas Hausler for preliminary investigations of catalyst imine complexes and Dr Fabio Morana for establishing in the beginning of our ion pair investigations the syntheses of catalysts and substrates.Notes and references
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2017, 117, 10608–10620 CrossRef CAS PubMed.
- T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 Search PubMed.
- D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 Search PubMed.
- M. Rueping, E. Sugiono, C. Azap, T. Theissmann and M. Bolte, Org. Lett., 2005, 7, 3781–3783 CrossRef CAS PubMed.
- S. Hoffmann, A. M. Seayad and B. List, Angew. Chem., Int. Ed., 2005, 44, 7424–7427 CrossRef CAS PubMed.
- R. I. Storer, D. E. Carrera, Y. Ni and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 84–86 Search PubMed.
- K. Saito and T. Akiyama, Chem. Commun., 2012, 48, 4573–4575 RSC.
- M. Yamanaka, J. Itoh, K. Fuchibe and T. Akiyama, J. Am. Chem. Soc., 2007, 129, 6756–6764 Search PubMed.
- X. Xu, Y. Qian, L. Yang and W. Hu, Chem. Commun., 2010, 47, 797–799 RSC.
- E. P. Ávila, R. M. S. Justo, V. P. Gonçalves, A. A. Pereira, R. Diniz and G. W. Amarante, J. Org. Chem., 2015, 80, 590–594 Search PubMed.
- K. Shen, X. Liu, Y. Cai, L. Lin and X. Feng, Chem.–Eur. J., 2009, 15, 6008–6014 CrossRef CAS PubMed.
- C. Gharui, S. Singh and S. C. Pan, Org. Biomol. Chem., 2017, 15, 7272–7276 RSC.
- S. Saha and C. Schneider, Chem.–Eur. J., 2015, 21, 2348–2352 Search PubMed.
- S. G. Ouellet, A. M. Walji and D. W. C. Macmillan, Acc. Chem. Res., 2007, 40, 1327–1339 CrossRef CAS PubMed.
- C. Min and D. Seidel, Chem. Soc. Rev., 2017, 46, 5889–5902 Search PubMed.
- Q. Kang, Z.-A. Zhao and S.-L. You, Org. Lett., 2008, 10, 2031–2034 CrossRef CAS PubMed.
- K. Saito, K. Horiguchi, Y. Shibata, M. Yamanaka and T. Akiyama, Chem.–Eur. J., 2014, 20, 7616–7620 CrossRef CAS PubMed.
- R. Maji, S. C. Mallojjala and S. E. Wheeler, Chem. Soc. Rev., 2018, 47, 1142–1158 Search PubMed.
- A. Milo, A. J. Neel, F. D. Toste and M. S. Sigman, Science, 2015, 347, 737–743 Search PubMed.
- J. P. Reid and J. M. Goodman, J. Am. Chem. Soc., 2016, 138, 7910–7917 CrossRef CAS PubMed.
- J. P. Reid, L. Simón and J. M. Goodman, Acc. Chem. Res., 2016, 49, 1029–1041 CrossRef CAS PubMed.
- L. Simón and J. M. Goodman, J. Org. Chem., 2011, 76, 1775–1788 CrossRef PubMed.
- J. P. Reid and J. M. Goodman, Chem. Commun., 2019, 55, 1778–1781 RSC.
- C. B. Santiago, J. Y. Guo and M. S. Sigman, Chem. Sci., 2018, 9, 2398–2412 RSC.
- A. J. Neel, A. Milo, M. S. Sigman and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 3863–3875 Search PubMed.
- M. Orlandi, J. A. S. Coelho, M. J. Hilton, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 6803–6806 Search PubMed.
- M. Sickert, F. Abels, M. Lang, J. Sieler, C. Birkemeyer and C. Schneider, Chem.–Eur. J., 2010, 16, 2806–2818 CrossRef CAS PubMed.
- J. Greindl, J. Hioe, N. Sorgenfrei, F. D. Morana and R. M. Gschwind, J. Am. Chem. Soc., 2016, 49, 15965–15971 CrossRef PubMed.
- W. Tang, S. Johnston, J. A. Iggo, N. G. Berry, M. Phelan, L. Lian, J. Bacsa and J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 1668–1672 CrossRef CAS PubMed.
- L. Liu, M. Leutzsch, Y. Zheng, M. W. Alachraf, W. Thiel and B. List, J. Am. Chem. Soc., 2015, 137, 13268–13271 CrossRef CAS PubMed.
- N. Sorgenfrei, J. Hioe, J. Greindl, K. Rothermel, F. Morana, N. Lokesh and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 16345–16354 Search PubMed.
- L. Simón and J. M. Goodman, J. Am. Chem. Soc., 2008, 130, 8741–8747 CrossRef PubMed.
- T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 Search PubMed.
- E. R. P. Zuiderweg and S. W. Fesik, Biochemistry, 1989, 28, 2387–2391 Search PubMed.
- J. Schleucher, M. Schwendinger, M. Sattler, P. Schmidt, O. Schedletzky, S. J. Glaser, O. W. Sørensen and C. Griesinger, J. Biomol. NMR, 1994, 4, 301–306 CrossRef CAS PubMed.
- M. Foroozandeh, R. W. Adams, M. Nilsson and G. A. Morris, J. Am. Chem. Soc., 2014, 136, 11867–11869 CrossRef CAS PubMed.
- J. Mauhart, S. Glanzer, P. Sakhaii, W. Bermel and K. Zangger, J. Magn. Reson., 2015, 259, 207–215 CrossRef CAS PubMed.
- K. Zangger, Prog. Nucl. Magn. Reson. Spectrosc., 2015, 86–87, 1–20 CrossRef CAS PubMed.
- D. Uhrín, J. Bramham, S. J. Winder and P. N. Barlow, J. Biomol. NMR, 2000, 18, 253–259 CrossRef.
- A. Verma and B. Baishya, J. Magn. Reson., 2016, 266, 51–58 Search PubMed.
- S. Akoka and P. Giraudeau, Magn. Reson. Chem., 2015, 53, 986–994 CrossRef CAS PubMed.
- R. J. Lichtenecker, N. Coudevylle, R. Konrat and W. Schmid, ChemBioChem, 2013, 14, 818–821 CrossRef CAS.
- C. Prasanna, A. Dubey and H. S. Atreya, Methods in Enzymology, Academic Press, 2015, vol. 565, pp. 167–189 Search PubMed.
- D. P. Frueh, A. C. Goodrich, S. H. Mishra and S. R. Nichols, Curr. Opin. Struct. Biol., 2013, 23, 734–739 CrossRef CAS.
- H. Kessler, H. Oschkinat, C. Griesinger and W. Bermel, J. Magn. Reson., 1986, 70, 106–133 CAS.
- K. Stott, J. Stonehouse, J. Keeler, T.-L. Hwang and A. J. Shaka, J. Am. Chem. Soc., 1995, 117, 4199–4200 CrossRef CAS.
- D. Jeannerat and J. Furrer, Comb. Chem. High Throughput Screening, 2012, 15, 15–35 CrossRef CAS.
- C. Yu and G. C. Levy, J. Am. Chem. Soc., 1984, 106, 6533–6537 CrossRef CAS.
- P. S. Pregosin, P. G. Anil Kumar and I. Fernández, Chem. Rev., 2005, 105, 2977–2998 CrossRef CAS.
- K. Shikii, S. Sakurai, H. Utsumi, H. Seki and M. Tashiro, Anal. Sci., 2004, 20, 1475–1477 CrossRef CAS.
- D. P. Cistola and K. B. Hall, J. Biomol. NMR, 1995, 5, 415–419 CrossRef CAS.
- L. Fusaro, E. Locci, A. Lai and M. Luhmer, J. Phys. Chem. B, 2010, 114, 3398–3403 CrossRef CAS PubMed.
- R. Mitra, H. Zhu, S. Grimme and J. Niemeyer, Angew. Chem., Int. Ed., 2017, 56, 11456–11459 CrossRef CAS PubMed.
- M. Sickert and C. Schneider, Angew. Chem., Int. Ed., 2008, 47, 3631–3634 CrossRef CAS PubMed.
- R. Pollice, M. Bot, I. J. Kobylianskii, I. Shenderovich and P. Chen, J. Am. Chem. Soc., 2017, 139, 13126–13140 CrossRef CAS.
- S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01044k |
| ‡ Authors contributed equally to this work. |
| § Due to chemical exchange between TRIM/imine, [TRIM/imine]2 and free imine and the resulting line broadening as well as signal overlap, only the complex TRIM/7 could be analysed in detail, as it gave adequate line widths. |
| ¶ Significant line broadening for distinct proton signals of TRIPs isopropyl and TiPSYs phenyl-moieties at 180 K caused by medium chemical exchange between different substituent conformations demonstrated a high degree of conformational freedom for these catalysts. |
| This journal is © The Royal Society of Chemistry 2019 |