
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-step multivalent capture assay for nucleic acid detection with dual-affinity regulation using mutation inhibition and allosteric activation†
Xing
Lu‡
,
Guobao
Zhou‡
 *,
Yanbo
Zeng
,
Zhengzhi
Yin
,
Zulei
Zhang
,
Liping
Guo
,
Yunyun
Zhai
,
Yiwen
Yang
,
Hailong
Wang
and
Lei
Li
*
*,
Yanbo
Zeng
,
Zhengzhi
Yin
,
Zulei
Zhang
,
Liping
Guo
,
Yunyun
Zhai
,
Yiwen
Yang
,
Hailong
Wang
and
Lei
Li
*
College of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing 314001, China. E-mail: lei.li@mail.zjxu.edu.cn; gbzhou@mail.zjxu.edu.cn
First published on 5th April 2019
Abstract
The rational modulation of receptor affinity through distal-site mutation and allosteric control is valuable in biosensor designing to tune the useful dynamic range. Our ability to programmatically engineer dual-affinity regulation into diverse affinities of target binding and activities of hybridization chain reaction, however, remains limited. By programmable engineering of the switching equilibria of the recognition hairpin using distal-site mutation inhibition and allosteric activation, we obtained a set of receptors varying significantly in affinities of target binding and activities of the hybridization chain reaction. For the first time, we developed an electrocatalytic biosensor for nucleic acid detection with a tunable dynamic range based on a conformational switch triggered bidirectional hybridization chain reaction and blocker assisted multivalent binding. This designable biosensor thus enables single-step incubation, diverse affinities of target binding, diverse efficiencies of signal amplification and diverse single nucleotide discrimination for quantitative analyses of nucleic acids of various lengths in serum, which holds great potential as a compelling platform suitable for liquid biopsy.
Introduction
Mutation and allosteric control are two main strategies for biomolecules regulating their affinities in many naturally occurring biology processes.1–5 The underlying mechanisms that regulate molecular binding affinities are increasingly providing inspiration in biosensors to tune, extend and narrow the useful detection range. Mutation and allosteric control are often employed in biosensors to regulate diverse affinities of receptors responding to their target molecules, which efficiently transfer a given input to an altered output.6–10 Biomimetic sensors that employ mutation or allosteric regulation have a broad control of the dynamic range, but most of them have low sensitivity of signal transduction.11,12 The biosensors based on precise valence control of enzymes have high enzymatic amplification for signal transduction. However, they have a limited adjustable dynamic range, which is only dependent on the number of linked enzymes.13,14 The dual-affinity regulation of receptors with diverse affinities of target binding and activities of the hybridization chain reaction are ideally suited to both extend the dynamic range and amplify signal transduction. This unique property of dual-affinity regulation meets the desirable demand of biosensors with a tunable dynamic range for the analysis of biomarkers in clinical samples with different concentrations. For example, the levels of the overall cell-free DNA expression profile vary from the aM level to the pM level in peripheral blood samples in both healthy controls and cancer patients.15,16The detection of cell-free DNA expression profiles at the genetic level enables broad liquid biopsy approaches in infection diagnostics,17 early childhood wheezing,18 fetal chromosome abnormalities,19 cancer management,20–25etc. As cf-DNA is more chemically and bioenvironmentally stable than RNA, cf-DNA is particularly informative, suitable as a molecular biomarker.26,27 Sensitive, robust and specific detection of DNA plays an important role in efficient diagnosis, prognostic analysis of cancer metastasis and cancer therapies. However, sensitive and single nucleotide resolution detection of DNA remains challenging because of the low abundance of targets in peripheral blood.
In the past few decades, some popular and potent technologies using signal amplification strategies have been developed to increase the nucleic acid detection sensitivity. Typically, DNA nanostructure based signal amplification methods mainly include the rolling circle reaction (RCA),28–34 hybridization chain reaction (HCR),35–44 entropy-driven catalysis (EDC),45–48 catalyzed hairpin assembly (CHA),49–54etc. The hybridization chain reaction is even widely employed in biosensors to amplify the signal. Fan and coworkers had integrated the hybridization chain reaction amplification and tetrahedral DNA nanostructure (with outstanding hybridization conditions) to build an ultrasensitive nucleic acid detection platform.38 However, it suffers from extensive sample processing with multiple steps of incubation and washes, which restrain its applications in on-site, real time analytical devices suitable for point of care (POC) detection.
Existing hybridization chain reaction amplified biosensors have several disadvantages. First, the efficiency of the hybridization chain reaction that occurred at interfaces is low due to the limited mass transfer of the hairpins to the surface. Second, the hairpin probes used in the hybridization chain reaction are not universal due to the sequences of the hairpins varied with the change of the initiator sequence. Third, the stabilities of the hairpins that will affect the signal to noise (S/N) ratio are greatly dependent on the target sequence. Fourth, although ultrasensitive nucleic acid detection performance has been obtained by using hybridization chain reaction amplification, multiple processing steps are always required. The cancer cell capture and analysis employing the multibranched hybridization chain reaction proposed by Zuo and coworkers55 had solved some of the above problems. However, the excess unreacted hairpins incubated with cancer cells should be removed by using several centrifugation and washing steps. These processes will lead to serious loss of cancer cells. In this work, excess unreacted hairpins can't be separated easily from target induced multibranched hybridization chain reaction products. So we proposed the blocker assisted multivalent capture strategy to prohibit the excess hairpin competitive binding with capture probes on the surface (Fig. 1, bottom).
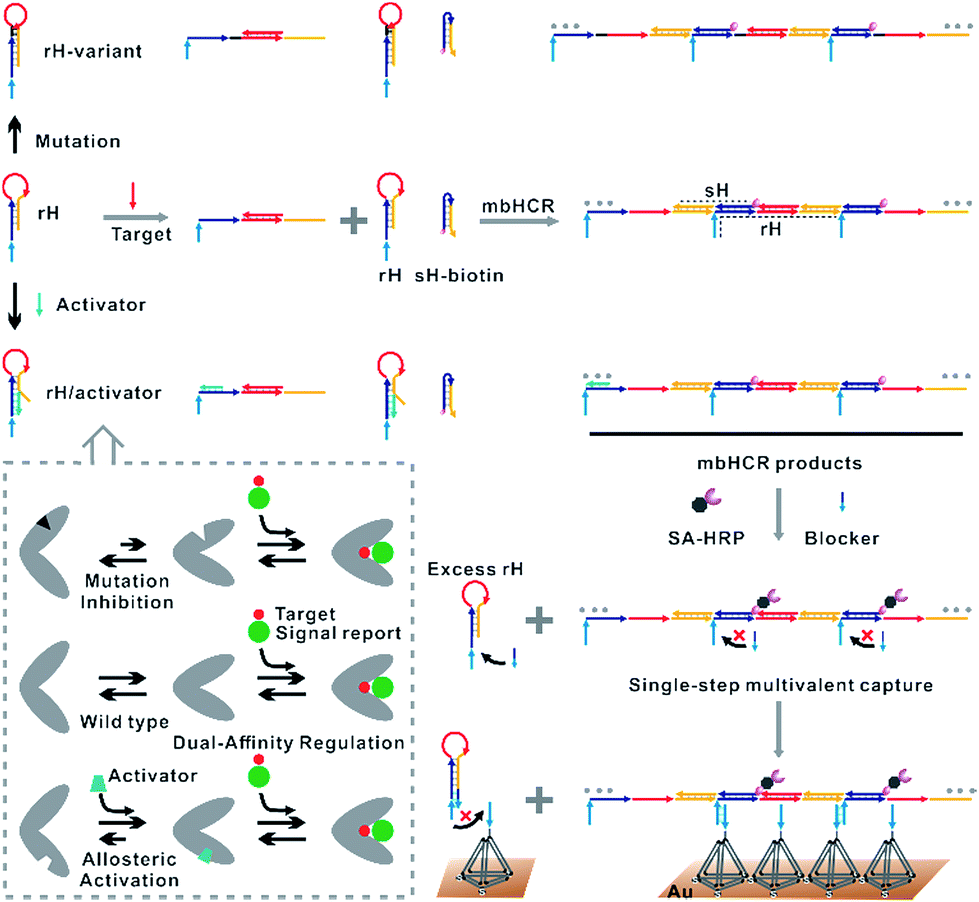 | ||
| Fig. 1 Top: dual-affinity regulation through distal-site mutation inhibition and allosteric activation was designed here to tune target binding affinity and hybridization chain reaction activity. Bottom: schematic of blocker assisted single-step electrocatalytic assay for nucleic acid detection based on a conformational switch induced multibranched and bidirectional hybridization chain reaction. First, the binding of the target to rH initiated the hybridization chain reaction in both directions. HCR products were then pretreated with SA-HRP and the blocker. The blocker was used to prevent the competitive capture of unreacted rH. | ||
Here, we verified the utility of varying the switching equilibrium constant of the receptor to modulate both the target binding affinity and hybridization chain reaction activity. The useful detection range of the constructed electrocatalytic biosensor was regulated through distal-site mutation inhibition and allosteric activation (Fig. 1, top). The dual-affinity regulation applied in our biosensor not only obtained a broad dynamic range but also amplified the signal transduction. In addition, we re-engineered the traditional toehold initiated hybridization chain reaction to a conformational switch induced multibranched and bidirectional hybridization chain reaction (mbHCR), which can produce long chain products with multiple branched arms for multivalent capture and multiple biotins linked with SA-HRP for further signal amplification. A blocker was employed to prevent the binding of unreacted hairpins with the probes on the electrode. Thus, the target binding, signal amplification and multivalent binding can be finished in single-step incubation. Furthermore, by employing the allosteric activation strategy, we could sensitively detect nucleic acids of diverse lengths with excellent selectivity using a universal signal reporter in diluted serum.
Results and discussion
The design of the single-step electrocatalytic assay for the analysis of nucleic acids is depicted in Fig. 1. Our re-engineered multibranched and bidirectional hybridization chain reaction contains two monomer hairpins: recognition hairpin (rH) with a single-stranded extension at the 5′ termination and signal hairpin (sH-biotin). The sequences of the two hairpins used in this work followed the sequences designed by Pierce and coworkers56 except that a section of the sequence was inserted in the rH for target recognition. In the absence of the target, the rH and sH do not hybridize each other because the hairpins are protected by long stems, and therefore the signal output is suppressed. Binding of a specific target DNA sequence to the loop of rH breaks the stem and triggers the hybridization chain reaction in both directions by the two hairpins into the multibranched double helix. Long mbHCR products are produced for efficient electrochemical signal amplification and multivalent capture. In order to obtain target binding, the hybridization chain reaction and multivalent capture at single-step incubation, the mbHCR products are pretreated with SA-HRP and the blocker. With optimized design, the blocker is employed to hybridize with unreacted rH and thereby it helps to prevent the capture of excess rH on the gold electrode surface. However, the blocker cannot hybridize easily with the rH in the mbHCR products due to the low equilibrium stability. It is important to note that the adding of active-2 for enhancing rH binding affinity will not disrupt the hybridization of rH with the blocker. This assumption was estimated via NUPACK at 25 °C (Fig. S1†).The useful dynamic range of the DNA binding switch and hybridization chain reaction are controlled by using distal-site mutation inhibition and allosteric activation, in which target binding affinity and hybridization chain reaction activity are controlled by either destabilizing or stabilizing the receptor (Fig. 1, top). To create this situation, we tuned the switching equilibria of rH to alter its dual-affinity by reducing or extending the length of the stem in rH. The free energy of rH was estimated via NUPACK at 25 °C (Fig. S2†). It decreased from −26.07 kcal mol−1 (rH, original hairpin with 18 base pairs of stems) to −30.63 kcal mol−1 (rH-v1, 21 base pairs of stems) and −35.41 kcal mol−1 (rH-v2, 24 base pairs of stems) indicating the progressive reduction of the dual-affinity of the receptors through mutation inhibition. When adding allosteric effector activator 1 or activator 2 (see sequences listed in Table S1†) to help open the hairpin structure, the equilibrium probability of the stem in rH decreased suggesting the easier opening of rH-a1 and rH-a2 by target DNA than that of original rH.
To demonstrate the feasibility of tuning the target binding affinity and hybridization chain reaction activity of mbHCR, gel electrophoresis analysis was first performed. The product of receptor hybridized with the target increased with the reduced switching equilibrium constant of rH (Fig. 2A). The product of mbHCR was also apparently related to the stability of rH. The molecular weight of the multibranched nicked double helix became larger by decreasing the stability of the rH (Fig. 2B), which demonstrated the negative and positive modulation of the target binding affinity and hybridization chain reaction activity though mutation inhibition and allosteric activation.
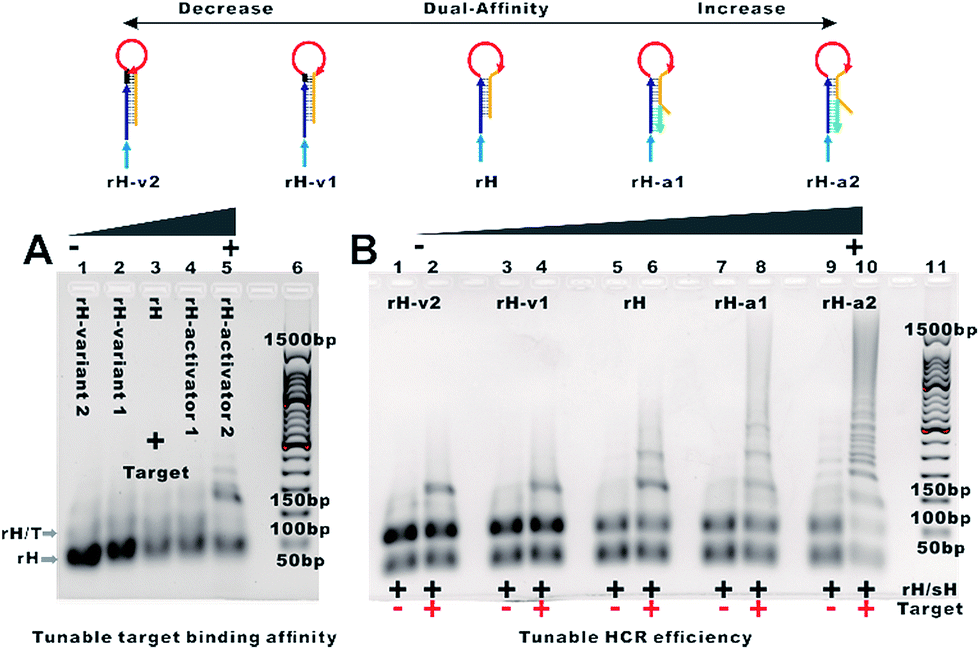 | ||
| Fig. 2 (A) Analysis by AGE of reactions of receptors with the target. Lane 1, 1 μM rH-v2 + 0.2 μM target-24; lane 2, 1 μM rH-v1 + 0.2 μM target-24; lane 3, 1 μM rH + 0.2 μM target-24; lane 4, 1 μM rH-a1 + 0.2 μM target-24; lane 5, 1 μM rH-a2 + 0.2 μM target-24; lane 6, 50 bp DNA ladder markers. (B) Analysis by AGE of the mutation inhibition and allosteric activation controlled multibranched hybridization chain reaction. Lane 1, 1 μM rH-v2/sH. Lane 2, 1 μM rH-v2/sH and 0.2 μM target-24. Lane 3, 1 μM rH-v1/sH. Lane 4, 1 μM rH-v1/sH and 0.2 μM target-24. Lane 5, 1 μM rH/sH. Lane 6, 1 μM rH/sH and 0.2 μM target-24. Lane 7, 1 μM rH-a1/sH. Lane 8, 1 μM rH-a1/sH and 0.2 μM target-24. Lane 9, 1 μM rH-a2/sH. Lane 10, 1 μM rH-a2/sH and 0.2 μM target-24. Lane 11, 50 bp DNA ladder markers. | ||
We next characterized the kinetics of the dual-affinity controlled mbHCR through monitoring the fluorescence intensity change initiated by 0 or 100 nM target DNA. By employing mutation inhibition and allosteric activation to modulate the target binding affinity and hybridization chain reaction activity of the receptors, the kinetic rate of fluorescence intensity increased with the increasement of dual-affinity (Fig. 3A–E). We further demonstrated the bidirectional initiation of mbHCR in the following experiments. 5′ termination (obtain 3′ trigger) and 3′ termination (obtain 5′ trigger) were employed to block one direction initiator. Then we monitored the real time fluorescence intensity of the hybridization chain reaction induced by the same amount of 3′ trigger and 5′ trigger (Fig. 3F and G). We observed a relatively low rate constant for the 5′ directional HCR because the loop-based displacement in the 5′ directional HCR was harder (much higher energy barrier to open the hairpin) than the toehold-based displacement in the 3′ directional HCR. The AGE analysis further proved the lower efficiency of the 5′ directional HCR than the 3′ directional HCR (Fig. 3H).
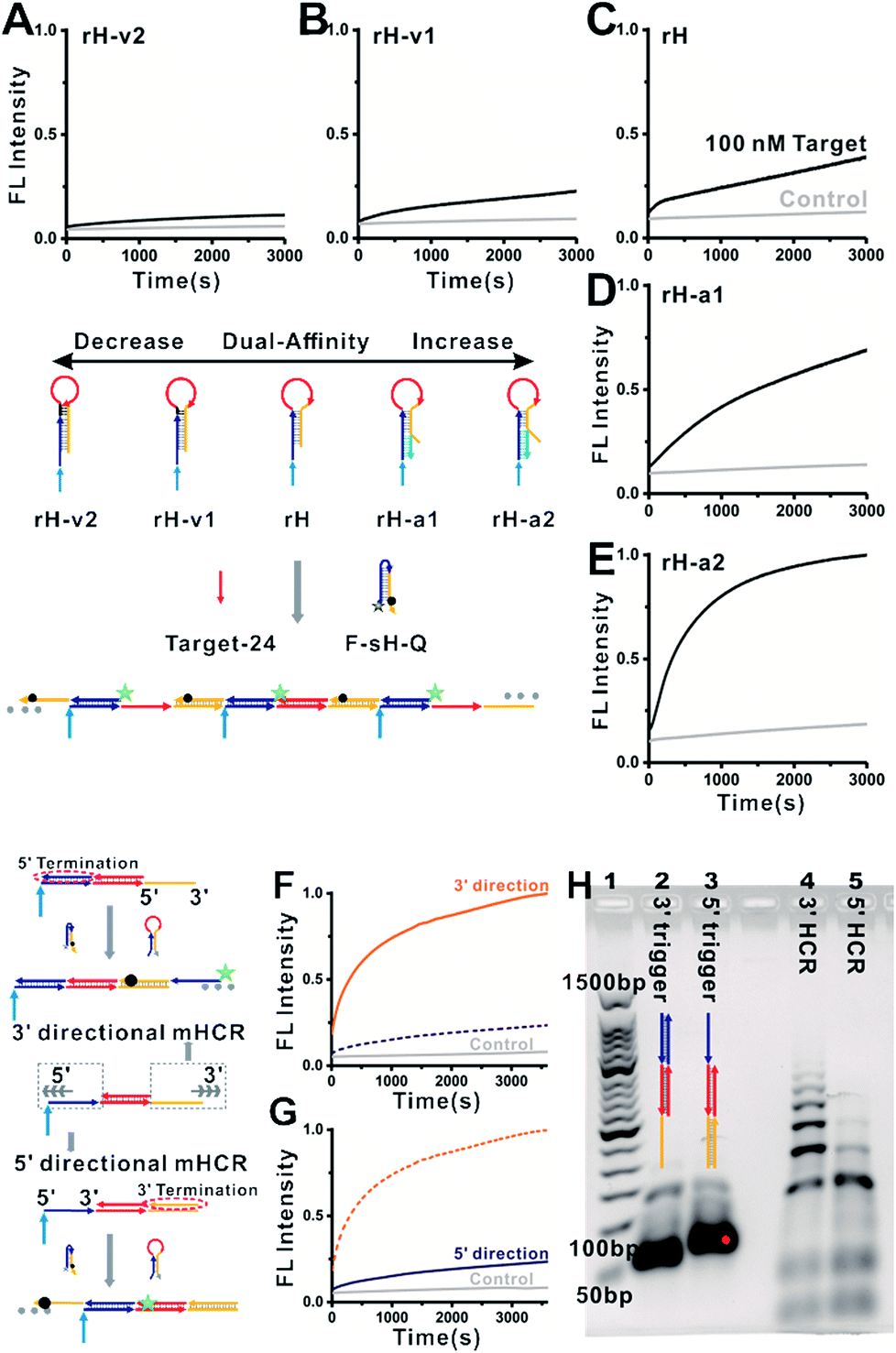 | ||
| Fig. 3 Kinetics of mbHCR with dual-affinity regulation. 0 or 100 nM target-24 was added into the system at t ≈ 0. Fluorescence intensity of each sample was monitored within 10 s of mixing by pipetting. Here, [rH] = [F-sH-Q] = 50 nM. The control trace (light gray) and reaction trace (green) show that mbHCR was initiated with 0 or 100 nM target-24. The kinetics of mbHCR was hindered by mutation inhibition (A, B) or accelerated by adding allosteric effectors (D, E). (F, G) kinetics of the hybridization chain reaction initiated in two directions. Same amount of 3′ trigger (F) or 5′ trigger (G) was added into the system at t ≈ 0. Increasing traces of fluorescence indicated that the loop-toehold initiated hybridization chain reaction is slower than the end-toehold initiated hybridization chain reaction. (H) Analysis by AGE of the two directional hybridization chain reactions. Lane 1, 50 bp DNA ladder markers; lane 2, 3 μM 3′ trigger (3 μM rH + 3 μM target-24 + 3 μM 5′ termination); lane 3, 3 μM 5′trigger (3 μM rH + 3 μM target-24 + 3 μM 3′ termination); lane 4, 3′mbHCR (1 μM rH + 1 μM sH-biotin + 0.2 μM 3′trigger); lane 5, 5′mbHCR (1 μM rH + 1 μM sH-biotin + 0.2 μM 5′trigger). | ||
Having validated the facile dual-affinity regulation of mbHCR, we next attempted to detect nucleic acids through an electrochemical method. Our previous work has proved that the multivalent hybridization capture efficiency on the surface is higher than monomer hybridization.55 However, a limitation existing here is that the unreacted monomer hairpin can't be removed easily from mbHCR products. In order to obtain the DNA analysis with single-step incubation and wash, we first employed the gap hybridization strategy57 to diminish the binding efficiency of monomer hybridization on the surface. The monomer hybridization is destabilized because a 10 nt gap is reserved between the capture probe and rH stem. Tetrahedron DNA nanostructures (Fig. S3† validates the successful self-assembly of TDN) with a capturing probe on the vertex were employed to capture the target induced mbHCR products and generate the electrocatalytic i–t current of the enzymatic reaction. We investigated the influence of the capture reaction temperature and the length of single-stranded extension in rH. We found that the reactions were taking place well at 37 °C (Fig. S4†). At the optimal incubation temperature, the S/N was enhanced with the increased length of single-stranded extension in rH (Fig. S5†). However, the influence of the 10 nt gap generated instability of hybridization will be weakened by increasing the extension length of rH, which means that the competitive hybridization of excess rH with the capture probe will dramatically disrupt the detection of target nucleic acids at low concentrations. The sensitivity of target-24 analysis using rH-target-24 (4-10) was better than that using rH-target-24 (4-12) (data not shown).
To overcome the above limitations, we then employed the blocker to hybridize prior with excess rH prohibiting its capture on the surface. The hybridization cases of diverse lengths of the blocker with the mbHCR product and rH were estimated via NUPACK at 25 °C (Fig. S6 and S7†). The hybridization probabilities of the blocker with the mbHCR product and rH are both enhanced with the increased length of the blocker (Fig. 4, top). The detection performance influenced by the length of the blocker was then investigated. We found that a 12 nt blocker was optimum to produce the best S/N (Fig. 4, bottom). This is because the 14 nt blocker binding to mbHCR products with high efficiency will prevent the multivalent capture. The 10 nt blocker has the lowest probability binding to mbHCR products. However, the 10 nt extension in rH-target-24 (4-6) has relatively lower efficiency of multivalent capture when compared with 12 nt extension in rH-target-24 (6-6). The results also show that the background signal produced at 25 °C was lower than that produced at 37 °C (37 °C used in Fig. S5† and 25 °C used in Fig. 4).
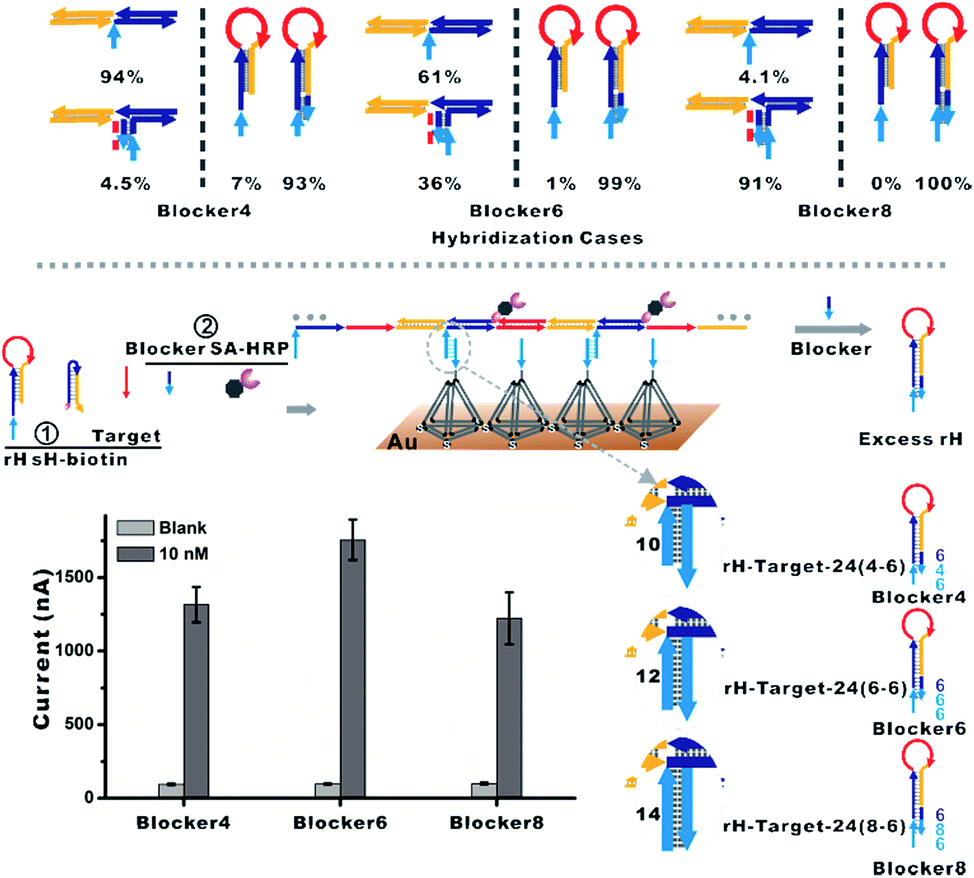 | ||
| Fig. 4 (Top) Hybridization cases of diverse lengths of the blocker with the mbHCR product and rH were estimated via NUPACK. The percentages were calculated from Fig. S6 and S7.† (Bottom) The blocker was employed to hybridize prior with excess rH, which will prohibit the hybridization of rH with the capture probe on the surface. The influence of the length of the blocker on the detection performance was investigated. | ||
Our previous work also demonstrated that the multivalent capture obtained nearly saturated hybridization efficiency in 15 minutes at room temperature.55 A multivalent binding reaction was carried out within 30 minutes in this work. As predicted, when we used five rH with different target binding affinities and hybridization chain reaction activities for nucleic acid analysis, the detection limits shifted in intervals by almost one order of magnitude. With the increase in the dual-affinity of rH, the detection limit was enhanced from 5 nM (rH-v2) to 10 fM (rH-a2) (Fig. 5). With mutation inhibition and allosteric activation, the detection sensitivity of the constructed electrocatalytic biosensor could be tuned about 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold. The hybridization chain reaction performed in solution and the multivalent capture performed on the surface are both efficient. Thus the nucleic acid analysis performances (detection sensitivity and detection speed) are outstanding (Table S2†). Further optimization (such as optimizing the concentration of the activator) can probably lead to even higher sensitivity.
000-fold. The hybridization chain reaction performed in solution and the multivalent capture performed on the surface are both efficient. Thus the nucleic acid analysis performances (detection sensitivity and detection speed) are outstanding (Table S2†). Further optimization (such as optimizing the concentration of the activator) can probably lead to even higher sensitivity.
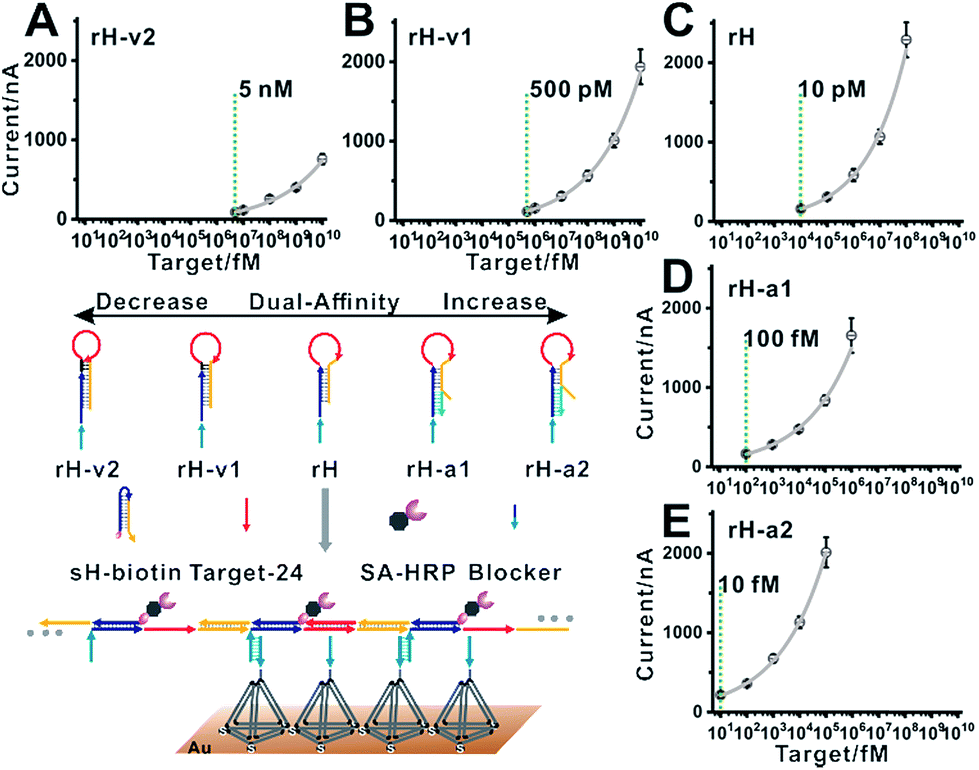 | ||
| Fig. 5 Blocker assisted single-step electrocatalytic assay. The detection limits were 5 nM, 500 pM, 10 pM, 100 fM, and 10 fM for (A) rH-v2, (B) rH-v1, (C) rH, (D) rH-a1, and (E) rH-a2, respectively. | ||
The developed single-step electrocatalytic assay using rH-a2 with high sensitivity could also be adapted for the analysis of nucleic acids of diverse lengths in diluted serum. The detection performance in serum with or without dilution is shown in Fig. S8.† We obtained an optimized S/N in 75% serum and 25% 2× SPSC buffer. The reduced signal observed in serum is possibly due to the nonspecific interaction of the added biomolecules with target DNA and the decrease of salt concentration in serum, which will decrease the efficiency of target binding and the hybridization chain reaction. The sequences of rH and sH used in this work were designed by Pierce and coworkers, excepting a section of recognition sequence was inserted in the rH for target binding. Fig. 6A–C show the excellent performance for the analysis of the DNA targets of three diverse lengths with detection limits of 50 fM (target-DNA-24), 50 fM (target-DNA-21), and 100 fM (target-DNA-18), respectively. The AGE analysis, typical CV curves and i–t curves all validated that the mbHCR triggered by target-DNA-18 was less efficient than that triggered by target-DNA-24 and target-DNA-21 (Fig. S9–S11†). Five samples (S1 to S5) containing different amounts of nucleic acids (lines indicate the known concentrations) were measured. The results (Fig. 6D) show an impressive validation of the good accuracy of our single-step electrocatalytic assay in 75% serum and 25% 2× SPSC buffer.
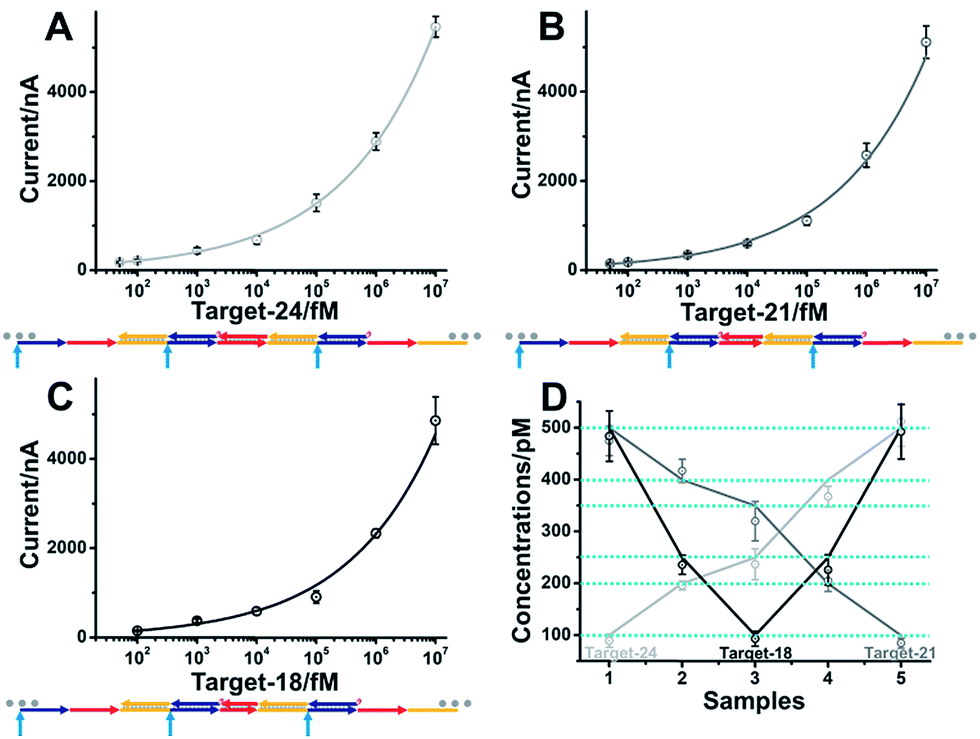 | ||
| Fig. 6 Analysis of nucleic acids of diverse lengths in 75% serum and 25% 2× SPSC buffer. Logarithmic plot of amperometric current versus target-24 (A) (light gray), target-21 (B) (gray) and target-18 (C) (black) concentrations using the electrocatalytic mbHCR strategy. (D) Five samples (1–5) containing varying amounts of target-24 (light gray), target-21 (gray) and target-18 (black) (lines indicate the known concentrations) were measured. The results deviated from the lines are due to experimental errors. | ||
The sequence-specificity of our assay with single mismatch targets in various locations was considered finally. Clearly, perfectly matched targets could be easily differentiated from other mismatches at the same concentration of 10 nM in buffer by using rH-a2 as the receptor, revealing a good single nucleotide resolution (Fig. S12†). The location of single-base mismatches did not affect significantly the signal output. It is easy to distinguish a perfectly matched target from five single-base mismatched targets at different locations (Fig. S13†). Besides, the selectivity can be increased by using rH-v2 as the receptor because a higher energy barrier has to be broken to open the recognition hairpin (Fig. S14†). These results successfully demonstrate the high potential of the electrocatalytic assay as a single nucleotide resolution biosensor for nucleic acid detection in clinical samples.
Conclusions
In summary, a novel single-step electrocatalytic strategy, called blocker assisted mbHCR, has been proposed. This method enables the detection of nucleic acids with a tunable detection range by programmable modulation of the target binding affinity and hybridization chain reaction activity using mutation inhibition and allosteric activation. The dual-affinity regulation demonstrated here both extended the dynamic range and amplified the signal transduction. The blocker assisted single-step strategy and dual-affinity regulation can be applied in other solid-phase biosensors. Besides, the detection cost can be reduced as the signal hairpin is universal. The selectivity of single nucleotide polymorphism also can be tuned through dual-affinity regulation. The analysis of DNA of multiple lengths in serum with high precision revealed the high potential of the developed strategy as a reliable and sensitive electrocatalytic biosensor for the detection of nucleic acids in complex matrices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC Grants 21677060 and 21507041) and the Zhejiang Provincial Natural Science Foundation of China (Grants LQ19B050002, LY16B050007 and Y18B050014).Notes and references
- V. J. Hilser, J. O. Wrabl and H. N. Motlagh, Annu. Rev. Biophys., 2012, 41, 585–609 CrossRef CAS PubMed.
- A. del Sol, C.-J. Tsai, B. Ma and R. Nussinov, Structure, 2009, 17, 1042–1050 CrossRef CAS PubMed.
- S. A. Benner, Chem. Rev., 1989, 89, 789–806 CrossRef CAS.
- C. J. Tsai, A. del Sol and R. Nussinov, Mol. BioSyst., 2009, 5, 207–216 RSC.
- C. Y. Huang and J. E. Ferrell, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10078–10083 CrossRef CAS.
- A. J. Simon, A. Vallee-Belisle, F. Ricci and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15048–15053 CrossRef CAS PubMed.
- J. S. Marvin, E. E. Corcoran, N. A. Hattangadi, J. V. Zhang, S. A. Gere and H. W. Hellinga, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4366–4371 CrossRef CAS.
- J. L. Vinkenborg, N. Karnowski and M. Famulok, Nat. Chem. Biol., 2011, 7, 519–527 CrossRef CAS PubMed.
- Y. Xiang, A. Tong and Y. Lu, J. Am. Chem. Soc., 2009, 131, 15352–15357 CrossRef CAS PubMed.
- F. Ricci, A. Vallee-Belisle, A. Porchetta and K. W. Plaxco, J. Am. Chem. Soc., 2012, 134, 15177–15180 CrossRef CAS PubMed.
- A. Vallee-Belisle, F. Ricci and K. W. Plaxco, J. Am. Chem. Soc., 2012, 134, 2876–2879 CrossRef CAS PubMed.
- A. Porchetta, A. Vallee-Belisle, K. W. Plaxco and F. Ricci, J. Am. Chem. Soc., 2012, 134, 20601–20604 CrossRef CAS PubMed.
- Y. L. Xianyu, J. Wu, Y. P. Chen, W. S. Zheng, M. X. Xie and X. Y. Jiang, Angew. Chem., Int. Ed., 2018, 57, 7503–7507 CrossRef CAS PubMed.
- Q. Liu, Z. L. Ge, X. H. Mao, G. B. Zhou, X. L. Zuo, J. W. Shen, J. Y. Shi, J. Li, L. H. Wang, X. Q. Chen and C. H. Fan, Angew. Chem., Int. Ed., 2018, 57, 7131–7135 CrossRef CAS PubMed.
- L. Cabel, C. Proudhon, E. Romano, N. Girard, O. Lantz, M. H. Stern, J. Y. Pierga and F. C. Bidard, Nat. Rev. Clin. Oncol., 2018, 15, 639–650 CrossRef CAS PubMed.
- J. Das, I. Ivanov, L. Montermini, J. Rak, E. H. Sargent and S. O. Kelley, Nat. Chem., 2015, 7, 569–575 CrossRef CAS PubMed.
- N. R. Y. Ho, G. S. Lim, N. R. Sundah, D. Lim, T. P. Loh and H. Shao, Nat. Commun., 2018, 9, 3238 CrossRef PubMed.
- B. A. Turturice, D. R. Gold, A. A. Litonjua, E. Oken, S. Rifas-Shiman, D. L. Perkins and P. W. Finn, J. Allergy Clin. Immunol., 2019, 143, 419–421 CrossRef PubMed.
- D. W. Bianchi and R. W. K. Chiu, N. Engl. J. Med., 2018, 379, 464–473 CrossRef CAS PubMed.
- L. D. Mattos-Arruda, B. Weigelt, J. Cortes, H. H. Won, C. K. Y. Ng, P. Nuciforo, F. C. Bidard, C. Aura, C. Saura, V. Peg, S. Piscuoglio, M. Oliveira, Y. Smolders, P. Patel, L. Norton, J. Tabernero, M. F. Berger, J. Seoane and J. S. Reis-Filho, Ann. Oncol., 2018, 25, 1729–1735 CrossRef PubMed.
- W. K. J. Lam, P. Jiang, K. C. A. Chan, S. H. Cheng, H. Zhang, W. Peng, O. Y. O. Tse, Y. K. Tong, W. Gai, B. C. Y. Zee, B. B. Y. Ma, E. P. Hui, A. T. C. Chan, J. K. S. Woo, R. W. K. Chiu and Y. M. D. Lo, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5115–E5124 CrossRef CAS PubMed.
- P. Hu, S. Zhang, T. Wu, D. Ni, W. Fan, Y. Zhu, R. Qian and J. Shi, Adv. Mater., 2018, 30, 1801690 CrossRef PubMed.
- L. Cabel, C. Proudhon, E. Romano, N. Girard, O. Lantz, M.-H. Stern, J. Y. Pierga and F. C. Bidard, Nat. Rev. Clin. Oncol., 2018, 15, 639–650 CrossRef CAS PubMed.
- A. M. Newman, A. F. Lovejoy, D. M. Klass, D. M. Kurtz, J. J. Chabon, F. Scherer, H. Stehr, C. L. Liu, S. V. Bratman, C. Say, L. Zhou, J. N. Carter, R. B. West, G. W. Sledge Jr, J. B. Shrager, B. W. Loo Jr, J. W. Neal, H. A. Wakelee, M. Diehn and A. A. Alizadeh, Nat. Biotechnol., 2016, 34, 547–555 CrossRef CAS PubMed.
- S.-J. Dawson, D. W. Y. Tsui, M. Murtaza, H. Biggs, O. M. Rueda, S.-F. Chin, M. J. Dunning, D. Gale, T. Forshew, B. Mahler-Araujo, S. Rajan, S. Humphray, J. Becq, D. Halsall, M. Wallis, D. Bentley, C. Caldas and N. Rosenfeld, N. Engl. J. Med., 2013, 368, 1199–1209 CrossRef CAS PubMed.
- J. Das, I. Ivanov, E. H. Sargent and S. O. Kelley, J. Am. Chem. Soc., 2016, 138, 11009–11016 CrossRef CAS PubMed.
- S. K. Silverman, Acc. Chem. Res., 2009, 42, 1521–1531 CrossRef CAS PubMed.
- X. Li, X. W. Xu, J. Song, Q. W. Xue, C. Z. Li and W. Jiang, Biosens. Bioelectron., 2017, 91, 631–636 CrossRef CAS PubMed.
- Y. Q. Chen, Y. Y. Song, F. Wu, W. T. Liu, B. S. Fu, B. K. Feng and X. Zhou, Chem. Commun., 2015, 51, 6980–6983 RSC.
- J. Hu, M. H. Liu and C. Y. Zhang, Chem. Sci., 2018, 9, 4258–4267 RSC.
- L. Tang, Y. Liu, M. M. Ali, D. K. Kang, W. Zhao and J. H. Li, Anal. Chem., 2012, 84, 4711–4717 CrossRef CAS PubMed.
- R. Deng, L. Tang, Q. Tian, Y. Wang, L. Lin and J. H. Li, Angew. Chem., Int. Ed., 2014, 126, 2421–2425 CrossRef.
- R. Deng, K. Zhang and J. H. Li, Acc. Chem. Res., 2017, 50, 1059–1068 CrossRef CAS PubMed.
- X. Tang, R. Deng, Y. Sun, X. Ren, M. Zhou and J. H. Li, Anal. Chem., 2018, 90, 10001–10008 CrossRef CAS PubMed.
- J. Huang, H. Wang, X. Yang, K. Quan, Y. Yang, L. Ying, N. Xie, M. Ou and K. M. Wang, Chem. Sci., 2016, 7, 3829–3835 RSC.
- J. Huang, Y. Wu, Y. Chen, Z. Zhu, X. Yang, C. J. Yang, K. M. Wang and W. H. Tan, Angew. Chem., Int. Ed., 2011, 50, 401–404 CrossRef CAS PubMed.
- M. Ou, J. Huang, X. Yang, X. He, K. Quan, Y. Yang, N. Xie, J. Li and K. M. Wang, ChemBioChem, 2018, 19, 147–152 CrossRef CAS PubMed.
- Z. L. Ge, M. H. Lin, P. Wang, H. Pei, J. Yan, J. Y. Sho, Q. Huang, D. N. He, C. H. Fan and X. L. Zuo, Anal. Chem., 2014, 86, 2124–2130 CrossRef CAS PubMed.
- H. M. T. Choi, V. A. Beck and N. A. Pierce, ACS Nano, 2014, 8, 4284–4294 CrossRef CAS PubMed.
- S. Bi, S. Yue and S. Zhang, Chem. Soc. Rev., 2017, 46, 4281–4298 RSC.
- Z. Wu, G. Q. Liu, X. L. Yang and J. H. Jiang, J. Am. Chem. Soc., 2015, 137, 6829–6836 CrossRef CAS PubMed.
- J. Wei, X. Gong, Q. Wang, M. Pan, X. Liu, J. Liu, F. Xia and F. Wang, Chem. Sci., 2018, 9, 52–61 RSC.
- F. Wang, J. Elbaz, R. Orbach, N. Magen and I. Willner, J. Am. Chem. Soc., 2011, 133, 17149–17151 CrossRef CAS PubMed.
- P. Liu, X. Yang, S. Sun, Q. Wang, K. M. Wang, J. Huang, J. Liu and L. He, Anal. Chem., 2013, 85, 7689–7695 CrossRef CAS PubMed.
- Y. Wang, L. Tang, Z. Li, Y. Lin and J. H. Li, Nat. Protoc., 2014, 9, 1944 CrossRef CAS PubMed.
- C. P. Liang, P. Q. Ma, H. Liu, X. Guo, B. C. Yin and B. C. Ye, Angew. Chem., Int. Ed., 2017, 56, 9077–9081 CrossRef CAS PubMed.
- D. Y. Zhang, A. J. Turberfield, B. Yurke and E. Winfree, Science, 2007, 318, 1121–1125 CrossRef CAS PubMed.
- Y. Lv, L. Cui, R. Peng, Z. Zhao, L. Qiu, H. Chen, C. Jin, X. B. Zhang and W. H. Tan, Anal. Chem., 2015, 87, 11714–11720 CrossRef CAS.
- Q. Wei, J. Huang, J. Li, J. Wang, X. Yang, J. Liu and K. M. Wang, Chem. Sci., 2018, 9, 7802–7808 RSC.
- K. Quan, J. Huang, X. Yang, Y. Yang, L. Ying, H. Wang, N. Xie, M. Ou and K. M. Wang, Anal. Chem., 2016, 88, 5857–5864 CrossRef CAS PubMed.
- C. Wu, S. Cansiz, L. Zhang, I. T. Teng, L. Qiu, J. Li, Y. Liu, C. Zhou, R. Hu, T. Zhang, C. Cui, L. Cui and W. Tan, J. Am. Chem. Soc., 2015, 137, 4900–4903 CrossRef CAS PubMed.
- R. Deng, K. Zhang, Y. Sun, X. Ren and J. H. Li, Chem. Sci., 2017, 8, 3668–3675 RSC.
- C. Jung, P. B. Allen and A. D. Ellington, Nat. Nanotechnol., 2015, 11, 157 CrossRef PubMed.
- J. Liu, P. Du, J. Zhang, H. Shen and J. Lei, Chem. Commun., 2018, 54, 2550–2553 RSC.
- G. B. Zhou, M. H. Lin, P. Song, X. Q. Chen, J. Chao, L. H. Wang, Q. Huang, W. Huang, C. H. Fan and X. L. Zuo, Anal. Chem., 2014, 86, 7843–7848 CrossRef CAS PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed.
- C. Poehlmann and M. Sprinzl, Anal. Chem., 2010, 82, 4434–4440 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, table of detailed DNA sequences and supporting figures. See DOI: 10.1039/c9sc01199d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |