
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-efficiency blue thermally activated delayed fluorescence from donor–acceptor–donor systems via the through-space conjugation effect†
Feifei
Gao‡
,
Ruiming
Du‡
,
Chunmiao
Han
,
Jing
Zhang
,
Ying
Wei
,
Guang
Lu
and
Hui
Xu
 *
*
Key Laboratory of Functional Inorganic Material Chemistry, Ministry of Education & School of Chemistry and Material Science, Heilongjiang University, 74 Xuefu Road, Harbin 150080, People's Republic of China. E-mail: hxu@hlju.edu.cn; ywei@hlju.edu.cn
First published on 25th April 2019
Abstract
The photophysical optimization of donor (D)–acceptor (A) molecules is a real challenge because of the intrinsic limitation of their charger transfer (CT) excited states. Herein, two D–A–D molecules featuring blue thermally activated delayed fluorescence (TADF) are developed, in which a homoconjugated acceptor 5,10-diphenyl-5,10-dihydrophosphanthrene oxide (DPDPO2A) is incorporated to bridge four carbazolyl or 3,6-di-t-butyl-carbazolyl groups for D–A interaction optimization without immoderate conjugation extension. It is shown that the through-space conjugation effect of DPDPO2A can efficiently enhance intramolecular CT (ICT) and simultaneously facilitate the uniform dispersion of the frontier molecular orbitals (FMO), which remarkably reduces the singlet–triplet splitting energy (ΔEST) and increases FMO overlaps for radiation facilitation, resulting in the 4–6 fold increased rate constants of reverse intersystem crossing (RISC) and singlet radiation. The maximum external quantum efficiency beyond 20% and the state-of-the-art efficiency stability from sky-blue TADF OLEDs demonstrate the effectiveness of the “conjugation modulation” strategy for developing high-performance optoelectronic D–A systems.
1. Introduction
Organic donor–acceptor (D–A) molecules feature charge-transfer (CT) excited states and ambipolar characteristics,1,2 and therefore are widely used in optoelectronic applications, such as organic light-emitting diodes (OLEDs),3–25 organic solar cells (OSCs),26–34 photodetectors,35,36 memory devices,37–39 and so on.40–44 The electrical properties of D–A systems are readily modulated on the basis of the inherent relationship between molecules and D/A groups regarding electron gain and loss.45 In contrast, the photophysical properties of D–A molecules are more difficult to optimize due to the strong dependence of the excited-state characteristics on the intramolecular CT (ICT) interaction.46 For instance, thermally activated delayed fluorescence (TADF) materials emerge recently for OLED applications, which should have near-zero singlet–triplet splitting energy (ΔEST) to facilitate reverse intersystem crossing (RISC) for delayed fluorescence (DF).47–51 It is known that ΔEST is twice the electron exchange energy (J) expressed as:52,53 | (1) |
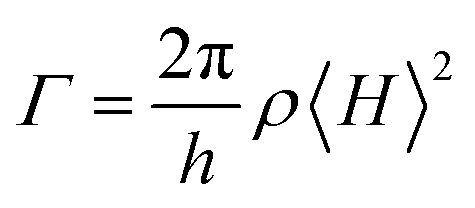 | (2) |
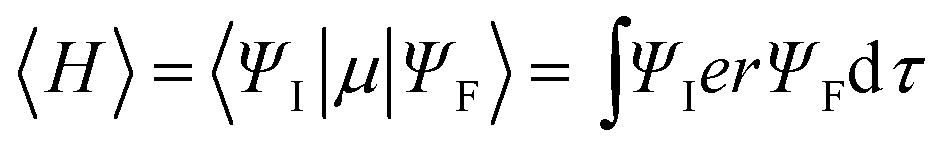 | (3) |
Obviously, the conflict between ΔEST and Γ could be resolved by finding the best compromise for 〈ΨH|ΨL〉.10,55 Although it was shown that the moderate HOMO–LUMO overlap is an essential condition for improving the radiative efficiency of TADF molecules, the optimal frontier molecular orbital (FMO) distribution is still ambiguous, which limits the targeted molecular design. From another perspective, Adachi et al. demonstrated that the delocalization of the HOMO and the LUMO can enhance the radiative rate of CT compounds through increasing the oscillator strength of their 1CT radiation despite small 〈ΨH|ΨL〉, which can be realized through either dendronizing the D group56 or increasing the density of D groups linked on the same π-bridge.10 The former more or less increases the synthesis complexity, whereas the latter leads to a remarkable bathochromic shift of the emission. Actually, because of the high sensitivity of emission color to the intensity of D–A interactions, the excited-state characteristics of blue TADF materials are even more difficult to modify.57,58
As alternatives, D–A–D and D–A–A type molecules seem promising, owing to their advantage of accurate ICT modulation.59,60 The D–A–D structure could be regarded as the combination of two D–A units with doubled but dispersed D and A groups. Lee et al. constructed a D–A–D dye through the single-bond linkage of two isophthalonitrile acceptors, which resulted in the LUMO delocalization, 1.5-fold photoluminescence quantum yield (PLQY, ϕPL) and twofold external quantum efficiency (EQE, ηEQE) of its devices, however, accompanied by a remarkable emission red shift (∼50 nm).61 In contrast, through incorporating P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O as the insulating acceptor, we realized a pure-blue D–A–D TADF dye, but its HOMO and LUMO were still localized on single D/A groups.62 Obviously, the conjugation between D–A units should be precisely modulated to optimize electronic interplays.63 In this sense, systems based on homoconjugation would be the desired alternatives, whose through-space conjugation effect on intramolecular electronic communications is between the through-bond conjugation and insulating effects.64 Therefore, they could not only support sufficient D–A interactions, but also avert immoderate conjugation extension, and are “ideal” for developing high-efficiency blue TADF dyes (Scheme 1).
O as the insulating acceptor, we realized a pure-blue D–A–D TADF dye, but its HOMO and LUMO were still localized on single D/A groups.62 Obviously, the conjugation between D–A units should be precisely modulated to optimize electronic interplays.63 In this sense, systems based on homoconjugation would be the desired alternatives, whose through-space conjugation effect on intramolecular electronic communications is between the through-bond conjugation and insulating effects.64 Therefore, they could not only support sufficient D–A interactions, but also avert immoderate conjugation extension, and are “ideal” for developing high-efficiency blue TADF dyes (Scheme 1).
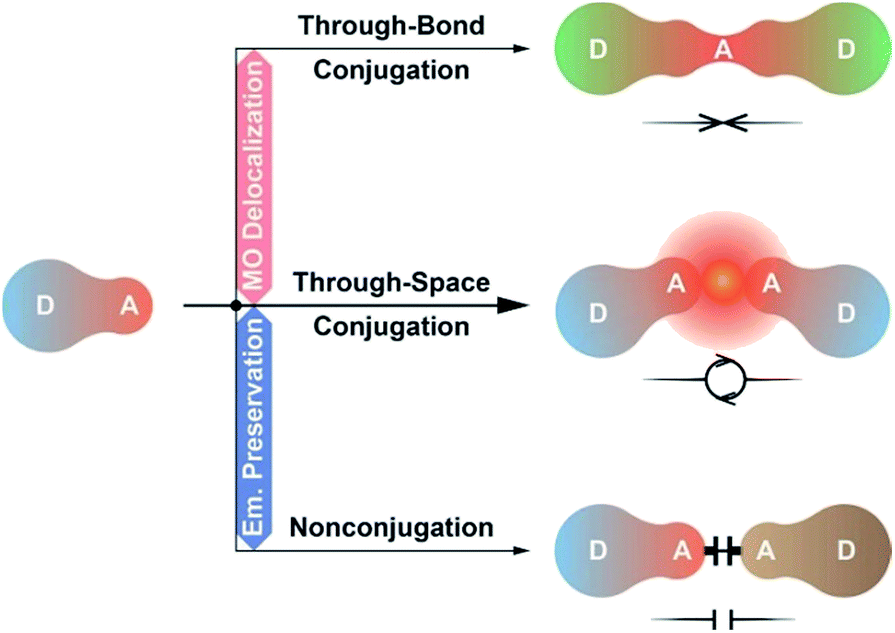 | ||
| Scheme 1 Molecular design of D–A–D systems. Two D–A type molecules can be combined to form (i) a uniform D–A–D system based on through-bond conjugation, (ii) a united D–A–D system based on through-space conjugation or (iii) a separated D–A–A–D system based on insulating linkage. | ||
It is noteworthy that the lengths of homoconjugated units are commonly larger than diphenylene, which would weaken D–A interactions, if used as π-linkers.65 In such a case, a homoconjugated acceptor is desired, since it can directly combine with D groups to provide strong enough ICT effects and simultaneously suppress excessive intramolecular electronic interactions. As a proof of concept, in this contribution, we incorporate a homoconjugated phosphine oxide (PO) acceptor 5,10-diphenyl-5,10-dihydrophosphanthrene oxide (DPDPO2A) into D–A–D systems 9,9′,9′′,9′′′-(5,10-diphenyl-5,10-dihydrophosphanthrene-2,3,7,8-tetrayl)tetrakis(9H-carbazole) oxides (4CzDPDPO2A) and 9,9′,9′′,9′′′-(5,10-diphenyl-5,10-dihydrophosphanthrene-2,3,7,8-tetrayl)tetrakis(3,6-di-tert-butyl-9H-carbazole) oxides (4tBCzDPDPO2A), collectively named 4ArDPDPO2A (Ar = Cz or tBCz), in which carbazoles (Cz) and 3,6-di-tert-butyl-carbazoles (tBCz) serve as D groups, respectively (Fig. 1a). In contrast to their insulated congeners 4ArPPOPO and 4ArPPODPO with completely localized HOMOs and LUMOs, the homoconjugated DPDPO2A acceptor dramatically facilitates intramolecular electronic interplays, providing HOMOs and LUMOs uniformly dispersed on D and A groups, respectively, which shortens the centroid distances and increases the overlap integrals between the HOMOs and the LUMOs, accompanied by enhanced ICT effects. As a result, the doped films of 4CzDPDPO2A and 4tBCzDPDPO2A reveal pure-blue and sky-blue emissions with 4–6 fold increased rate constants of prompt fluorescence (PF, kPF) and singlet radiation (kSr) and 2–4 fold increased DF rate constants (kDF), as well as dramatically improved RISC efficiencies (ϕRISC) of 85 and 94%, respectively. As expected, 4CzDPDPO2A and 4tBCzDPDPO2A achieve photoluminescence (PL, ϕPL) and electroluminescence (EL, ηEQE) quantum efficiencies of 65 and 81% and 11.5 and 23.7%, respectively, which are 2–10 times higher in comparison to 4ArPPOPO and 4ArPPODPO, making them favorable among the blue TADF dyes reported to date. This work demonstrates a facile strategy to optimize the electronic effect in D–A molecules through conjugation modulation, which provides a new way to overcome the bottlenecks of their optoelectronic applications.
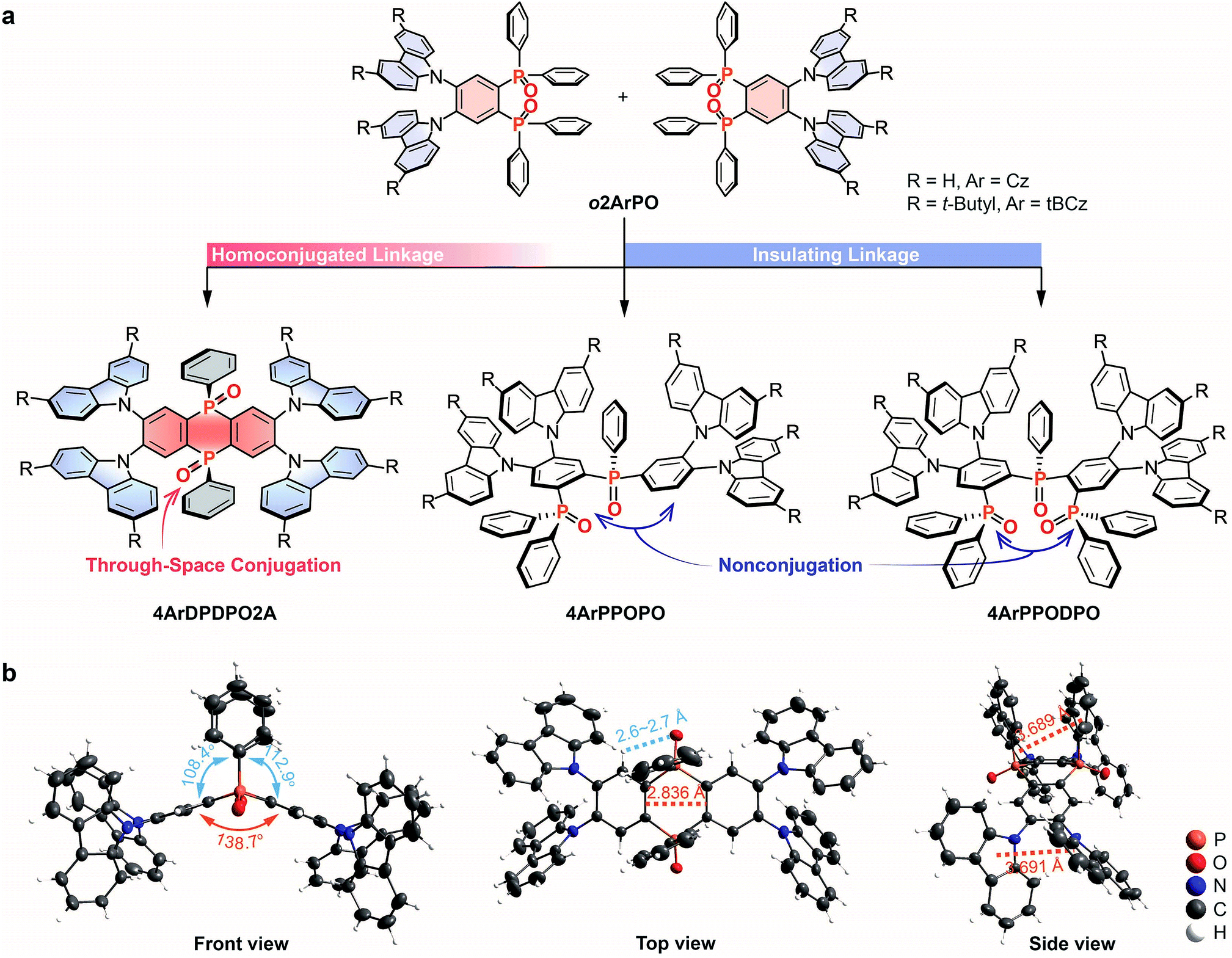 | ||
| Fig. 1 Design and structure of 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO. (a) Molecular design of 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO as the dimers of o2ArP2PO on the basis of homoconjugated and insulating linkages, respectively; (b) Front, top and side views of the single-crystal structure for 4CzDPDPO2A. | ||
2. Results and discussion
2.1. Design and structures
We choose our previously reported blue TADF molecules o2ArPO as the D–A unit, whose simple D–π–A structures consist of Cz/tBCz and diphenylphosphine oxide (DPPO) as D and A groups, respectively.66 Although their HOMOs are uniformly dispersed on the Cz/tBCz groups, their LUMOs are localized on the single phenyls. Despite the pure-blue emissions, the limited D–A interactions result in relatively low ϕPL and ηEQE less than 50 and 10%, respectively, for o2ArPO. From the D–π–A structures of o2ArPO to the D–A–D structures, when two PO groups are used to simultaneously link two Cz/tBCz substituted phenyls at ortho positions, the cyclic DPDPO2A acceptor with the feature of through-space conjugation and the homoconjugated 4ArDPDPO2A are formed; when the single PO group is used as an insulating linkage between two D–A units, 4ArPPOPO and 4ArPPODPO with nonconjugated D–A–D structures are formed. The D/A ratios in 4ArPPOPO and 4ArDPDPO2A are equal, but the D–A interaction intensity in each D–A unit of 4ArPPODPO is comparable to that in 4ArDPDPO2A (Scheme S1†). Actually, the basic structure of these molecules is the same as that of a PO-based acceptor substituted with four carbazole-based donors (Scheme S2†). In this case, the only structural difference between 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO is the conjugation extent, which should be the main reason leading to their diverse photophysical properties.
The single-crystal structure of 4CzDPDPO2A indicates a nearly coplanar DPDPO2A ring with a large dihedral angle of ∼140°, which should be ascribed to the strong intramolecular hydrogen bonds between PO groups and H atoms at both-side ortho positions with H⋯O distances less than 3 Å (Fig. 1b). The intramolecular hydrogen bonds also give rise to the cis configuration of DPDPO2A with coplanar PO groups, in which two same-side phenyls reveal the intramolecular π–π interaction with a centroid–centroid distance of ∼3.8 Å. Similarly, the ortho-substitution enables intramolecular π–π interactions between adjacent Cz groups with a centroid–centroid distance of ∼3.7 Å. More importantly, the distance between two CC bonds in the 1,4-diphosphinine oxide unit is 2.84 Å, which is short enough for the p-orbital overlap.67 These intramolecular interplays not only result in the planar phosphanthrene ring with potential homoconjugation, but also enhance the molecular rigidity to increase ϕPL.59 In the packing diagram, 4CzDPDPO2A reveals two different orientations, which are alternately aligned along the axis (Fig. S1†). The centroid-to-edge distance between Cz and DPDPO2A groups in adjacent molecules within 3.7 Å indicates a weak intermolecular charge transfer interaction, which in turn evidences the predominant influence of ICT effect on photophysical properties.
Although the crystals of 4tBCzDPDPO2A cannot be obtained, on account of the negligible intramolecular steric hindrance for 3,6-tert-butyls of the peripheral tBCz groups, the configurations of 4ArDPDPO2A would be similar, which is further demonstrated by the density functional theory (DFT) simulation (Fig. S2–S4†). In contrast, due to the C–P bond rotation of their PO linkers, the structures of 4ArPPOPO and 4ArPPODPO are more flexible, twisted and asymmetrical, which not only induces differences between two D–A units in spatial orientation, but also suppresses intermolecular interactions.21,68 As a result, the thermal stability of 4ArDPDPO2A is remarkably higher than that of 4ArPPOPO and 4ArPPODPO, but all the materials reveal a stable amorphous phase in the solid state (Fig. S5†). Because 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO are comparable regarding intermolecular interactions, it is rational to attribute their optical properties to their single molecule characteristics.
2.2. DFT and TDDFT simulations
DFT and time-dependent DFT (TDDFT) calculations were performed to figure out the frontier molecular orbital (FMO) distributions and the excitation characteristics. As depicted in Fig. 2a, the HOMOs of 4ArDPDPO2A are almost equally dispersed on their four Cz/tBCz groups, accompanied by minor contributions from phosphanthrene rings. Their HOMO+1s and HOMO+2s also reveal similar distributions (Fig. S2 and S3†). Meanwhile, the LUMOs, LUMO+1s and LUMO+2s of 4ArDPDPO2A are uniformly dispersed on their DPDPO2A groups. It should be noticed that for the LUMOs and LUMO+1s, the electronic wave functions of p-orbitals of sp2-hybrid C atoms at two sides of P atoms are effectively and directly overlapped without the incorporation of orbitals from P atoms, convincingly evidencing through-space conjugation between the “PO-separated” phenylenes in phosphanthrene rings (Fig. S4†). In contrast, the HOMOs and LUMOs of 4ArPPOPO and 4ArPPODPO are basically localized at one-to-two Cz/tBCz groups on one side of the molecules and the phenyls linked with PO groups. Furthermore, FMOs of 4ArPPODPO are more centralized than those of 4ArPPOPO, due to the stronger D–A interactions in the former. Therefore, as expected, in contrast to insulating linkage, the homoconjugated acceptors in D–A–D molecules can dramatically facilitate FMO delocalization, especially leading to uniform HOMO dispersion on all D groups at both sides of the acceptors.
 | ||
| Fig. 2 Theoretical simulation of 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO. (a) Contours and energy levels of the HOMOs and LUMOs for the S0 states of 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO simulated with the DFT method (B3LYP/6-31g*); (b) contours of the “hole” and “particle” and the constants of the singlet and triplet excitations for 4ArDPDPO2A simulated with the TDDFT method (ωB97XD/6-31g*). S1 and T1 refer to the first singlet and triplet energy levels. f is the oscillator strength of S0 → S1 excitation. For comparison, the NTO results of 4ArPPOPO and 4ArPPODPO are shown in Fig. S4;† (c) correlations between the centroid distance (nd(ΨH − ΨL)) and overlap integral (n〈ΨH|ΨL〉) of the frontier orbitals for the S0, S1 and T1 states of 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO. The values 0, 1 and 3 of superscript n refer to the S0, S1 and T1 states, respectively. The data of 4ArDPDPO2A are distinguished with dashed circles. | ||
The HOMO energy levels of the compounds are basically in accord with their D groups whose HOMO values are equal to ∼−5.4 eV for Cz and ∼−5.2 eV for tBCz (Fig. 2a, S2, S3 and Table S1†). Differently, the LUMOs of 4ArDPDPO2A are remarkably deeper than those of 4ArPPOPO and 4ArPPODPO by 0.15 and 0.25 eV, respectively, which reflects the stronger electron-withdrawing effect of the DPDPO2A group. Furthermore, it is noteworthy that the HOMO–LUMO energy gaps of 4ArDPDPO2A are 0.1 and 0.2 eV smaller than those of 4ArPPOPO and 4ArPPODPO, respectively, which is perfectly consistent with the results of cyclic voltammetry (CV) analysis (Fig. S6 and Table S2†). The measured HOMO energy levels of 4ArDPDPO2A are about 0.05 eV smaller than those of their analogues, while the HOMO energy levels of 4ArPPOPO and 4ArPPODPO are equivalent. Therefore, in comparison to 4ArPPOPO, the doubled DPPO groups at both sides of 4ArPPODPO can hardly influence the HOMOs contributed by the D groups, which further manifests their localized ICT interactions only contributed by single D–A units. In opposite, the experimental values of the LUMO energy levels for 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO gradually decrease from ∼3.3, ∼3.1 to ∼3.0 eV. It is rational that the larger steric hindrance of two ortho-DPPO groups in 4ArPPODPO provides a more twisted configuration and further reduces the ICT effect, leading to their highest LUMO energy levels. Besides the stronger electron-withdrawing effect of DPDPO2A acceptors, the deepest LUMOs and HOMOs of 4ArDPDPO2A should be actually attributed to the strongest ICT interactions between their DPDPO2A and Cz/tBCz groups. This is further evidenced by their smallest experimental HOMO–LUMO energy gaps of ∼2.8 eV, which is as much as 0.2 and 0.3 eV smaller than those of 4ArPPOPO and 4ArPPODPO, respectively. Consequently, the incorporation of homoconjugated DPDPO2A as the acceptor can indeed improve the ICT effect, owing to its through-space conjugation effect facilitated D–A interactions.
Natural transition orbital (NTO) analysis of singlet and triplet excitations was performed to identify the excited-state characteristics (Fig. 2b and S7†).69,70 For S0 → S1 excitations of 4ArDPDPO2A, similar to the FMO locations of their ground states (S0), the “holes” are uniformly dispersed on their four D groups, accompanied by minor contributions from phenylenes of DPDPO2A rings, while the “particles” are mainly distributed on their DPDPO2A groups, manifesting their CT-predominant first singlet excited states (S1). In contrast, the S1 states of 4ArPPOPO and 4ArPPODPO are completely localized with the “holes” on single D groups and the “particles” on single phenylenes linked with PO groups. The significantly delocalized S1 states of 4ArDPDPO2A undoubtedly originate from the through-space conjugation effect of DPDPO2A for the uniformity of D–A interactions in whole molecules. The insulating PO linkages in 4ArPPOPO and 4ArPPODPO in turn impede the electronic communications between two D–A units, resulting in their locally confined S1 states. It is noteworthy that comparable to their insulated analogues, the S1 energy levels of 4ArDPDPO2A (∼3.0 eV) are only slightly lower by 0.1 eV, still corresponding to blue emissions, which manifests the limited influence of the homoconjugated linkage on emission color. On the other hand, for S0 → T1 excitations, all the molecules reveal LE T1 states with the “holes” and “particles” localized on their Cz groups, which benefits the RISC transitions from the T1 to the S1.71 Although the planar geometry of the DPDPO2A acceptor is a requirement, its homoconjugation is the main reason for the preserved excited-state energy levels and enhanced ICT effect of 4ArDPDPO2A.
The overlap integrals (n〈ΨH|ΨL〉) and the centroid distances (nd(ΨH − ΨL)) of the FMOs for the S0, S1 and T1 states, respectively corresponding to n = 0, 1 and 3, are estimated to quantitatively investigate the influence of conjugation on orbital distribution (Fig. 2c and Table S1†).72 In accordance with their LE T1 states, all the molecules have comparable 3〈ΨH|ΨL〉 as large as 0.8, accompanied by negligible 3d(ΨH − ΨL). However, 0〈ΨH|ΨL〉 values of 4ArDPDPO2A reach 0.43, which is 2 and 4 fold those of 4ArPPOPO and 4ArPPODPO. Especially, 0d(ΨH − ΨL) values of 4ArDPDPO2A are extremely small and are equal to 2.8 Å, which is extraordinary for D–A systems and only one seventh of those of 4ArPPOPO and 4ArPPODPO. Such small 0d(ΨH − ΨL) values undoubtedly benefit from the uniformly and symmetrically dispersed HOMOs and LUMOs of 4ArDPDPO2A due to the through-space conjugation effect of DPDPO2A. Similarly, in comparison to their analogues, 4ArDPDPO2A reveal remarkably larger 1〈ΨH|ΨL〉 and smaller 0d(ΨH − ΨL). It is noteworthy that compared to 4ArPPOPO, the more centralized FMOs in more twisted 4ArPPODPO by the steric effect dramatically decrease their 0〈ΨH|ΨL〉 and 1〈ΨH|ΨL〉, and enlarge their 0d(ΨH − ΨL) and 1d(ΨH − ΨL), which is the origin of the conflict between ΔEST and Γ for the insulating D–A–D systems. Obviously, as expected, the employment of homoconjugated DPDPO2A can realize the best compromise 〈ΨH|ΨL〉 through simultaneous ICT enhancement due to the electronic effect and FMO delocalization due to the spatial effect.
In consequence, among these molecules, 4ArDPDPO2A simultaneously achieve comparable S1 energy levels, the smallest ΔEST and the largest 〈ΨH|ΨL〉 (Table S2†), which are ascribed to their strongest ICT effects and the uniformly delocalized FMOs, respectively. Therefore, it is convincing that the homoconjugated acceptor-based D–A–D systems would be one of the alternatives with efficient blue TADF emission.
2.3. Optical properties
It is known that the emission color of CT molecules is strongly dependent on the solvent polarity, namely so-called “solvatochromism”, which is ascribed to the large polarity difference between their S0 and S1 states.73 Accordingly, the PL spectra of these PO materials are measured in different solvents (Fig. 3a). In high-polarity solvents, the emissions from all the materials are remarkably red-shifted, manifesting their effective ICT interactions. Nevertheless, it is noted that the bathochromic shifts for 4ArDPDPO2A are the biggest, being 130 nm for the maxima, which is 30 and 40 nm larger than those for 4ArPPOPO and 4ArPPODPO, respectively. As a result, along with the increase of solvent polarity (f), the Stokes shift (δ) of 4ArDPDPO2A increases more sharply than their analogues, which is quantitatively evidenced by the bigger slopes for the former (Fig. 3b). Consistent with theoretical simulation results, by virtue of the through-space conjugation effect, the ICT intensities in 4ArDPDPO2A are the strongest among these molecules, whereas the smallest red shifts for 4ArPPODPO are in accord with their weakest ICT interactions restrained by their twisted configurations. Therefore, with respect to the ICT effect, the linkages in multiple D–A systems are equally important, besides the electronic effects for D and A groups.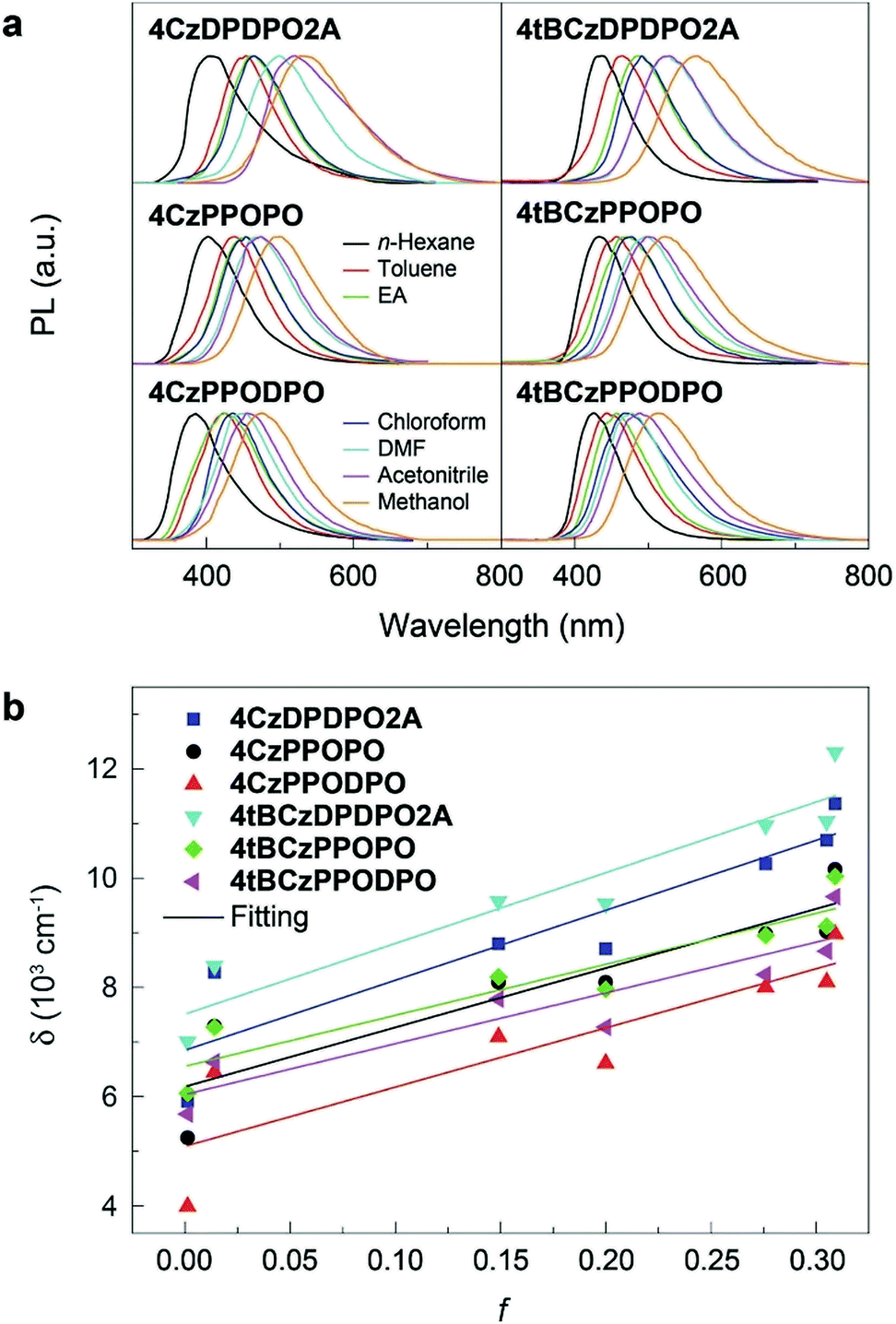 | ||
| Fig. 3 Solvatochromic properties of 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO. (a) Spectral variation of 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO in solvents with different polarities; (b) relationship between solvent orientation polarizability (f) and Stokes shift (δ). | ||
The ground-state dipole moment (μ0) of the molecules is evaluated by DFT calculations (Table S1†). The μ0 of 4ArDPDPO2A is ∼4.5–5.1 Debye, which is about 2 Debye smaller than that of 4ArPPOPO due to the thoroughly symmetrical and coplanar DPDPO2A groups in the former. In the same way, the additional PO and the twisted configuration provide the biggest μ0 for 4ArPPODPO. According to the Lippert–Mataga relationship,1 the singlet excited-state dipole moment (μe) is estimated (Fig. 3b and Table S1†). Because of the reverse order for their ICT intensity, the μe differences between 4ArDPDPO2A and their analogues decrease. Nevertheless, the μe of ArDPDPO2A which is ∼9 Debye is still 1.5–3.5 Debye smaller than that of their analogues, and even 1.2 and 6.2 Debye smaller than those of their D–A units, respectively, which should be attributed to the uniformly dispersed FMOs on the S1 states of ArDPDPO2A.66 The small μ0 of TADF emitters would facilitate their RISC through ΔEST reduction by the host field effect,74 while their small μe is beneficial to suppress dipole–dipole interaction induced quenching.22 In this sense, ArDPDPO2A would be superior in simultaneous TADF improvement and quenching suppression.
The electronic absorption spectra of all the compounds in dilute chloroform solutions (10−6 mol L−1) contain three bands around 230, 285 and 340 nm, corresponding to π → π* transitions of phenyl and Cz groups and n → π* transitions of the latter, respectively (Fig. 4a and b). More importantly, different from the absorption tails for 4ArPPOPO and 4ArPPODPO around 350 nm, 4ArDPDPO2A reveal additional and distinct CT absorption bands centered at 360 and 375 nm, respectively, further displaying their homoconjugation-enhanced ICT. Meanwhile, all the absorption bands are preserved in the spectra of the neat films, further indicating their limited intermolecular interactions (Fig. S8†). The absorption, fluorescence (FL) and phosphorescence (PH) spectra of these molecules in dilute toluene solutions were further measured to determine their intrinsic excited-state energy levels (Fig. S9†). In accord with TDDFT results, the intramolecular electronic and vibrational couplings facilitate red-shifts of PH spectra for these compounds by 30 nm in comparison to the carbazole molecule. As expected, the red-shifted fluorescence and preserved phosphorescence of 4ArDPDPO2A give rise to their near-zero ΔEST of 0.09 and 0.06 eV, respectively, which are remarkably lower by 0.07–0.23 eV in contrast to their nonconjugated analogues (Table S2†).
 | ||
| Fig. 4 Photophysical properties of 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO in the DBFDPO matrix. (a) Electronic absorption spectra in dilute CHCl3 (10−6 mol L−1), room-temperature fluorescence (FL) and low-temperature time-resolved phosphorescence (PH) spectra in DBFDPO-hosted vacuum-evaporated films of 4CzDPDPO2A, 4CzPPOPO and 4CzPPODPO and (b) 4tBCzDPDPO2A, 4tBCzPPOPO and 4tBCzPPODPO; (c) transient emission spectra of 4CzDPDPO2A, 4CzPPOPO and 4CzPPODPO and (d) 4tBCzDPDPO2A, 4tBCzPPOPO and 4tBCzPPODPO. | ||
The vacuum-evaporated films of 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO as dopants in the 4,6-bis(diphenylphosphoryl)dibenzofuran (DBFDPO) matrix were prepared to get an insight into the photophysical properties of the emissive layers (EML) in their OLEDs. The emitters can be homogenously dispersed in DBFDPO, resulting in a small Root-Mean-Square roughness around 1 nm (Fig. S11†). All the films reveal blue/sky-blue fluorescence. It is shown that in comparison to Cz-based analogues, the stronger electron-donating effect of the tBCz group results in moderate emission red shifts of ∼10 nm (Fig. 4a and b). The FL peak wavelengths of 4ArDPDPO2A are 470 and 479 nm, respectively, corresponding to sky-blue emissions, which are only 15 and 10 nm larger than those of 4ArPPOPO and 4ArPPODPO, respectively. Therefore, as designed, the emission bathochromic shifts of 4ArDPDPO2A should mainly originate from their enhanced ICT effect rather than the limited conjugation extension by DPDPO2A. The PH spectrum of the 4CzDPDPO2A-based film at 77 K shows the 0 → 0 transition at 480 nm, giving rise to a small ΔEST of 0.05 eV, which is only half of ∼0.1 eV for its analogues (Table S3†). Therefore, compared to insulating linkages, homoconjugated DPDPO2A can indeed effectively reduce ΔEST. The PH emission of the 4tBCzDPDPO2A-based film peaking at 484 nm is almost overlapped with its FL, which further reduces its ΔEST to 0.03 eV. Nevertheless, 4tBCzPPOPO and 4tBCzPPODPO also give ΔEST as small as 0.04 eV for their films. Since D groups are on both sides of D–A–D systems, D groups with stronger electron-donating ability can enhance the local ICT interaction. In this sense, the comparable ΔEST values of 4CzDPDPO2A, 4tBCzPPOPO and 4tBCzPPODPO actually in turn manifest the effectiveness of through-space conjugation in ICT enhancement. Differently, despite the one more PO acceptor in 4ArPPODPO, its ΔEST is equivalent to that of 4ArPPOPO, since their insulating linkages restrain D–A interactions in single D–A units rather than entire molecules. Nevertheless, all the molecules reveal typical TADF emissions combined with one short-lifetime PF component (Fig. S10†) and one long-lifetime DF component (Fig. 4c and d).
The rate constants (k) and efficiencies (ϕ) of the key transitions during the TADF process are estimated according to the transient characteristics and ϕPF and ϕDF (Fig. 5 and Table S3†).53 The 4tBCzDPDPO2A-based film reveals an extremely high kPF at the level of 108 s−1, which is among the highest values for blue TADF dyes to date.56 The kPF of the 4CzDPDPO2A-based film also reaches 7 × 107 s−1 which is about half of that of 4tBCzDPDPO2A, however, it is still 2–3 fold those of 4ArPPOPO and 4ArPPODPO. Furthermore, 4ArDPDPO2A endow their films with an improved kDF of 2–3 × 104 s−1, which is 2–4 fold those of 4ArPPOPO and 4ArPPODPO. Obviously, the incorporation of the DPDPO2A acceptor in 4ArDPDPO2A simultaneously facilitates their PF and DF processes, among which 4tBCzDPDPO2A with stronger D groups is more superior. In this sense, the ICT interaction would be the key influencing factor for the overall photophysical properties of these emitters.
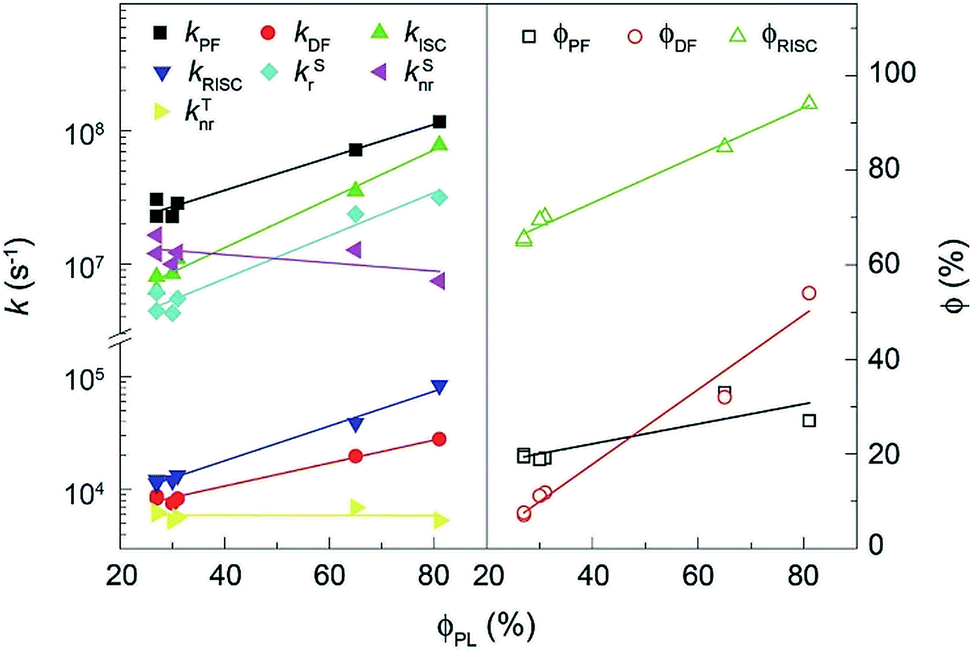 | ||
| Fig. 5 Linear correlations between ϕPL of 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO and the rate constants (k) and efficiencies (ϕ) of the key transitions involved in the TADF process. | ||
Considering RISC as the core transition for the DF process, the k and ϕ of RISC (kRISC and ϕRISC) are further evaluated. The kRISC of the 4tBCzDPDPO2A-based film is beyond 8 × 104 s−1, which is 2 fold that of 4CzDPDPO2A and as high as 8 fold those of 4ArPPOPO and 4ArPPODPO, respectively. At the same time, 4tBCzDPDPO2A realizes the highest ϕRISC of 94%, which is beneficial to RISC for overcoming its inferior k in contrast to intersystem crossing (ISC). On the other hand, the rate constants (kSr) of singlet radiation for 4ArDPDPO2A-based films are as high as 2–3 × 106 s−1, which is dramatically higher by 4–6 fold in comparison to 4ArPPOPO and 4ArPPODPO. Moreover, 4tBCzDPDPO2A also achieves the lowest rate constant of singlet nonradiation (kSnr). Nevertheless, the kSnr values of 4ArDPDPO2A and 4ArPPODPO based films are comparable, which are 50% smaller than those of 4ArPPOPO. The highly rigid coplanar structures of 4ArDPDPO2A and the larger steric hindrance in 4ArPPODPO with one more ortho-DPPO group would restrain the nonradiation paths, e.g. excited-state structural relaxation. More significantly, the kSrs of 4ArDPDPO2A are 2–4 fold larger than their kSnrs, but the situation for 4ArPPOPO and 4ArPPODPO is just opposite, which indicates the intrinsic superiority of 4ArDPDPO2A in luminescence. In contrast, the rate constants of triplet nonradiation (kTnr) for these emitters are nearly equivalent, ascribed to their similar T1 locations. Nonetheless, with respect to triplet utilization, triplet nonradiation directly competes with RISC. Since the kTnrs are more than half of the kRISCs for 4ArPPOPO and 4ArPPODPO, but only one seventh or tenth for 4ArDPDPO2A, the facilitated RISC of 4ArDPDPO2A actually in turn suppresses triplet nonradiation.
The increased DF emissions from 4ArDPDPO2A-based films are indicated by their transient emission spectra (Fig. 4c and d). The DF lifetime (τDF) of 4CzDPDPO2A reaches 16.2 μs, which is about two fold those of its analogues (Table S3†). The τDFs of 4tBCzPPOPO and 4tBCzPPODPO are comparable to that of 4CzDPDPO2A, but remarkably shorter than that of 4tBCzDPDPO2A which is 19.3 μs. The longer τDF of 4ArDPDPO2A should be ascribed to the enhanced locally excited feature of their T1 states and the superiority of their rigid cyclic structures in exited-state relaxation suppression. In contrast, the PF lifetimes (τPF) of 4ArDPDPO2A-based films are significantly shorter than those of their analogues, which reflects the accelerated singlet radiative transitions for the former. As a consequence, the ϕPLs of 4ArDPDPO2A are dramatically improved to 65% and 81%, respectively, which are 2–3 fold those of their analogues. According to the PF and DF ratios in the transient emission spectra, the PF efficiencies (ϕPF) of 4ArDPDPO2A are about 30%, which is 50% higher than those of other emitters. More importantly, 4ArDPDPO2A reveal the state-of-the-art DF efficiencies (ϕDF) of 32% and 54%, respectively, which are higher by ∼4 fold in contrast to their analogues. Thus, the high ϕPLs of 4ArDPDPO2A are mainly contributed by their significantly improved DF emissions, which should be the combined results of their simultaneously facilitated RISC and radiation.
In consequence, 4ArDPDPO2A successfully demonstrate the feasibility of solving the conflict between ΔEST and Γ by virtue of the through-conjugation effect of the homoconjugated acceptors, e.g. DPDPO2A, which can simultaneously enhance ICT interactions, corresponding to ΔEST reduction and RISC facilitation, and singlet radiation for efficient TADF emissions. In opposite, it can be noticed that the k and ϕ values of 4ArPPOPO and 4ArPPODPO are basically comparable, except for structure-related kSnr, which is ascribed to the limited intramolecular electronic communications between two D–A units in nonconjugated D–A–D systems. Therefore, the electronic effect is localized by insulating linkage, but uniformized by homoconjugated acceptors, which is the main reason resulting in the dramatically improved photophysical properties of 4ArDPDPO2A.
2.4. OLED performance
To confirm the superiority of the homoconjugated systems in optoelectronic applications, these molecule doped DBFDPO films were used as EMLs to fabricate OLEDs with a structure of ITO|MoOx (6 nm)|N,N′-di(1-naphthyl)-N,N′-diphenyl-(1,1′-biphenyl)-4,4′-diamine (NPB, 60 nm)|1,3-bis(N-carbazolyl)benzene (mCP, 5 nm)|DBFDPO:PO emitters (30 wt%, 30 nm)|DBFDPO (45 nm)|LiF (1 nm)|Al (100 nm), in which NPB and mCP served as hole-transporting and exciton-blocking layers, respectively, and DBFDPO was the host matrix and electron-transporting layer (Fig. 6a). The FMO energy levels of the employed materials can be mutually well matched to provide favorable carrier injection. The exciton confinement and carrier capture on the emitters can be further supported by DBFDPO with a high T1 energy level of 3.16 eV and suitable FMO energy levels.75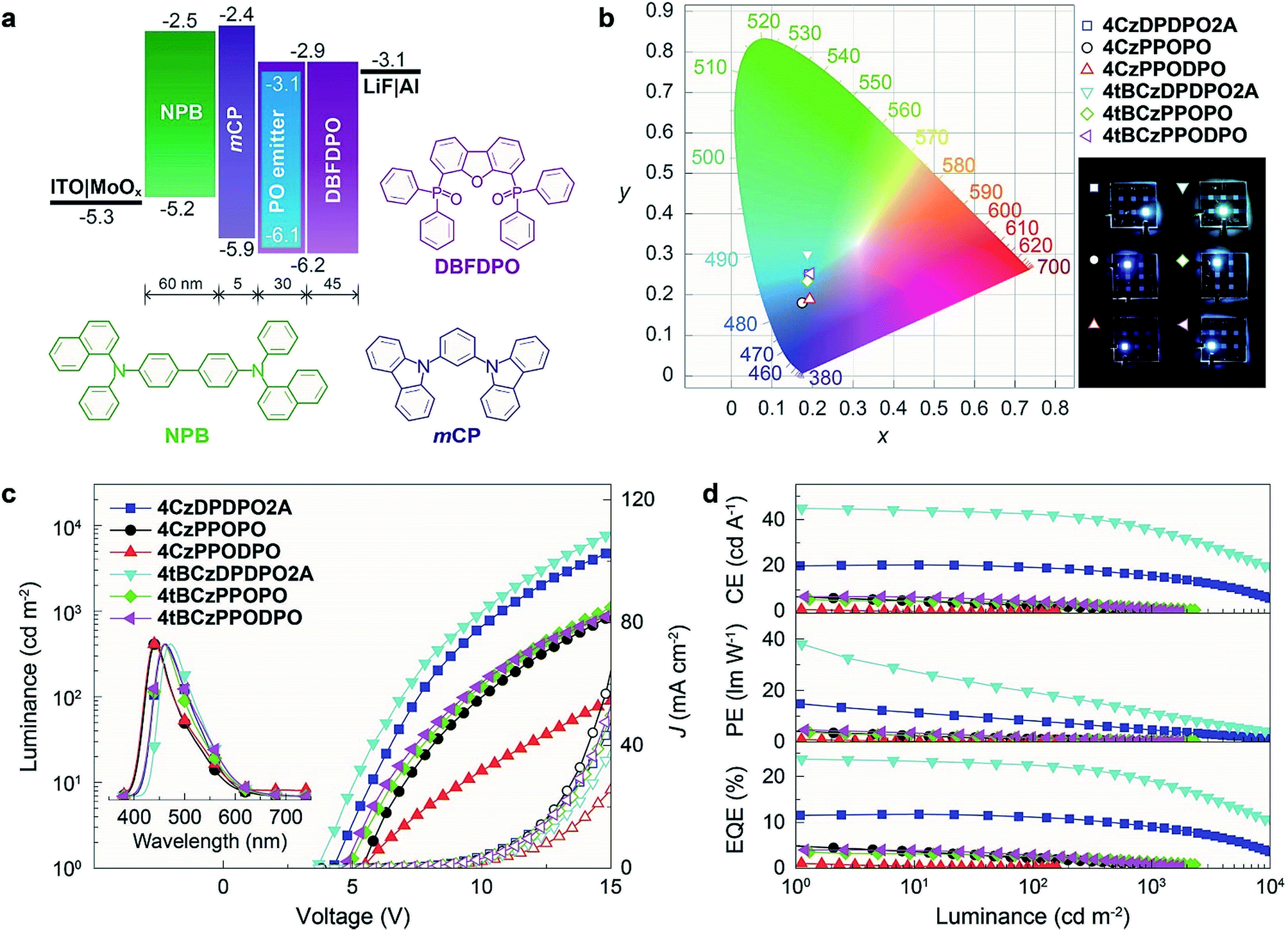 | ||
| Fig. 6 EL performance of 4ArDPDPO2A, 4ArPPOPO and 4ArPPODPO-based blue OLEDs. (a) Configuration and energetic diagram of the devices and the chemical structures of the employed DBFDPO as the host, NPB as the hole transporting layer and mCP as the exciton-blocking layer; (b) photos and the CIE chromaticity coordinates of the devices; (c) luminance–current density–voltage (L–J–V) characteristics and EL spectra (insets) of the devices; (d) efficiency–luminance relationships of the devices. | ||
4ArDPDPO2A realize blue and sky-blue electroluminescence (EL) emissions with Commission Internationale de L'Eclairage (CIE) coordinates of (0.17, 0.24) and (0.18, 0.30), respectively (Fig. 6b and Table 1). It is shown that the CIE coordinates of 4CzDPDPO2A-based devices are comparable to those of 4tBCzPPOPO and 4tBCzPPODPO, in accord with their nearly overlapped EL spectra with blue emissions peaking at 460 nm, which are slightly red-shifted in contrast to deep-blue-emitting 4CzPPOPO and 4CzPPODPO-based devices with ∼0.05 smaller y coordinates and ∼440 nm EL peaks (inset of Fig. 6c and Table 1). The CIE coordinates of 4tBCzDPDPO2A-based devices further increase to (0.18, 0.30), accompanied by an EL peak at 472 nm, which corresponds to sky-blue emission. Thus, as designed, the through-space conjugation reveals its superiority in restraining the conjugation-induced bathochromic shift, making homoconjugated groups “ideal” for constructing blue TADF emitters.
| Emitter | V (V) | L max (cd m−2) | η | λ EL /CIE (x, y) | ||
|---|---|---|---|---|---|---|
| η CE (cd A−1) | η PE (lm W−1) | η EQE (%) | ||||
| a At 1, 100 and 1000 cd m−2. b The maximum luminance. c EL efficiencies at the maximum, 100 and 1000 cd m−2. d EL peak wavelength. | ||||||
| 4CzDPDPO2A | 4.3, 7.8, 10.8 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 020 020 |
20.2, 19.3, 15.9 | 14.8, 7.8, 4.6 | 11.5, 11.0, 9.0 | 460/0.17, 0.24 |
| 4CzPPOPO | 5.3, 10.3, 15.8 | 1344 | 7.1, 3.1, 1.1 | 4.2, 0.9, 0.2 | 4.9, 2.1, 0.8 | 444/0.17, 0.18 |
| 4CzPPODPO | 5.5, 16.0, — | 157 | 1.6, 0.3, — | 0.9, 0.1, — | 1.1, 0.2, — | 440/0.19, 0.19 |
| 4tBCzDPDPO2A | 3.7, 7.3, 10.3 | 18765 |
44.8, 41.8, 35.0 | 38.0, 18.0, 10.7 | 23.7, 22.2, 18.5 | 472/0.18, 0.30 |
| 4tBCzPPOPO | 5.0, 9.5, 15.0 | 2322 | 5.8, 4.3, 2.2 | 3.7, 1.4, 0.5 | 3.6, 2.6, 1.3 | 460/0.18, 0.23 |
| 4tBCzPPODPO | 4.8, 9.3, 16.3 | 1761 | 7.0, 5.1, 1.3 | 4.6, 1.7, 0.2 | 4.0, 2.9, 0.7 | 460/0.19, 0.25 |
The driving voltages of 4ArDPDPO2A-based blue devices are the lowest, being about 4, 7 and 10 V at 1, 100 and 1000 nits, which are remarkably lower by 1, 3 and 5 V, respectively, in contrast to their analogues (Fig. 6c and Table 1). On account of the similar current density (J) of these devices, the higher luminance of 4ArDPDPO2A-based blue devices at the same J should be the main reason for their reduced operating voltages, rather than electrical performance,21,76 which is further evidenced by their dramatically improved maximum luminance beyond 104 nits which is at least 5 fold those for 4ArPPOPO and 4ArPPODPO based devices. For the same reason, compared to 4CzDPDPO2A, 4tBCzDPDPO2A reduced the driving voltages of its devices by 0.5 V, accompanied by 70% increased maximum luminance.
Inherited from their remarkably higher ϕPL, 4ArDPDPO2A significantly elevate EL efficiencies of their blue devices (Fig. 6d and Table 1). The maximum efficiencies of 4tBCzDPDPO2A-based devices reach 44.8 cd A−1 for current efficiency (CE, ηCE), 38.0 lm W−1 for power efficiency (PE, ηPE) and 23.7% for external quantum efficiency (EQE, ηEQE), which are favorable for blue TADF diodes reported so far. Considering the outcoupling ratio of ITO glass which is 25%, the exciton utilization efficiency and the internal quantum efficiency of 4tBCzDPDPO2A-based devices already reached 100%. More importantly, 4tBCzDPDPO2A reveals the state-of-the-art EL efficiency stability. Its devices realize an ηEQE of 22.2 and 18.5% at 100 and 1000 nits, corresponding to EQE roll-offs as low as 6 and 22%, respectively, which is equivalent to the lowest values reported so far for blue TADF diodes.77 By contrast, the maximum EL efficiencies of 4CzDPDPO2A-based blue devices are reduced by about 50%, but its maximum ηEQE of 11.5% is still favorable among pure-blue TADF diodes. Especially, most of the pure-blue TADF devices suffer from the serious roll-offs due to the high-energy excitons more sensitive to quenching effects. In contrast, 4CzDPDPO2A supports its devices with an ηEQE of 11.0 and 9.0% at 100 and 1000 nits, corresponding to the impressive EQE roll-offs of 4 and 22%, respectively. In comparison to 4CzDPDPO2A, the maximum EL efficiencies of 4ArPPOPO and 4ArPPODPO based devices are further halved, accompanied by sharply worsened roll-offs which are more than 30 and 60% at 100 and 1000 nits, respectively.
It is noteworthy that the maximum ηEQEs of these devices reveal a perfect linear dependence on the k and ϕ of the TADF transitions for these emitters, which demonstrates the consistency between photophysical and EL performance modulation (Fig. S12†). Significantly, in comparison to the common focus on kPF and kDF, the maximum ηEQE is more sensitive to the variations of kSr and kRISC, as indicated by the larger slopes of their fitting lines. Therefore, as predicted, the simultaneous enhancement of the singlet radiation and triplet-to-singlet conversion is crucial to improve the EL performance of TADF emitters. Furthermore, in contrast to ϕPF, the triplet-correlated ϕDF and ϕRISC are more influential to the maximum ηEQE, which is in accord with the statistical advantage and dominant contribution of triplet excitons for EL. Therefore, it is convincing that the success of 4ArDPDPO2A in TADF OLEDs originates from the superiority of homoconjugated acceptors in optimizing the photophysical properties of D–A–D systems by the through-space conjugation effect, namely simultaneously enhancing ICT and singlet radiation through FMO optimization.
The excellent EL performance of 4ArDPDPO2A manifests the great potential of homoconjugated D–A systems as TADF emitters, especially for blue OLEDs. Although the exciton utilization efficiencies (EUE) of 4ArDPDPO2A approach 100%, the homoconjugated TADF dye still has a huge space for development with respect to D–A combination and optimization.
3. Experimental section
3.1. Fabrication and characterization of OLEDs
Before loading into a deposition chamber, the ITO substrate was cleaned with detergents and deionized water, dried in an oven at 120 °C for 4 h, and treated with oxygen plasma for 3 min. Devices were fabricated by evaporating organic layers at a rate of 0.1–0.2 nm s−1 onto the ITO substrate sequentially at a pressure below 4 × 10−4 Pa. Onto the electron-transporting layer, a layer of LiF with 1 nm thickness was deposited at a rate of 0.1 nm s−1 to improve electron injection. Finally, a 100 nm-thick layer of Al was deposited at a rate of 0.6 nm s−1 as the cathode. The emission area of the devices was 0.09 cm2 as determined by the overlap area of the anode and the cathode. After fabrication, the devices were immediately transferred to a glove box for encapsulation with glass cover slips using epoxy glue. The EL spectra and CIE coordinates were measured using a PR655 spectra colorimeter. The current–density–voltage and brightness–voltage curves of the devices were measured using a Keithley 4200 source meter and a calibrated silicon photodiode. All the measurements were carried out at room temperature under ambient conditions. For each structure, four devices were fabricated in parallel to confirm the performance repeatability. The device results reported were the data most close to the average values.4. Conclusions
We demonstrate the feasibility of optimizing the photophysical properties of D–A–D molecules via the through-space conjugation effect of homoconjugated acceptors. In contrast to insulating linkage and through-bond conjugation, through-space conjugation provides sufficient intramolecular electronic communication without immoderate conjugated extension. This feature makes it “ideal” for selectively adjusting optoelectronic properties. As evidenced by 4ArDPDPO2A, besides preserved blue emissions, the employment of homoconjugated DPDPO2A groups uniformly delocalizes FMOs for simultaneous ICT and 〈ΨH|ΨL〉 enhancement, giving rise to their dramatically increased kRISC and kSr by 4–6 times. The comprehensively optimized TADF transitions endow 4tBCzDPDPO2A with ϕPL and ηEQE reaching 81 and 23.7%. This work not only develops a new kind of blue TADF emitter, but also indicates the significance of conjugation optimization for D–A systems, which would accelerate their realistic applications in diverse optoelectronic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Changjiang Scholar Program of Chinese Ministry of Education (Q2016208), National Natural Science Foundation of China (21672056, 61605042, 21602048, 51873056 and B5182900), Natural Science Foundation of Heilongjiang Province (QC2016072 and QC2017008) and National Postdoctoral Program for Innovative Talents (BX201600048 and BX20180092).Notes and references
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- A. Chaskar, H.-F. Chen and K.-T. Wong, Adv. Mater., 2011, 23, 3876–3895 CrossRef CAS PubMed.
- D. Yu, F. Zhao, C. Han, H. Xu, J. Li, Z. Zhang, Z. Deng, D. Ma and P. Yan, Adv. Mater., 2012, 24, 509–514 CrossRef CAS PubMed.
- C. Han, Z. Zhang, H. Xu, J. Li, G. Xie, R. Chen, Y. Zhao and W. Huang, Angew. Chem., Int. Ed., 2012, 51, 10104–10108 CrossRef CAS PubMed.
- C. Han, Z. Zhang, H. Xu, S. Yue, J. Li, P. Yan, Z. Deng, Y. Zhao, P. Yan and S. Liu, J. Am. Chem. Soc., 2012, 134, 19179–19188 CrossRef CAS PubMed.
- D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2011, 133, 17895–17900 CrossRef CAS PubMed.
- S. Gong, Q. Fu, W. Zeng, C. Zhong, C. Yang, D. Ma and J. Qin, Chem. Mater., 2012, 24, 3120–3127 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- D. M. E. Freeman, A. J. Musser, J. M. Frost, H. L. Stern, A. K. Forster, K. J. Fallon, A. G. Rapidis, F. Cacialli, I. McCulloch, T. M. Clarke, R. H. Friend and H. Bronstein, J. Am. Chem. Soc., 2017, 139, 11073–11080 CrossRef CAS PubMed.
- G. Xie, X. Li, D. Chen, Z. Wang, X. Cai, D. Chen, Y. Li, K. Liu, Y. Cao and S.-J. Su, Adv. Mater., 2016, 28, 181–187 CrossRef CAS.
- Z. Weixuan, L. Hsin-Yu, L. Wei-Kai, J. Min, S. Yi-Jiun, Z. Cheng, G. Shaolong, Z. Tao, X. Guohua, S. Monima, W. Ken-Tsung, W. Chung-Chih and Y. Chuluo, Adv. Mater., 2018, 30, 1704961 CrossRef PubMed.
- H. Wang, L. Meng, X. Shen, X. Wei, X. Zheng, X. Lv, Y. Yi, Y. Wang and P. Wang, Adv. Mater., 2015, 27, 4041–4047 CrossRef CAS PubMed.
- Q. Zhang, H. Kuwabara, W. J. Potscavage, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS PubMed.
- C. Li, R. Duan, B. Liang, G. Han, S. Wang, K. Ye, Y. Liu, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2017, 56, 11525–11529 CrossRef CAS PubMed.
- L.-S. Cui, Y.-M. Xie, Y.-K. Wang, C. Zhong, Y.-L. Deng, X.-Y. Liu, Z.-Q. Jiang and L.-S. Liao, Adv. Mater., 2015, 27, 4213–4217 CrossRef CAS PubMed.
- H. G. Kim, K. H. Kim and J. J. Kim, Adv. Mater., 2017, 1702159, DOI:10.1002/adma.201702159.
- X.-K. Liu, Z. Chen, C.-J. Zheng, M. Chen, W. Liu, X.-H. Zhang and C.-S. Lee, Adv. Mater., 2015, 27, 2025–2030 CrossRef CAS PubMed.
- D. Zhang, X. Song, M. Cai and L. Duan, Adv. Mater., 2018, 30, 1705250 CrossRef PubMed.
- J. Zhang, D. Ding, Y. Wei, F. Han, H. Xu and W. Huang, Adv. Mater., 2016, 28, 479–485 CrossRef CAS PubMed.
- C. Han, Z. Zhang, D. Ding and H. Xu, Chem, 2018, 4, 2154–2167 CAS.
- Y. Tao, L. Xu, Z. Zhang, R. Chen, H. Li, H. Xu, C. Zheng and W. Huang, J. Am. Chem. Soc., 2016, 138, 9655–9662 CrossRef CAS PubMed.
- C. Han, C. Duan, W. Yang, M. Xie and H. Xu, Sci. Adv., 2017, 3, e1700904 CrossRef PubMed.
- H. Liu, Q. Bai, L. Yao, H. Zhang, H. Xu, S. Zhang, W. Li, Y. Gao, J. Li, P. Lu, H. Wang, B. Yang and Y. Ma, Chem. Sci., 2015, 6, 3797–3804 RSC.
- W.-Y. Wong and C.-L. Ho, Acc. Chem. Res., 2010, 43, 1246–1256 CrossRef CAS PubMed.
- S. Dai, T. Li, W. Wang, Y. Xiao, T. K. Lau, Z. Li, K. Liu, X. Lu and X. Zhan, Adv. Mater., 2018, 30, 1706571 CrossRef PubMed.
- J. D. Chen, Y. Q. Li, J. Zhu, Q. Zhang, R. P. Xu, C. Li, Y. X. Zhang, J. S. Huang, X. Zhan, W. You and J. X. Tang, Adv. Mater., 2018, 30, 1706083 CrossRef PubMed.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, F. E. CurchodBasile, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, K. NazeeruddinMd and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 593–597 CAS.
- Q. Zhang, B. Kan, F. Liu, G. Long, X. Wan, X. Chen, Y. Zuo, W. Ni, H. Zhang, M. Li, Z. Hu, F. Huang, Y. Cao, Z. Liang, M. Zhang, T. P. Russell and Y. Chen, Nat. Photonics, 2015, 9, 35–41 CrossRef CAS.
- Y. Sun, G. C. Welch, W. L. Leong, C. J. Takacs, G. C. Bazan and A. J. Heeger, Nat. Mater., 2012, 11, 44–48 CrossRef CAS PubMed.
- M. Qian, R. Zhang, J. Hao, W. Zhang, Q. Zhang, J. Wang, Y. Tao, S. Chen, J. Fang and W. Huang, Adv. Mater., 2015, 27, 3546–3552 CrossRef CAS PubMed.
- H. Zhang, Y. Wu, W. Zhang, E. Li, C. Shen, H. Jiang, H. Tian and W.-H. Zhu, Chem. Sci., 2018, 9, 5919–5928 RSC.
- H. Dong, H. Zhu, Q. Meng, X. Gong and W. Hu, Chem. Soc. Rev., 2012, 41, 1754–1808 RSC.
- K.-J. Baeg, M. Binda, D. Natali, M. Caironi and Y.-Y. Noh, Adv. Mater., 2013, 25, 4267–4295 CrossRef CAS PubMed.
- R. H. Kim, H. J. Kim, I. Bae, S. K. Hwang, D. B. Velusamy, S. M. Cho, K. Takaishi, T. Muto, D. Hashizume, M. Uchiyama, P. André, F. Mathevet, B. Heinrich, T. Aoyama, D.-E. Kim, H. Lee, J.-C. Ribierre and C. Park, Nat. Commun., 2014, 5, 3583 CrossRef PubMed.
- T. Sekitani, T. Yokota, U. Zschieschang, H. Klauk, S. Bauer, K. Takeuchi, M. Takamiya, T. Sakurai and T. Someya, Science, 2009, 326, 1516–1519 CrossRef CAS PubMed.
- V. K.-M. Au, D. Wu and V. W.-W. Yam, J. Am. Chem. Soc., 2015, 137, 4654–4657 CrossRef CAS PubMed.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384–387 CrossRef CAS PubMed.
- X. Xiong, F. Song, J. Wang, Y. Zhang, Y. Xue, L. Sun, N. Jiang, P. Gao, L. Tian and X. Peng, J. Am. Chem. Soc., 2014, 136, 9590–9597 CrossRef CAS PubMed.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- H. N. Kim, Z. Guo, W. Zhu, J. Yoon and H. Tian, Chem. Soc. Rev., 2011, 40, 79–93 RSC.
- X. Guo, M. Zhang, J. Tan, S. Zhang, L. Huo, W. Hu, Y. Li and J. Hou, Adv. Mater., 2012, 24, 6536–6541 CrossRef CAS PubMed.
- H. Meier, Angew. Chem., Int. Ed., 2005, 44, 2482–2506 CrossRef CAS PubMed.
- M. W. Wolf, K. D. Legg, R. E. Brown, L. A. Singer and J. H. Parks, J. Am. Chem. Soc., 1975, 97, 4490–4497 CrossRef CAS.
- J. Partee, E. L. Frankevich, B. Uhlhorn, J. Shinar, Y. Ding and T. J. Barton, Phys. Rev. Lett., 1998, 82, 3673–3676 CrossRef.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- G. X. Yifan Li, S. Gong, K. Wu and C. Yang, Chem. Sci., 2016, 7, 5441–5447 RSC.
- X. Cai, X. Li, G. Xie, Z. He, K. Gao, K. Liu, D. Chen, Y. Cao and S.-J. Su, Chem. Sci., 2016, 7, 4264–4275 RSC.
- M. Klessinger, Angew. Chem., Int. Ed., 1995, 34, 549–551 CrossRef CAS.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- E. Fermi, Nuclear Physics: A Course Given by Enrico Fermi at the University of Chicago, University of Chicago Press, Chicago, 1950 Search PubMed.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- P. Rajamalli, N. Senthilkumar, P. Gandeepan, P.-Y. Huang, M.-J. Huang, C.-Z. Ren-Wu, C.-Y. Yang, M.-J. Chiu, L.-K. Chu, H.-W. Lin and C.-H. Cheng, J. Am. Chem. Soc., 2016, 138, 628–634 CrossRef CAS PubMed.
- P. Rajamalli, N. Senthilkumar, P. Y. Huang, C. C. Ren-Wu, H. W. Lin and C. H. Cheng, J. Am. Chem. Soc., 2017, 139, 10948–10951 CrossRef CAS PubMed.
- Y. J. Cho, S. K. Jeon, B. D. Chin, E. Yu and J. Y. Lee, Angew. Chem., Int. Ed., 2015, 54, 5201–5204 CrossRef CAS PubMed.
- C. Duan, J. Li, C. Han, D. Ding, H. Yang, Y. Wei and H. Xu, Chem. Mater., 2016, 28, 5667–5679 CrossRef CAS.
- C. Li, C. Duan, C. Han and H. Xu, Adv. Mater., 2018, 30, 1804228 CrossRef PubMed.
- J. F. Coetzee and G. R. Padmanabhan, J. Phys. Chem., 1965, 69, 3193–3196 CrossRef CAS.
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. Van Voorhis, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 11908–11911 CrossRef CAS PubMed.
- Q. Liang, C. Han, C. Duan and H. Xu, Adv. Magn. Opt. Mater., 2018, 6, 1800020 CrossRef.
- L. T. Scott, Pure Appl. Chem., 1986, 58, 105–110 CAS.
- J. Li, D. Ding, Y. Wei, J. Zhang and H. Xu, Adv. Magn. Opt. Mater., 2016, 4, 522–528 CrossRef CAS.
- Y. Pan, W. Li, S. Zhang, L. Yao, C. Gu, H. Xu, B. Yang and Y. Ma, Adv. Magn. Opt. Mater., 2014, 2, 510–515 CrossRef CAS.
- T. Chen, L. Zheng, J. Yuan, Z. An, R. Chen, Y. Tao, H. Li, X. Xie and W. Huang, Sci. Rep., 2015, 5, 10923 CrossRef PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- S. Manzetti and T. Lu, J. Phys. Org. Chem., 2013, 26, 473–483 CrossRef CAS.
- R. Ishimatsu, S. Matsunami, K. Shizu, C. Adachi, K. Nakano and T. Imato, J. Phys. Chem. A, 2013, 117, 5607–5612 CrossRef CAS PubMed.
- G. Méhes, K. Goushi, W. J. Potscavage Jr and C. Adachi, Org. Electron., 2014, 15, 2027–2037 CrossRef.
- C. Han, G. Xie, J. Li, Z. Zhang, H. Xu, Z. Deng, Y. Zhao, P. Yan and S. Liu, Chem.–Eur. J., 2011, 17, 8947–8956 CrossRef CAS PubMed.
- Z. Zhang, Z. Zhang, D. Ding, Y. Wei, H. Xu, J. Jia, Y. Zhao, K. Pan and W. Huang, J. Phys. Chem. C, 2014, 118, 20559–20570 CrossRef CAS.
- H. Yang, Q. Liang, C. Han, J. Zhang and H. Xu, Adv. Mater., 2017, 29, 1700553 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, single-crystal packing diagrams, theoretical simulation results, optical properties, thermal stability, and electrochemical analysis. CCDC 1869741. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01240k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |