
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative insights into charge-separated states from one- and two-pulse laser experiments relevant for artificial photosynthesis†
Svenja
Neumann
,
Christoph
Kerzig
 * and
Oliver S.
Wenger
*
* and
Oliver S.
Wenger
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: christoph.kerzig@unibas.ch; oliver.wenger@unibas.ch
First published on 9th May 2019
Abstract
Charge-separated states (CSSs) are key intermediates in photosynthesis and solar energy conversion. However, the factors governing the formation efficiencies of CSSs are still poorly understood, and light-induced electron–hole recombinations as deactivation pathways competing with desired charge accumulations are largely unexplored. This greatly limits the possibility to perform efficient multi-electron transfer, which is essential for artificial photosynthesis. We present a systematic investigation of two donor–sensitizer–acceptor triads (with different donor–acceptor distances) capable of storing as much as 2.0 eV in their CSSs upon the absorption of a visible photon. Using quantitative one- and two-pulse laser flash photolysis, we provide deep insights into both the CSS formation quantum yield, which can reach up to 80%, and the fate of the CSS upon further (secondary) excitation with green photons. The triad with shorter intramolecular distances shows a remarkable excitation wavelength dependence of the CSS formation quantum yield, and the CSS of this triad undergoes more efficient light-induced charge recombination than the longer equivalent by about one order of magnitude, whilst thermal charge recombination shows the exact opposite behavior. The unexpected results of our detailed photophysical study can be rationalized by detrimental singlet charge transfer states or structural considerations, and could significantly contribute to the future design of CSS precursors for accumulative multi-electron transfer and artificial photosynthesis.
1 Introduction
Encouraged by the efficiency of natural photosynthesis,1 numerous research groups have focused on mimicking its key steps. In particular, a great many studies explored molecular donor–acceptor compounds,2–27 in which the primary photoinduced charge separation was frequently enabled by a covalently linked photosensitizer.5,6,10–16,19,24 Optimizing the primary charge separation is without any doubt one basic requirement for the realization of much more complex artificial systems allowing solar fuel production through multiphoton-driven charge accumulation.28–31 However, the great majority of previous studies on charge-separated states merely investigated their qualitative formation after light absorption and their lifetimes, but systematic quantum yield investigations12,14,32 are scarce. In direct consequence, the factors governing the charge-separated state (CSS) formation efficiencies are still poorly understood. Even less explored are the processes that occur upon further excitation of CSSs with a second photon, such as detrimental light-induced charge recombination,33,34 although their deep understanding is of pivotal importance for the efficient accumulation of redox equivalents.29,30,35The molecular design of our compounds (Scheme 1a) is such that about 2.0 eV are stored (in CH3CN) in the final CSS after absorption of a single visible photon (energies of the photons used for excitation, 2.6 to 3.0 eV). A comparison with CSS energies of other triads revealed that the stored energy typically amounts to about 1.5 eV upon visible light excitation,9–12,36 and in two recent CSS studies stored energies between 1.7 and 1.8 eV were declared as highly energetic.4,5 Recently, we reported on the distance dependence of thermal charge recombination in the compounds from Scheme 1a in comparison to two series of other molecular triads.16 The unusually high CSS energy of the two specific triads in Scheme 1a prompted us to perform a complete quantitative photophysical investigation including new multi-photon excitation studies, geared at understanding light-induced charge recombination. We optimized our laser setup such that it permits one- and two-pulse laser experiments with sensitive and quantitative detection of reaction intermediates, as well as homogeneous excitation in the detection volume (a detailed description of our setup is given in the ESI†). That setup allows us to gain insights that are far beyond those obtained in former investigations of photoinduced electron transfer in donor–acceptor compounds and CSS formation.
 | ||
| Scheme 1 (a) Structure of molecular triads. (b) Schematic representation of the mechanism for light-induced (hν1) formation of a CSS in a molecular electron donor-photosensitizer-electron acceptor triad together with thermal (τ) and light-induced (hν2) charge recombination. | ||
The triads under study contain a triarylamine (TAA) donor and an anthraquinone (AQ) moiety as an acceptor. Symmetrical addition of p-xylene units to the central bipyridine moiety enabled us to elongate the triad and to perform distance-dependent studies (Scheme 1a). After visible-light excitation of the iridium photosensitizer, a CSS is formed. As indicated in Scheme 1b, charge recombination between oxidized donor and reduced acceptor can either occur via well-known thermal charge recombination with characteristic lifetimes (τ; central part)16 or through poorly-understood light-induced recombination pathways (hν2; left part). Since it is desirable to use high-energy CSSs for demanding (photo)chemical processes,4 we considered the following aspects in our study: (i) the photoactive system should store as much energy as possible (Section 2.1), (ii) long lifetimes of the CSS are essential for the applicability of the photoactive system for subsequent (photo)reactions (Section 2.2), (iii) high quantum yields for light-induced charge separation are desirable (Section 2.3), (iv) the energy efficiency of photon usage is an important factor, and it is desirable to convert a very large fraction of the photon energy into chemical energy (Sections 2.1 and 2.3), and (v) the absorption of a second visible photon should ideally not induce light-driven charge recombination, which usually represents a detrimental CSS decay pathway (Section 2.4). In particular, that last aspect is underexplored given its importance for accumulative multi-electron transfer,37–44 and, as noted above, quantitative factors of CSS formation are often neglected in studies of donor–sensitizer–acceptor compounds.29,30,35
2 Results and discussion
We recently reported the synthesis and characterization of the triads TAA-ph1-Ir-ph1-AQ and TAA-ph2-Ir-ph2-AQ focusing exclusively on the influence of driving forces on the distance dependences of thermal charge recombination rates. Our present study substantially extends the earlier investigation of the triads and contains the following new photophysical experiments and insights: (i) excitation wavelength dependent quantum yields for charge separation were measured and insight into the importance of intraligand charge-transfer excitations was obtained; (ii) two-pulse experiments were performed to determine the quantum yield for light-induced charge recombination, to explore whether the distance dependence of that poorly understood side reaction obeys conventional electron transfer theory. The extent of quantitative information gained from these studies is unmatched by earlier investigations of molecular donor–acceptor compounds, and our two-color two-pulse (pump–pump–probe) experiments might inspire new types of photophysical studies.2.1 CSS formation and energetic considerations
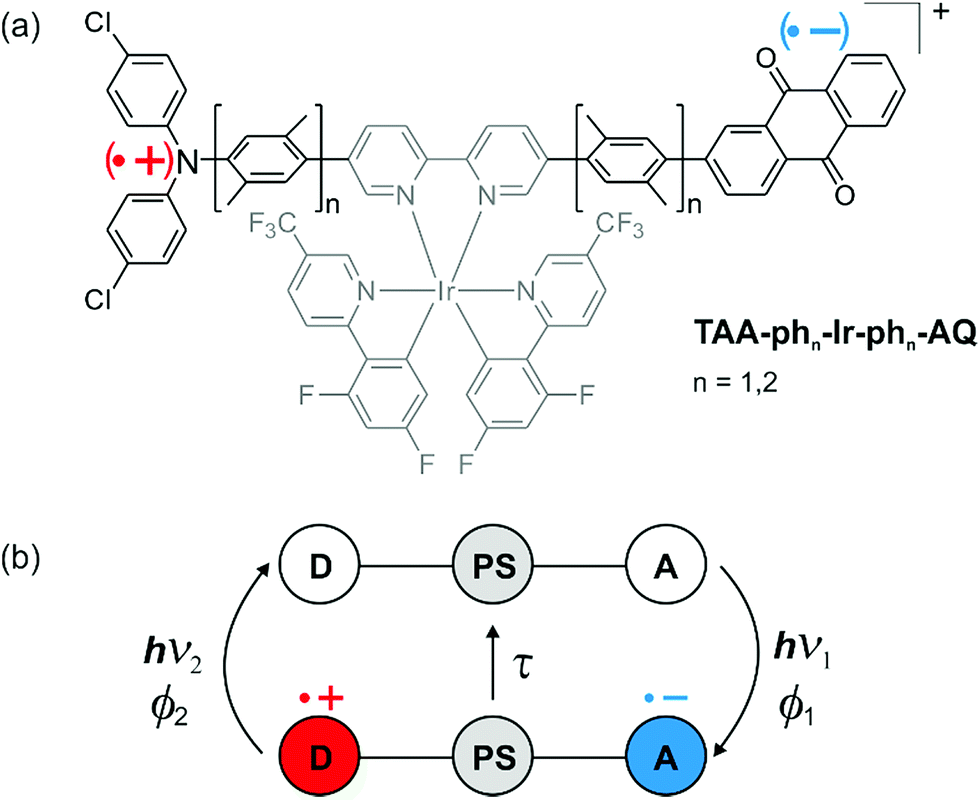
Photoexcitation of our Ir-based sensitizer into its metal-to-ligand charge transfer (MLCT) absorption band between 390 and 450 nm produces, after intersystem crossing, its triplet state.16 However, comparison of the UV-Vis spectrum of TAA-ph1-Ir-ph1-AQ (Fig. 1a, green trace) with TAA-ph2-Ir-ph2-AQ (Fig. 1a, violet trace) and Ir-Ref (Fig. 1a, red trace; see ESI page S8† for molecular structure) shows an unexpectedly high extinction coefficient in the blue for the shorter triad. That observation suggests that this triad exhibits not only an MLCT transition in this spectral range, but there is also an intraligand charge transfer (CT). We postulated earlier that this intraligand CT occurs between the TAA donor and the bipyridine ligand of the photosensitizer and that the increase of the distance between those two moieties in the TAA-ph2-Ir-ph2-AQ triad decreases the contribution of the intraligand CT significantly,16 which is now further substantiated by DFT-calculations (see ESI, page S6†). The population of additional CT transitions is obviously not limited to our system, as the comparison of the UV-Vis absorption spectra within other series of triads suggests, where exclusively the molecules with the shortest distance show additional shoulders in the UV-Vis absorption spectra.11,45 The same effect can also be observed in dyads with a short TAA-bipyridine distance,46 while AQ-bipyridine dyads show no differences in the absorption behavior, even with short intramolecular distances.47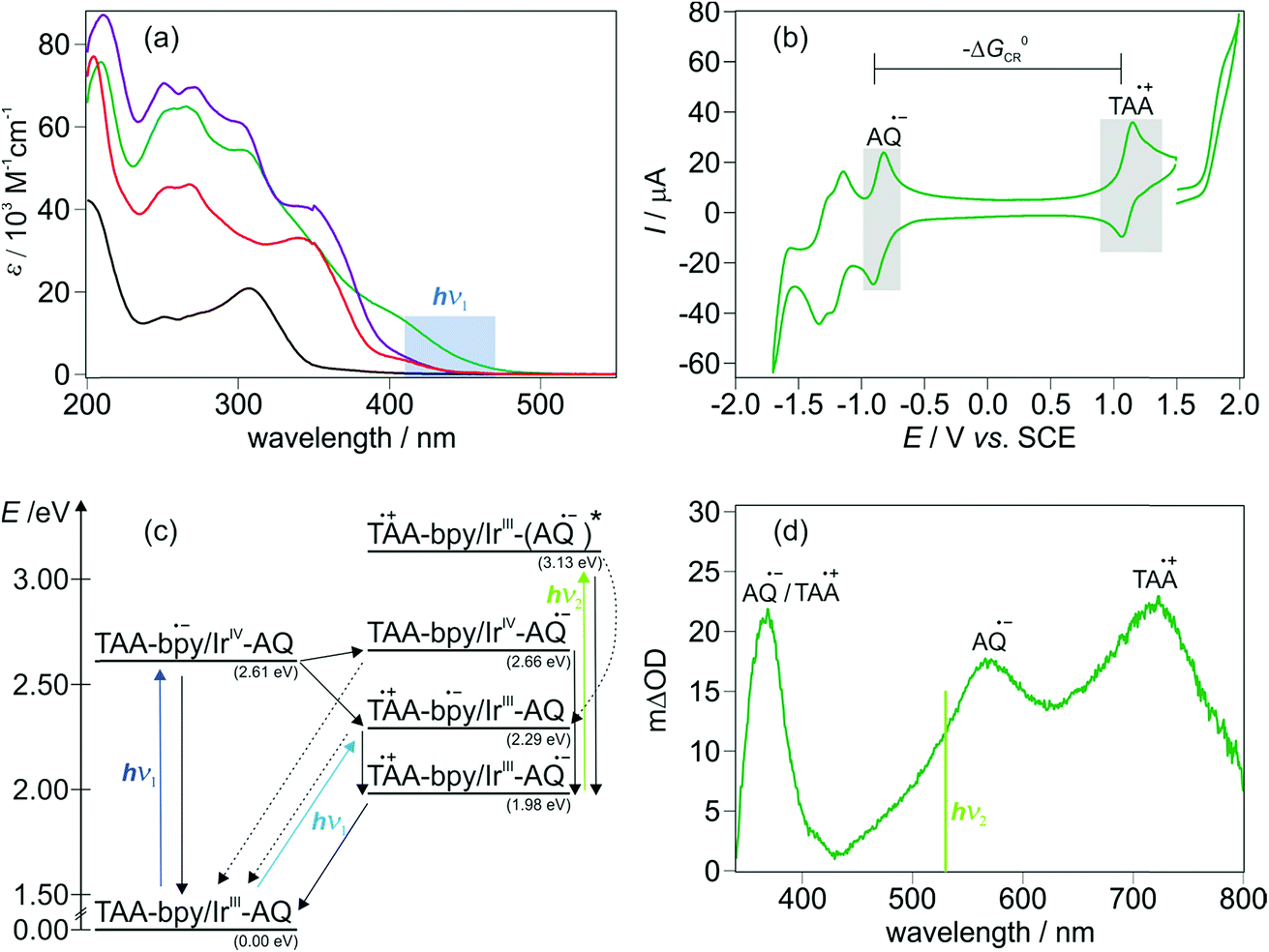 | ||
| Fig. 1 (a) UV-Vis absorption spectra of TAA-ph1-Ir-ph1-AQ (green), TAA-ph2-Ir-ph2-AQ (violet), Ir-Ref (red, see Fig. S3† for molecular structure) and the bipyridine-containing free ligand of the longer triad (black) in CH3CN at 293 K. The highlighted region indicates the wavelength range used for photoexcitation. (b) Cyclic voltammogram for TAA-ph1-Ir-ph1-AQ in dry and deoxygenated CH3CN. TBAPF6 (0.1 M) was used as an electrolyte and a scan rate of 0.1 V s−1 was applied. (c) Averaged energy of relevant excited states and relevant intermediates for TAA-ph1-Ir-ph1-AQ and TAA-ph2-Ir-ph2-AQ in CH3CN. Energies were estimated using the redox potentials from Table S1 and eqn (S1).†E00 for the Ir sensitizer is 2.61 eV (ref. 16) whereas E00 for AQ˙− was estimated from its long wavelength absorption band.49 (d) Transient UV-Vis absorption spectrum of TAA-ph1-Ir-ph1-AQ in deoxygenated CH3CN at 293 K. A 20 μM solution was excited at 440 nm with laser pulses of ca. 10 ns duration; detection occurred by integration over a period of 200 ns immediately afterwards. | ||
The amount of stored energy in the CSS (ECSS) is equivalent to the driving force (−ΔG0CR) of the thermal charge recombination process of the photoexcited triads under study. The driving force can be derived from the redox potentials of AQ˙− and TAA˙+ in the respective triad (Fig. 1b and ESI, page S5†), which results in a −ΔG0CR value of 1.99 eV for the shorter TAA-ph1-Ir-ph1-AQ triad and 1.96 eV for TAA-ph2-Ir-ph2-AQ, i.e., both triads can store ca. 2.0 eV in their CSSs (in CH3CN). The maximal excitation wavelength for detectable CSS formation in the TAA-ph2-Ir-ph2-AQ triad is 450 nm, whereas the CSS of TAA-ph1-Ir-ph1-AQ can be generated with wavelengths up to ca. 470 nm due to the higher extinction coefficients resulting from the superposition of Ir-based MLCT and intraligand CT transitions (see Fig. 1a and Section 2.3 for details). Excitation with longer wavelengths is preferred to maximize energy efficiencies (η), which we define as the ratios of ECSS and the photon energies at the maximum excitation wavelength usable for CSS formation under our conditions. With the photon energies at the above-mentioned limiting wavelengths, energy efficiencies of 71% (longer triad) and 75% (shorter triad) thus result. These values are close to the upper limit of already reported high-energy CSSs where a visible photon was absorbed to form the respective CSS.4,5,11,48 The energy efficiencies for the triads under study also compare very favorably with a reported two-photon process, which stores the same amount of energy as our triads.28 Based on the redox potentials from the cyclic voltammograms of the triads (Fig. 1b and ESI, page S5†), an energy level scheme for excited states and relevant intermediates of the triads TAA-ph1-Ir-ph1-AQ and TAA-ph2-Ir-ph2-AQ was calculated (Fig. 1c). The shown energies are averaged over both triads. Excitation of TAA-ph1-Ir-ph1-AQ (λ1 = 470 nm) mainly results in an intraligand CT from the TAA unit to the bipyridine ligand of the Ir sensitizer since there is a significant oscillator strength for this excitation (see ESI, page S6†). Electron transfer from the reduced bipyridine to the AQ unit finally gives the long-lived CSS. By contrast, the absorption of a higher energy photon (λ1 = 450 nm) by TAA-ph2-Ir-ph2-AQ mainly excites an MLCT transition. The final CSS can then be generated by (i) reductive quenching of the excited Ir chromophore by the TAA moiety, followed by an electron transfer from the reduced Ir photosensitizer to the AQ unit, or (ii) oxidative quenching of the excited Ir sensitizer by the AQ unit, followed by an electron transfer from the TAA moiety to the IrIV species. The formed final CSS in both triads can then undergo thermal charge recombination into the ground state. In addition, the final CSS can also be selectively photoexcited by absorption of a second visible photon (λ2 = 532 nm) inducing rapid reverse electron transfer as discussed in Section 2.4.
The formation of the final CSS was detected immediately after photoexcitation by transient absorption spectroscopy (Fig. 1d and S5 of the ESI†). The transient absorption spectrum of TAA-ph1-Ir-ph1-AQ (Fig. 1d) clearly shows the spectroscopic signatures of the anticipated CSS. The one-electron oxidized TAA unit exhibits bands at 370 and 720 nm whereas the AQ˙− species is characterized by bands at 370 and 570 nm.16 For TAA-ph2-Ir-ph2-AQ, the same spectroscopic signatures can be observed though the TAA˙+ signal shifts from 720 to ca. 700 nm (see ESI, Fig. S5†).
2.2 Lifetimes of charge-separated states
In a recent study we compared the distance dependence of charge recombination rates in TAA-ph1-Ir-ph1-AQ and TAA-ph2-Ir-ph2-AQ to two other series of triads with Ru-based sensitizers.16 For TAA-ph1-Ir-ph1-AQ and TAA-ph2-Ir-ph2-AQ we found that CSS lifetimes decrease with increasing donor–acceptor distance, and this somewhat counter-intuitive behavior is readily understood on the basis of Marcus theory as discussed earlier.11,16,45,50–52 In the present study, lower excitation densities were used (see also ESI page S3†) and lifetimes of the CSSs (τCSS) of ca. 1.2 μs for TAA-ph1-Ir-ph1-AQ and ca. 85 ns for TAA-ph2-Ir-ph2-AQ were determined in dry and deoxygenated CH3CN at 293 K. The instantaneous CSS formation within the duration of the exciting laser pulse (ca. 10 ns), which is further confirmed by kinetic emission measurements (see ESI page S8†), and thermal charge recombination in both triads are illustrated by Fig. 2.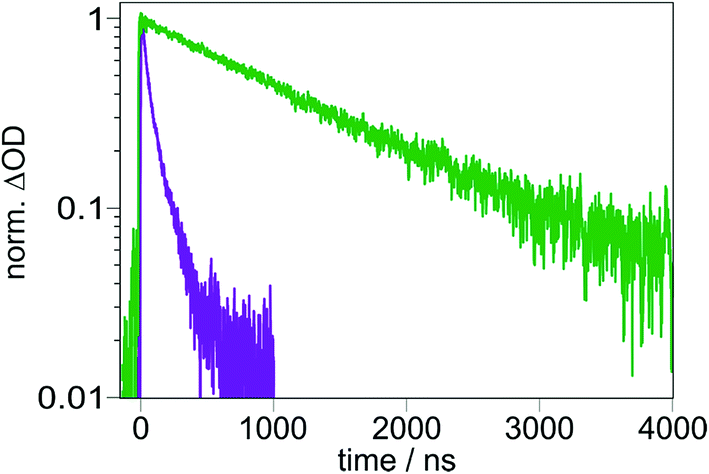 | ||
| Fig. 2 Formation and decay of the transient absorption signals for TAA-ph1-Ir-ph1-AQ (green) and TAA-ph2-Ir-ph2-AQ (violet) in dry and deoxygenated CH3CN at 293 K monitoring the TAA˙+ band at 720 nm (green) and 700 nm (violet). Excitation of both triads occurred at 433 nm with laser pulses of ca. 10 ns duration. | ||
Consideration of the relationship between a high driving force, i.e., a high ECSS, and a long donor–acceptor distance has an important impact on the future design of CSS precursors for artificial photosynthesis and charge accumulation. The capability of high energy storage is desirable, but too long donor–acceptor distances in such systems can counteract a long lifetime of the CSS (see Fig. 2),11,16,45,52 which is required to exploit the stored energy for subsequent (photo)reactions,4,53 and are therefore counterproductive for the application in solar energy storage and conversion.
2.3 Quantum yields and excitation wavelength dependences of CSS formation
In sophisticated molecular systems designed for multiphoton-driven charge accumulation, the quantum yield for formation of the primary CSS should ideally be close to unity. In this section, we will comprehensibly describe an experimental technique suitable for CSS quantum yield determinations using molecular triads as model compounds to study that process in isolation, and we will provide strategies to understand unexpected results. Our methodology is based on relative actinometry54 combined with laser flash photolysis (LFP). In principle the actinometry-LFP combo is a rather old approach,55–57 but many 21st century photochemists do not seem to be familiar with that powerful tool, perhaps due to the lack of tutorial reviews as recently published for other photochemical techniques.58,59Fig. 3 enlarges on our quantum yield determination methodology. As reference system suitable for laser flash photolysis actinometry,60–64 we selected tris(2,2′-bipyridine)ruthenium(II) [Ru(bpy)3]2+ in water (the favorable properties of that system are summarized on page S3 of the ESI†). First, solutions of [Ru(bpy)3]2+ and the respective Ir-containing triad were prepared with concentrations ensuring identical absorption values at the desired excitation wavelength. An example for TAA-ph1-Ir-ph1-AQ prior to exciting with 410 nm laser pulses is shown in Fig. 3a. Second, employing the very same excitation conditions for the reference system and the triad under study, transient absorption traces at 455 nm (maximum of the [Ru(bpy)3]2+ ground state bleach)63 and the CSS absorption maximum (TAA-ph1-Ir-ph1-AQ, 720 nm; TAA-ph2-Ir-ph2-AQ, 700 nm) were recorded (Fig. 3b).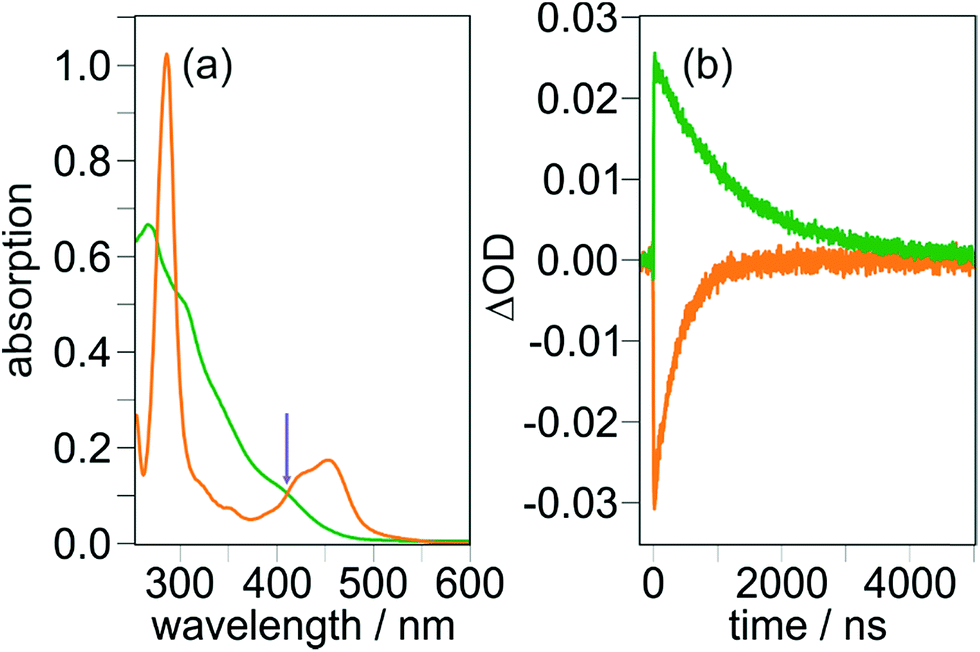 | ||
| Fig. 3 Raw data used for the quantum yield determination of CSS formation for TAA-ph1-Ir-ph1-AQ upon 410 nm laser excitation with 8 mJ laser pulses of about 10 ns duration. (a) UV-Vis absorption spectra of TAA-ph1-Ir-ph1-AQ (green, in CH3CN) and [Ru(bpy)3]2+ (orange, in water), which was used as reference compound for relative actinometry. (b) Transient absorption traces upon 410 nm excitation of the deoxygenated solutions shown in (a). CSS formation was monitored at 720 nm (green), [Ru(bpy)3]2+ ground state bleach at 455 nm (orange). For further explanation, see the main text and Section 1.3 of the ESI.† | ||
Third and finally, the transient absorption traces so obtained were converted into relative intermediate concentrations using the well-established (difference) molar absorption coefficients for the [Ru(bpy)3]2+ ground state bleach (together with the quantum yield for the [Ru(bpy)3]2+ excitation producing its triplet state, which is unity65) and the estimated extinction coefficients at the maximum of the respective CSS (see Section 1.3 of the ESI† for details). Surprisingly, the CSS quantum yields for the two triads differ by almost a factor of 3 under very similar conditions (Fig. 4). Control experiments with 410 nm excitation at significantly reduced laser intensity (to about 50%) gave essentially the same quantum yields for both triads compared to the results at higher intensities (8 mJ, see Fig. 3). We regard these observations as clear evidence for the absence of biphotonic side reactions, such as a two-photon ionization66 or light-induced charge recombination (compare, Section 2.4), and saturation effects.67 Moreover, all quantum yield determinations presented in this work were carried out with solutions having absorptions below 0.105 at the excitation wavelengths to avoid filter effects.68 A simplified procedure frequently used to estimate quantum yields is the analysis of the quenching efficiency of the photosensitizer as CSS precursor employing kinetic measurements.4,46 This easy-to-obtain quantity, which does not take chemically unproductive deactivation pathways into account, would predict quantitative CSS formation for both TAA-ph1-Ir-ph1-AQ and TAA-ph2-Ir-ph2-AQ (see Section 4.1 of the ESI†). Therefore, our results highlight the importance of direct quantum yield determinations with relative actinometry.
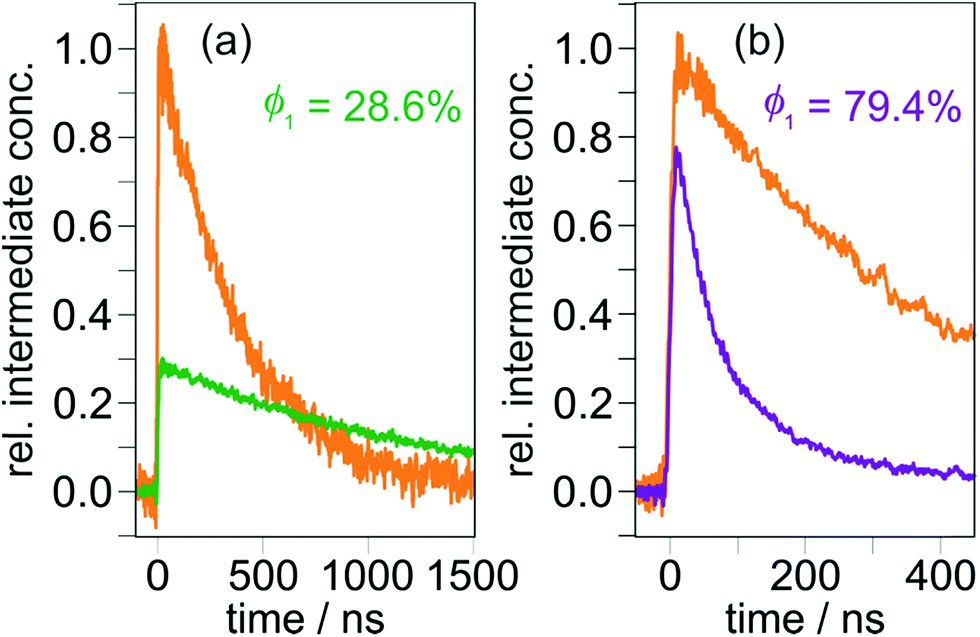 | ||
| Fig. 4 CSS formation quantum yields of TAA-ph1-Ir-ph1-AQ (a) and TAA-ph2-Ir-ph2-AQ (b) upon 410 nm laser excitation (8 mJ pulse energy). The CSS concentrations after the laser pulses were calculated relative to the amounts of absorbed photons as measured by means of relative actinometry with additional experiments on [Ru(bpy)3]2+ (orange reference traces) under identical excitation conditions. Further details are given in the main text and Section 1.3 of the ESI.† | ||
The widel y differing quantum yields obtained upon 410 nm excitation (28.6 vs. 79.4%, Fig. 4) prompted us to explore the influence of the excitation wavelength on CSS formation. Prior to carrying out additional kinetic measurements as in Fig. 4, we recorded transient absorption spectra using at least four different laser wavelengths for exciting our triads. Our experiments demonstrate that the spectral shapes and relative absorption signal intensities do not depend on the excitation wavelength (see Fig. S5 of the ESI†). Hence, in line with Kasha's rule,69,70 the respective CSS is the only intermediate observable on a nanosecond timescale. Furthermore, more than 500 laser flashes on the very same solution do neither change the CSS absorption spectra nor their intensities, indicating that no photochemical side reactions are taking place. All quantum yield determinations at the excitation wavelengths covering the experimentally accessible range (see Section 2.1) were carried out with the same methodology as at 410 nm (Fig. 3 and 4). To compensate small laser intensity fluctuations and obtain a suitable signal-to-noise ratio, at least 20 kinetic traces were averaged for every quantum yield measurement summarized in Table 1. The results presented in that table reveal a quasi-wavelength independence for the CSS formation quantum yield of TAA-ph2-Ir-ph2-AQ with a mean value as high as 82%, whereas for TAA-ph1-Ir-ph1-AQ, the CSS formation efficiencies differ by a factor of 5 over the explored excitation wavelength range, decreasing in a systematic manner at longer wavelengths.
| Excitation wavelength/nm | CSS formation quantum yieldb/% | |
|---|---|---|
| TAA-ph1-Ir-ph1-AQ | TAA-ph2-Ir-ph2-AQ | |
| a Measured in deoxygenated CH3CN at 293 K. The maximum relative error is expected to be less than 10%. b Details concerning the experimental methodology are given in the main text and in Section 1.2 of the ESI. c Cannot be determined owing to negligible absorption of the triad at the excitation wavelength. | ||
| 410 | 28.6 | 79.4 |
| 425 | 24.4 | 76.6 |
| 433 | 18.9 | 87.4 |
| 440 | 16.1 | 85.4 |
| 450 | 14.4 | 82.0 |
| 460 | 12.8 | —c |
| 470 | 6.0 | —c |
A plausible explanation for the markedly different behavior of the quantum yields in Table 1 is provided by different main pathways of CSS formation in the two triads. As discussed in Section 2.1, photoexcitation of TAA-ph1-Ir-ph1-AQ mainly occurs into an intraligand CT transition, whereas in the case of TAA-ph2-Ir-ph2-AQ, excitation into MLCT bands of the photosensitizer dominates when violet or blue photons are employed. Starting from these charge-transfer states, which are formed in their singlet states due to spin conservation,71 subsequent intersystem crossing (ISC) to the triplet manifold can occur. Ir-based excited 1MLCT states usually undergo very fast (<1 ps) and quantitative ISC,72,73 but intraligand CT states or CT states in purely organic systems are known for their ultrafast and unproductive (singlet) recombination in polar solvents (the recombination of 1TAA˙+-bpy˙−/IrIII-AQ in this case; see Fig. 1c for details).74–76 As a direct consequence, the quantum yields for the production of long-lived (triplet) CSSs via3MLCT states can approach values close to unity, while in the intermediate intraligand singlet CT states, ultrafast recombination competes with ISC and productive charge separation. In line with that reasoning, the CSS quantum yields of TAA-ph1-Ir-ph1-AQ increase with decreasing excitation wavelengths, i.e., when the admixture of direct MLCT excitation increases relative to intraligand CT excitation (see UV-Vis spectra in Fig. 1a).
2.4 Light-induced charge recombination
With the ultimate aim of achieving photoinduced charge accumulation in molecular systems without sacrificial additives, many sophisticated compounds have been prepared, but in the last decade only a handful of investigations succeeded in overcoming the associated difficulties.31,77–80 Starting from the first CSS, a key problem is the light-induced charge recombination upon excitation of intermediate electrons/holes with a second photon (see Scheme 1b), which is competitive with the desired accumulation of redox equivalents.29,30 Given that CSSs typically exhibit intense absorptions over a broad wavelength range, selective photosensitizer excitation is even more difficult under conditions of solar (broadband) irradiation than it is under well-defined lab (monochromatic) conditions. However, this is currently a heavily underexplored area given its importance for accumulative multi-electron transfer.29,33,35,78,81 The molecular triads investigated in this study do not permit charge accumulation, but they are well-suited for studying light-induced charge recombination processes and for quantum yield determinations of this undesired side reaction, which lay the grounds for structural optimizations to avoid that detrimental process.Two-pulse two-color laser flash photolysis is the ideal experimental technique to investigate consecutive photoreactions, both for purely intramolecular reactions34,60,82–84 and reaction sequences with intervening bimolecular processes.81,85–87 This holds also true for the mechanism shown in Scheme 1b: the first (violet or blue) pulse serves to produce the desired CSS, and the ensuing second (green) pulse88 investigates the fate of this intermediate upon absorption of another photon. In line with the absorption spectra displayed in Fig. 1a, the latter pulse (532 nm) alone has no effect on the ground states of the triads, which was verified by control experiments. Hence, that pulse selectively excites the CSSs in our systems with AQ˙− being the main absorbing species at 532 nm (see Fig. 1d and page S4 of the ESI†). The two-pulse experiments on TAA-ph1-Ir-ph1-AQ are summarized in Fig. 5. Initial experiments with an interpulse delay of 240 ns (the pulse sequence is shown at the top of Fig. 5b) and monitoring at the maximum of the CSS, 720 nm, revealed a green-light induced CSS bleach of about 24%, as is evidenced in the main plot of Fig. 5a. The rather long interpulse delay allows sensitive monitoring of the CSS absorption spectrum between the two pulses (200 ns integration time, green spectrum in Fig. 5b), and its comparison with the spectrum observed after the green pulse (gray spectrum in Fig. 5b). Comparing the spectra presented in Fig. 5b, we found that all (positive) absorption peaks in the gray spectrum are less intense by about 39%, and even the ground state bleach at 320 nm recovers accordingly; in other words, the normalized spectra are completely identical (normalization not shown). A correction of the ∼39% signal decrease observed in Fig. 5b for the thermal CSS recombination, which also takes place during spectral detection, gave essentially the same result for the light-induced recombination (23%) as extracted from kinetic traces (24%, see above). Moreover, the kinetics of thermal charge recombination are unaffected by the second pulse (Fig. S6 of the ESI†). Hence, all these two-pulse experiments on TAA-ph1-Ir-ph1-AQ unambiguously establish that charge recombination is the only green-light-induced process observable on the nanosecond timescale.
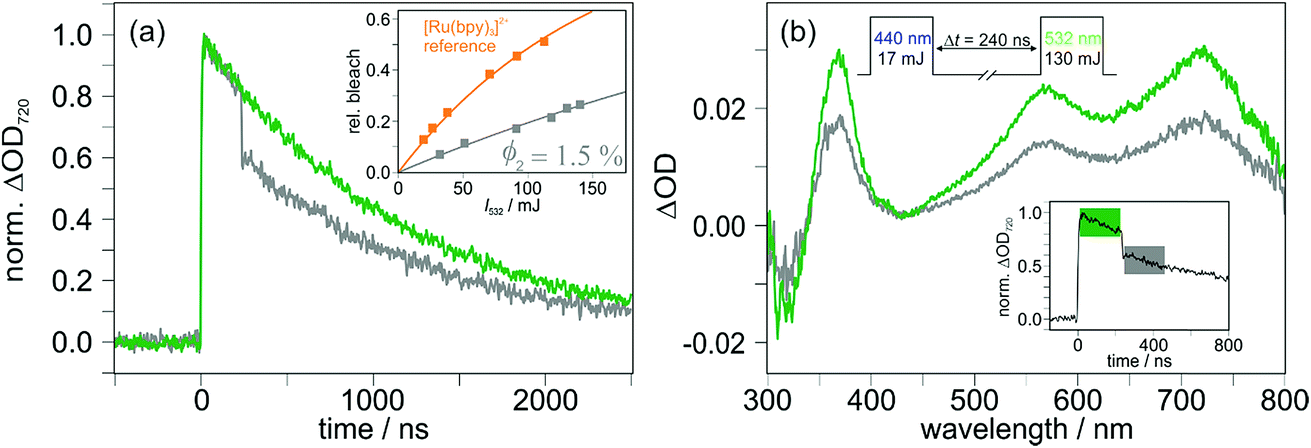 | ||
| Fig. 5 Observing and quantifying the light-induced charge recombination in TAA-ph1-Ir-ph1-AQ (20 μM in deoxygenated CH3CN) using two-pulse experiments with the general pulse sequence displayed at the top of panel (b). (a) Main plot: kinetic traces for the CSS of the triad at 720 nm with the second laser pulse blocked (green) or unblocked (gray). Inset: relative CSS bleaching at different intensities of the second laser (gray), together with the intensity dependence of the green-light induced [Ru(bpy)3]2+ bleach (orange) used as reference for the charge recombination quantum yield (ϕ2) determination. The fit parameters a for the saturation curves (1 − exp[−I532/a]) are 150 mJ (orange) and 463 mJ (gray). (b) Main plot: transient absorption spectrum of TAA-ph1-Ir-ph1-AQ before (green) and after (gray) the second laser pulse in a representative two-pulse experiment. The inset shows a kinetic transient absorption measurement under identical conditions (the detection windows used for recording the transient absorption spectra of the main plot have been highlighted). For details, see text. | ||
Given the sensitive and isolated detection of light-induced charge recombination, its quantum yield determination became a realistic target. Starting from the experimental conditions presented in Fig. 5a, we analyzed the CSS bleach at six different intensities of the second laser while keeping all other parameters unchanged. The intensity dependent relative recombination yields were extracted by independent fitting of pre- and post-pulse signals and extrapolation to the time of the second laser pulse, i.e., t = 240 ns (inset of Fig. 5a, gray data points). For a reliable quantum yield determination, we applied the same method as in a recent investigation on a second-pulse induced photoreaction.60 Briefly, we again employed [Ru(bpy)3]2+ in water as reference system (compare, Section 2.3) and quantified the relative green-light induced ground state bleach upon excitation with laser pulses spanning a similar intensity range (inset of Fig. 5a, orange data points). To minimize experimental uncertainties associated with single-point actinometry,61 we fitted both intensity dependencies with a saturation function that is valid for monophotonic photoreactions.60,89 By analyzing the fit parameters with the extinction coefficients (at 532 nm) of the respective absorbing species (CSS of TAA-ph1-Ir-ph1-AQ, 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 M−1 cm−1, see page S4 of the ESI† for details; [Ru(bpy)3]2+, 720 M−1 cm−1),62 a quantum yield for the light-induced charge recombination (ϕ2) of 1.5% was finally obtained.
500 M−1 cm−1, see page S4 of the ESI† for details; [Ru(bpy)3]2+, 720 M−1 cm−1),62 a quantum yield for the light-induced charge recombination (ϕ2) of 1.5% was finally obtained.
Similar two-pulse experiments were carried out with TAA-ph2-Ir-ph2-AQ (Section 4.2 of the ESI†). Owing to the shorter CSS lifetime of that triad (Section 2.2), single-wavelength transient absorption kinetic traces are much more sensitive than spectral measurements with (short) detection time windows. However, during kinetic traces recorded at both the AQ˙− and the TAA˙+ absorption maximum with a 45 ns interpulse delay, we did not observe any noticeable second-pulse induced absorption change, although at 532 nm the CSS of TAA-ph2-Ir-ph2-AQ absorbs slightly more strongly than that of TAA-ph1-Ir-ph1-AQ (see ESI page S4†). Taking into account the reduced detection sensitivity for these measurements on the short-lived CSS in this triad, we estimate an upper limit of 0.2% for the green-light induced charge recombination of this compound.
Given that the green pulse is mainly absorbed by the anthraquinone radical anion part of the CSS (page S4 of the ESI†), we assume that the following mechanism explains the widely differing quantum yields for light-induced charge recombination in our triads (1.5 vs. 0.2%). Excitation of AQ˙− at 532 nm (2.33 eV) produces – after ultrafast internal conversion – the lowest excited doublet state *AQ˙−, whose energy lies ∼1.15 eV above the electronic ground state of AQ˙− (estimated from its long wavelength absorption band).49 Isolated *AQ˙− is known to rapidly deactivate back to AQ˙−: averaging over the available literature values gave a lifetime as short as ∼40 ps,49,90,91 and we expect a similar natural lifetime of *AQ˙− in our triads. Based on structural and energetic considerations, we regard the electron transfer to the bipyridine (bpy) moiety as the most-likely reaction pathway competing with photophysical deactivation. Once the bipyridine radical anion is formed, recombination with the adjacent TAA˙+ could account for the observed ground-state regeneration through light-induced charge recombination (see also Fig. 1c).
With a driving force of about 0.85 eV for the reaction between *AQ˙− and bpy (see energies of the pertinent states in Fig. 1c), a conventional distance dependence of the electron transfer rates is expected, i.e., a rate constant decrease with increasing distances.9,92,93 With the above-mentioned quantum yields of light-induced charge recombination and a rate of ∼1/40 ps for photophysical deactivation, reasonable rate constants for the intermolecular electron transfer between *AQ˙− and bpy on the order of 4 × 108 s−1 (TAA-ph1-Ir-ph1-AQ) and <5 × 107 s−1 (TAA-ph2-Ir-ph2-AQ) can be estimated, if we simply assume that this intramolecular reaction is rate-limiting. The reasoning of the preceding paragraph could thus explain that green-light-induced charge recombination of TAA-ph1-Ir-ph1-AQ is more efficient than that of TAA-ph2-Ir-ph2-AQ by about one order of magnitude. Moreover, it is worth emphasizing that a quantum yield of 1.5% for an undesired pathway starting from a CSS is not negligible, since it could clearly outperform the quantum yield of desired charge accumulation (e.g., 0.5% in ref. 77). Although our triads are well-suited test systems to understand (biphotonic) light-induced recombination processes, we consider the probability of a (second) photon absorption by the CSSs of our triads with (sub)microsecond lifetimes as very unlikely under sunlight conditions. This is borne out by previous publications on intensity-dependent photoreactions with intermediates possessing nanosecond94 to microsecond60 lifetimes, whose efficient biphotonic ionization was only feasible with pulsed lasers or collimated laser diodes providing light power densities >1 kW cm−2.
The experiments of this section illustrate the wealth of information that is accessible by quantitative two-pulse experiments on molecular triads, and that electron transfer rate distance dependencies could also help understand light-induced recombination processes.
3 Conclusions
The results of our extensive photophysical investigations on two structurally closely related molecular triads clearly demonstrate the difficulty of optimizing all desirable properties for solar energy storage in a single molecular system at once (all pertinent results are summarized in Table 2). After excitation of an Ir-based sensitizer with a single visible photon, both triads are able to form an unusually highly energetic CSS (∼2.0 eV) comprising a triarylamine radical cation and an anthraquinone radical anion. Under optimized excitation conditions, up to 75% of the initial photon energy can be stored in the final CSS. The triad with the short bridge stores the energy 14 times longer than the counterpart with longer intramolecular distances.16 However, quantitative two-pulse laser experiments revealed that the CSS of the shorter triad (TAA-ph1-Ir-ph1-AQ) undergoes light-induced charge recombination about ten times more efficiently than that of the longer triad (TAA-ph2-Ir-ph2-AQ). Thus, the intriguing key finding is that thermal charge recombination is substantially slower in the shorter triad, but at the same time light-induced charge recombination is significantly more efficient in that compound. These observations can be understood in the framework of Marcus theory as pointed out in Sections 2.2 and 2.4. The CSS formation quantum yield of the shorter triad crucially depends on the laser excitation wavelength (anti-Vavilov behavior)70 and ranges from 6.0 to 28.9%, whereas CSS formation for the longer triad is largely wavelength-independent with a quantum yield as high as 82% under comparable conditions. To our knowledge, these observations have no precedent in the literature on charge-separation in donor-sensitizer-acceptor compounds. The wavelength-dependence and the low quantum yields for CSS formation in the short triad are most likely due to direct excitation into an additional intraligand CT state of singlet parentage (substantiated by DFT calculations), which undergoes ultrafast charge recombination competing with CSS formation. By contrast, the longer triad is predominantly excited into a 1MLCT excited state, and following rapid intersystem crossing to the 3MLCT state of the Ir sensitizer, productive charge separation outcompetes undesired charge recombination events. This shows that triplet excited states can lead to substantially higher quantum yield for charge separation than singlet excited states.| Triad | E CSS /eV | τ CSS /ns | ϕ 1 /% | ϕ 2 /% | η /% |
|---|---|---|---|---|---|
| a Energy of the respective CSS. b CSS lifetime in deoxygenated CH3CN at 293 K. c Quantum yield of CSS formation. d Quantum yield of light-induced charge recombination upon CSS excitation at 532 nm. e Upper limit for the efficiency of photon-to-chemical energy conversion. f Wavelength-dependent, see Table 1 for details. | |||||
| TAA-ph1-Ir-ph1-AQ | 1.99 | 1210 ± 20 | 6.0–28.9f | 1.5 | 75 |
| TAA-ph2-Ir-ph2-AQ | 1.96 | 85 ± 5 | 82.2 ± 4.4 | <0.2 | 71 |
As has emerged from this work, wavelength-dependent one- and two-pulse laser flash photolysis with quantitative detection of reaction intermediates is a powerful tool to investigate unexpected or even unexplored effects that are highly relevant for artificial photosynthesis. In particular, quantitative pump-probe and pump–pump–probe spectroscopy combined with electron transfer theory and simple DFT calculations provides a clear picture of important CSS properties. We anticipate that our approach might facilitate both the further optimization of CSS precursors and multi-electron storage in molecular systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Swiss National Science Foundation (grant number 200021_178760), from the Swiss Nanoscience Institute (SNI Project P1406) and from the German National Academy of Sciences Leopoldina (postdoctoral fellowship LPDS 2017-11). C. K. is grateful to the Research Fund of the University of Basel for a Novartis University of Basel Excellence Scholarship for Life Sciences.References
- Molecular Mechanisms of Photosynthesis, ed. R. E. Blankenship, Blackwell Science Ltd, Oxford, UK, 2002 Search PubMed.
- M. R. Wasielewski, M. P. Niemczyk, W. A. Svec and E. B. Pewitt, J. Am. Chem. Soc., 1985, 107, 1080–1082 CrossRef CAS.
- Y. Luo, M. Wächtler, K. Barthelmes, A. Winter, U. S. Schubert and B. Dietzek, Chem. Commun., 2018, 54, 2970–2973 RSC.
- G. N. Lim, C. O. Obondi and F. D'Souza, Angew. Chem., Int. Ed., 2016, 55, 11517–11521 CrossRef CAS PubMed.
- N. Zarrabi, C. Agatemor, G. N. Lim, A. J. Matula, B. J. Bayard, V. S. Batista, F. D'Souza and P. K. Poddutoori, J. Phys. Chem. C, 2019, 123, 131–143 CrossRef CAS.
- N. Zarrabi, C. O. Obondi, G. N. Lim, S. Seetharaman, B. G. Boe, F. D'Souza and P. K. Poddutoori, Nanoscale, 2018, 10, 20723–20739 RSC.
- R. M. Williams, M. Koeberg, J. M. Lawson, Y.-Z. An, Y. Rubin, M. N. Paddon-Row and J. W. Verhoeven, J. Org. Chem., 1996, 61, 5055–5062 CrossRef CAS.
- C. Luo, D. M. Guldi, H. Imahori, K. Tamaki and Y. Sakata, J. Am. Chem. Soc., 2000, 122, 6535–6551 CrossRef CAS.
- J. Wiberg, L. Guo, K. Pettersson, D. Nilsson, T. Ljungdahl, J. Mårtensson and B. Albinsson, J. Am. Chem. Soc., 2007, 129, 155–163 CrossRef CAS PubMed.
- H. Imahori, K. Tamaki, D. M. Guldi, C. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607–2617 CrossRef CAS PubMed.
- M. Kuss-Petermann and O. S. Wenger, J. Am. Chem. Soc., 2016, 138, 1349–1358 CrossRef CAS PubMed.
- H. Imahori, Y. Sekiguchi, Y. Kashiwagi, T. Sato, Y. Araki, O. Ito, H. Yamada and S. Fukuzumi, Chem.–Eur. J., 2004, 10, 3184–3196 CrossRef CAS PubMed.
- M. Borgström, N. Shaikh, O. Johansson, M. F. Anderlund, S. Styring, B. Åkermark, A. Magnuson and L. Hammarström, J. Am. Chem. Soc., 2005, 127, 17504–17515 CrossRef PubMed.
- J. Schäfer, M. Holzapfel, A. Schmiedel, U. E. Steiner and C. Lambert, Phys. Chem. Chem. Phys., 2018, 20, 27093–27104 RSC.
- L. Sun, L. Hammarström, B. Åkermark and S. Styring, Chem. Soc. Rev., 2001, 30, 36–49 RSC.
- S. Neumann and O. S. Wenger, Inorg. Chem., 2019, 58, 855–860 CrossRef CAS PubMed.
- M. M. Waskasi, G. Kodis, A. L. Moore, T. A. Moore, D. Gust and D. V. Matyushov, J. Am. Chem. Soc., 2016, 138, 9251–9257 CrossRef CAS PubMed.
- J. Sukegawa, C. Schubert, X. Zhu, H. Tsuji, D. M. Guldi and E. Nakamura, Nat. Chem., 2014, 6, 899–905 CrossRef CAS PubMed.
- K. Hu, A. D. Blair, E. J. Piechota, P. A. Schauer, R. N. Sampaio, F. G. L. Parlane, G. J. Meyer and C. P. Berlinguette, Nat. Chem., 2016, 8, 853–859 CrossRef CAS PubMed.
- M. Delor, T. Keane, P. A. Scattergood, I. V. Sazanovich, G. M. Greetham, M. Towrie, A. J. H. M. Meijer and J. A. Weinstein, Nat. Chem., 2015, 7, 689–695 CrossRef CAS PubMed.
- M. Natali, S. Campagna and F. Scandola, Chem. Soc. Rev., 2014, 43, 4005–4018 RSC.
- M. Krzeszewski, E. M. Espinoza, C. Červinka, J. B. Derr, J. A. Clark, D. Borchardt, G. J. O. Beran, D. T. Gryko and V. I. Vullev, Angew. Chem., Int. Ed., 2018, 57, 12365–12369 CrossRef CAS PubMed.
- D. S. Tyson, C. R. Luman and F. N. Castellano, Inorg. Chem., 2002, 41, 3578–3586 CrossRef CAS PubMed.
- M. S. Eberhart, L. M. R. Bowers, B. Shan, L. Troian-Gautier, M. K. Brennaman, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2018, 140, 9823–9826 CrossRef CAS PubMed.
- J.-J. Shen, J.-Y. Shao, Z.-L. Gong and Y.-W. Zhong, Inorg. Chem., 2015, 54, 10776–10784 CrossRef CAS PubMed.
- J. Zhou, Y. Wu, I. Roy, A. Samanta, J. F. Stoddart, R. M. Young and M. R. Wasielewski, Chem. Sci., 2019, 10, 4282–4292 RSC.
- E. Sundin and M. Abrahamsson, Chem. Commun., 2018, 54, 5289–5298 RSC.
- L. Favereau, A. Makhal, Y. Pellegrin, E. Blart, J. Petersson, E. Göransson, L. Hammarström and F. Odobel, J. Am. Chem. Soc., 2016, 138, 3752–3760 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, Coord. Chem. Rev., 2011, 255, 2578–2593 CrossRef CAS.
- L. Hammarström, Acc. Chem. Res., 2015, 48, 840–850 CrossRef PubMed.
- M. Orazietti, M. Kuss-Petermann, P. Hamm and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 9407–9410 CrossRef CAS PubMed.
- T. Higashino, T. Yamada, M. Yamamoto, A. Furube, N. V. Tkachenko, T. Miura, Y. Kobori, R. Jono, K. Yamashita and H. Imahori, Angew. Chem., Int. Ed., 2016, 55, 629–633 CrossRef CAS PubMed.
- M. Kuss-Petermann and O. S. Wenger, Helv. Chim. Acta, 2017, 100, e1600283 CrossRef.
- M.-H. Ha-Thi, V.-T. Pham, T. Pino, V. Maslova, A. Quaranta, C. Lefumeux, W. Leibl and A. Aukauloo, Photochem. Photobiol. Sci., 2018, 17, 903–909 RSC.
- A. Pannwitz and O. S. Wenger, Chem. Commun., 2019, 55, 4004–4014 RSC.
- M. Lauck, C. Förster, D. Gehrig and K. Heinze, J. Organomet. Chem., 2017, 847, 33–40 CrossRef CAS.
- R. Konduri, H. Ye, F. M. MacDonnell, S. Serroni, S. Campagna and K. Rajeshwar, Angew. Chem., Int. Ed., 2002, 41, 3185–3187 CrossRef CAS PubMed.
- B. Matt, J. Fize, J. Moussa, H. Amouri, A. Pereira, V. Artero, G. Izzet and A. Proust, Energy Environ. Sci., 2013, 6, 1504–1508 RSC.
- G. Knör, A. Vogler, S. Roffia, F. Paolucci and V. Balzani, Chem. Commun., 1996, 1643–1644 RSC.
- J.-F. Lefebvre, J. Schindler, P. Traber, Y. Zhang, S. Kupfer, S. Gräfe, I. Baussanne, M. Demeunynck, J.-M. Mouesca, S. Gambarelli, V. Artero, B. Dietzek and M. Chavarot-Kerlidou, Chem. Sci., 2018, 9, 4152–4159 RSC.
- D. Polyansky, D. Cabelli, J. T. Muckerman, E. Fujita, T. Koizumi, T. Fukushima, T. Wada and K. Tanaka, Angew. Chem., Int. Ed., 2007, 46, 4169–4172 CrossRef CAS PubMed.
- C. N. Valdez, A. M. Schimpf, D. R. Gamelin and J. M. Mayer, J. Am. Chem. Soc., 2016, 138, 1377–1385 CrossRef CAS PubMed.
- G. F. Manbeck and K. J. Brewer, Coord. Chem. Rev., 2013, 257, 1660–1675 CrossRef CAS.
- M. G. Pfeffer, B. Schäfer, G. Smolentsev, J. Uhlig, E. Nazarenko, J. Guthmuller, C. Kuhnt, M. Wächtler, B. Dietzek, V. Sundström and S. Rau, Angew. Chem., Int. Ed., 2015, 54, 5044–5048 CrossRef CAS PubMed.
- M. Kuss-Petermann and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 815–819 CrossRef CAS PubMed.
- J. Hankache, M. Niemi, H. Lemmetyinen and O. S. Wenger, Inorg. Chem., 2012, 51, 6333–6344 CrossRef CAS PubMed.
- J. Hankache, D. Hanss and O. S. Wenger, J. Phys. Chem. A, 2012, 116, 3347–3358 CrossRef CAS PubMed.
- B. Geiß and C. Lambert, Chem. Commun., 2009, 1670–1672 RSC.
- P. Brodard, A. Sarbach, J.-C. Gumy, T. Bally and E. Vauthey, J. Phys. Chem. A, 2001, 105, 6594–6601 CrossRef CAS.
- B. S. Brunschwig, S. Ehrenson and N. Sutin, J. Am. Chem. Soc., 1984, 106, 6858–6859 CrossRef CAS.
- M. Tachiya and S. Murata, J. Phys. Chem., 1992, 96, 8441–8444 CrossRef CAS.
- M. Kuss-Petermann and O. S. Wenger, Phys. Chem. Chem. Phys., 2016, 18, 18657–18664 RSC.
- R. N. Sampaio, L. Troian-Gautier and G. J. Meyer, Angew. Chem., Int. Ed., 2018, 57, 15390–15394 CrossRef CAS PubMed.
- H. J. Kuhn, S. E. Braslavsky and R. Schmidt, Pure Appl. Chem., 2004, 76, 2105–2146 CAS.
- R. Bensasson, C. R. Goldschmidt, E. J. Land and T. G. Truscott, Photochem. Photobiol., 1978, 28, 277–281 CrossRef CAS.
- I. Carmichael and G. L. Hug, J. Phys. Chem. Ref. Data, 1986, 15, 1–250 CrossRef.
- J. K. Hurley, H. Linschitz and A. Treinin, J. Phys. Chem., 1988, 92, 5151–5159 CrossRef CAS.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- C. Kerzig, X. Guo and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 2122–2127 CrossRef CAS.
- M. Brautzsch, C. Kerzig and M. Goez, Green Chem., 2016, 18, 4761–4771 RSC.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2018, 9, 6670–6678 RSC.
- M. Goez, C. Kerzig and R. Naumann, Angew. Chem., Int. Ed., 2014, 53, 9914–9916 CrossRef CAS PubMed.
- P. Müller and K. Brettel, Photochem. Photobiol. Sci., 2012, 11, 632–636 RSC.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117–214 CrossRef CAS.
- T. Kohlmann, R. Naumann, C. Kerzig and M. Goez, Photochem. Photobiol. Sci., 2017, 16, 185–192 RSC.
- C. Kerzig and M. Goez, Phys. Chem. Chem. Phys., 2014, 16, 25342–25349 RSC.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of photochemistry, CRC/Taylor & Francis, Boca Raton, 3rd edn, 2006 Search PubMed.
- A. P. Demchenko, J. Heldt, J. Waluk, P.-T. Chou, P. K. Sengupta, L. Brizhik and J. C. del Valle, Angew. Chem., Int. Ed., 2014, 53, 14316–14324 CrossRef CAS PubMed.
- A. P. Demchenko, V. I. Tomin and P.-T. Chou, Chem. Rev., 2017, 117, 13353–13381 CrossRef CAS PubMed.
- G. J. Kavarnos and N. J. Turro, Chem. Rev., 1986, 86, 401–449 CrossRef CAS.
- K.-C. Tang, K. L. Liu and I.-C. Chen, Chem. Phys. Lett., 2004, 386, 437–441 CrossRef CAS.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS.
- P. Hazra, D. Chakrabarty and N. Sarkar, Langmuir, 2002, 18, 7872–7879 CrossRef CAS.
- C. B. Larsen and O. S. Wenger, Angew. Chem., Int. Ed., 2018, 57, 841–845 CrossRef CAS PubMed.
- N. Banerji, G. Angulo, I. Barabanov and E. Vauthey, J. Phys. Chem. A, 2008, 112, 9665–9674 CrossRef CAS PubMed.
- J. Nomrowski and O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 5343–5346 CrossRef CAS PubMed.
- S. Karlsson, J. Boixel, Y. Pellegrin, E. Blart, H.-C. Becker, F. Odobel and L. Hammarström, J. Am. Chem. Soc., 2010, 132, 17977–17979 CrossRef CAS PubMed.
- S. Karlsson, J. Boixel, Y. Pellegrin, E. Blart, H.-C. Becker, F. Odobel and L. Hammarström, Faraday Discuss., 2012, 155, 233–252 RSC.
- S. Mendes Marinho, M.-H. Ha-Thi, V.-T. Pham, A. Quaranta, T. Pino, C. Lefumeux, T. Chamaillé, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2017, 56, 15936–15940 CrossRef CAS PubMed.
- T.-T. Tran, M.-H. Ha-Thi, T. Pino, A. Quaranta, C. Lefumeux, W. Leibl and A. Aukauloo, J. Phys. Chem. Lett., 2018, 9, 1086–1091 CrossRef CAS.
- Y. Oseki, M. Fujitsuka, M. Sakamoto and T. Majima, J. Phys. Chem. A, 2007, 111, 9781–9788 CrossRef CAS PubMed.
- M. P. Debreczeny, W. A. Svec, E. M. Marsh and M. R. Wasielewski, J. Am. Chem. Soc., 1996, 118, 8174–8175 CrossRef CAS.
- C. Bohne, M. G. Fan, Z. J. Li, Y. C. Liang, J. Lusztyk and J. C. Scaiano, J. Photochem. Photobiol. Chem., 1992, 66, 79–90 CrossRef CAS.
- M. Goez and C. Kerzig, Angew. Chem., Int. Ed., 2012, 51, 12606–12608 CrossRef CAS PubMed.
- R. W. Redmond, J. C. Scaiano and L. J. Johnston, J. Am. Chem. Soc., 1990, 112, 398–402 CrossRef CAS.
- M. Sakamoto, X. Cai, S. S. Kim, M. Fujitsuka and T. Majima, J. Phys. Chem. A, 2007, 111, 223–229 CrossRef CAS PubMed.
- For experimental reasons, the only wavelength used for the second laser pulse was 532 nm, because only one of our two Nd:YAG lasers is equipped with an OPO allowing the production of violet or blue photons that are indispensable for the initial CSS production pulse.
- C. Kerzig and M. Goez, Chem. Sci., 2016, 7, 3862–3868 RSC.
- J.-C. Gumy and E. Vauthey, J. Phys. Chem. A, 1997, 101, 8575–8580 CrossRef CAS.
- T. Kumpulainen, B. Lang, A. Rosspeintner and E. Vauthey, Chem. Rev., 2017, 117, 10826–10939 CrossRef CAS PubMed.
- P. P. Edwards, H. B. Gray, M. T. J. Lodge and R. J. P. Williams, Angew. Chem., Int. Ed., 2008, 47, 6758–6765 CrossRef CAS PubMed.
- M. Cordes and B. Giese, Chem. Soc. Rev., 2009, 38, 892–901 RSC.
- T. Kohlmann, R. Naumann, C. Kerzig and M. Goez, Photochem. Photobiol. Sci., 2017, 16, 1613–1622 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Comprehensive experimental details, further energetic considerations, quantum-mechanical computations and additional laser experiments. See DOI: 10.1039/c9sc01381d |
| This journal is © The Royal Society of Chemistry 2019 |