DOI:
10.1039/C9SC01386E
(Edge Article)
Chem. Sci., 2019,
10, 6254-6260
Mixed-carbene cyclometalated iridium complexes with saturated blue luminescence†
Received
21st March 2019
, Accepted 26th May 2019
First published on 27th May 2019
Abstract
Cyclometalated iridium complexes have emerged as top-performing emitters in organic light-emitting diodes (OLEDs) and other optoelectronic devices. A persistent challenge has been the development of cyclometalated iridium complexes with deep blue luminescence that have the requisite color purity, efficiency, and stability to function in color displays. In this work we report a new class of cyclometalated iridium complexes with saturated blue luminescence. These complexes have the general structure Ir(C^C:NHC)2(C^C:ADC), where C^C:NHC is an N-heterocyclic carbene (NHC) derived cyclometalating ligand and C^C:ADC is a different type of cyclometalating ligand featuring an acyclic diaminocarbene (ADC). The complexes are prepared by a cascade reaction that involves nucleophilic addition of propylamine to an isocyanide precursor followed by base-assisted cyclometalation of the ADC intermediate. All three emit deep blue light with good quantum efficiencies (ΦPL = 0.13–0.48) and color profiles very close to the ideal primary blue standards for color displays.
Introduction
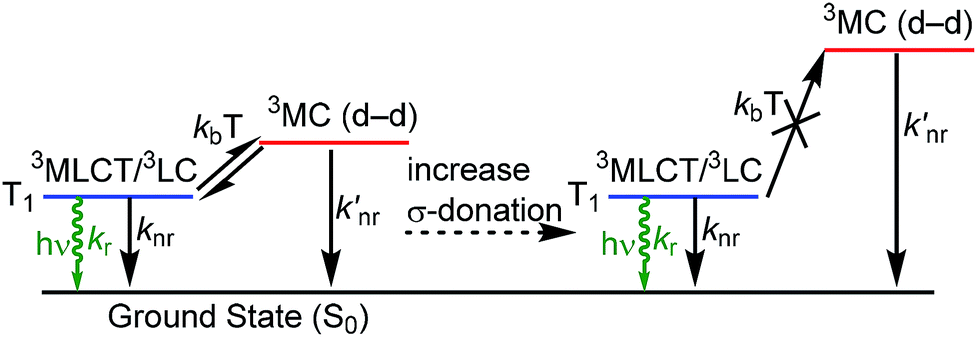
Organometallic compounds which luminesce from triplet excited states, i.e. via phosphorescence, have become prominent in a number of applications. Cyclometalated iridium complexes1 are a class of molecular phosphors that have achieved commercial success in organic light-emitting diodes (OLEDs)1–5 and have also been demonstrated to be effective in other contexts such as sensing6,7 and photocatalysis.8,9 Photoluminescence attributes of cyclometalated iridium complexes include high quantum yields, short lifetimes, and facile color tunability. A significant fundamental challenge in the design of luminescent iridium complexes, which is especially important in OLED research, is the lack of deep blue phosphorescent compounds with adequate performance metrics – i.e. color purity, quantum efficiency, and stability – to function in color displays or other optoelectronic applications. The physical origins of this challenge are summarized in Fig. 1. The HOMO in cyclometalated iridium complexes usually involves significant contribution from an Ir-centered dπ orbital, i.e. a t2g orbital in rigorously Oh complexes, whereas the LUMO is an unoccupied π* orbital on the cyclometalating ligands. These complexes luminesce from excited states that have contributions from triplet ligand-centered (3LC) states localized on the cyclometalating ligand and triplet metal-to-ligand charge transfer (3MLCT) states, which mix through configuration interaction in the low-energy T1 state.1 The lowest-energy ligand-field or metal-centered state, which involves transitions between Ir-centered d orbitals, is also a triplet state (3MC).10 One of the reasons bis-cyclometalated iridium complexes are very successful as triplet emitters is that the 3MC state is typically much higher in energy than the T1 state and thus cannot be populated. However, in deep-blue-emitting compounds with high-energy T1 states, the 3MC state is thermally accessible, and population of this nonradiative state decreases quantum yield and also promotes electrons into metal–ligand σ* orbitals, inducing ligand-loss decomposition pathways that limit photostability and lead to degradation of devices fabricated with these emitters.11
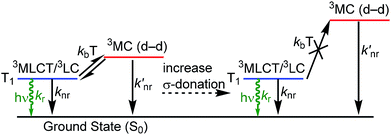 |
| | Fig. 1 Partial excited-state diagram for cyclometalated iridium complexes, showing partial occupation of 3MC states and destabilization of these states when stronger σ-donor ligands are incorporated. | |
As is also shown in Fig. 1, incorporation of strong σ-donor ligand sets can destabilize the deleterious 3MC state, allowing efficient luminescence from T1 and greater photostability. In an ideal case, where the σ-donor only perturbs the unoccupied dσ* orbitals and does not influence the frontier orbitals involved in the luminescent T1 state, this perturbation of the 3MC state would not influence the photoluminescence spectrum but should improve the quantum yield and photostability. As a result, N-heterocyclic carbenes (NHCs) have emerged as popular design elements in blue-emitting cyclometalated iridium complexes. Many recent works describe cyclometalated iridium complexes with NHC-containing C^C: cyclometalating ligands, chelated to iridium through the NHC and a phenyl group.12–21 In the context of blue OLEDs a majority of the recent breakthroughs involve homoleptic tris-cyclometalated Ir(C^C:NHC)3 complexes.12,14,15,17,19 The synthetic chemistry of heteroleptic compounds with only two C^C:NHC cyclometalating ligands is much less developed, although there are a couple of examples of blue-emitting bis-cyclometalated complexes of the type Ir(C^C:NHC)2(L^X), where L^X is a pyrazolate or triazolate-based ancillary ligand.16,18
Until recently NHCs were the only type of carbene ligand incorporated into blue-emitting iridium complexes. We recognized the possibility that acyclic diaminocarbenes (ADCs), which are known to be even stronger σ-donors than NHCs on account of the greater 2p character in their σ orbital,22–25 could potentially destabilize 3MC states to an even greater extent than is possible with NHCs and improve the photostability of blue-phosphorescent complexes. We have shown that ADCs can be installed onto cyclometalated iridium complexes via on-complex, nucleophilic addition to coordinated isocyanides. Versions of these complexes exhibit intense blue luminescence when immobilized in polymer films,26–28 with quantum efficiencies as high as 79% for a cyclometalated ADC complex.27 However, the biggest limitation of these first-generation compounds, which will preclude any practical device applications, is their rather poor color purity, with significant sky blue to blue-green coloration in the photoluminescence. We observed CIE coordinates29 (CIEx, CIEy) = (0.20, 0.41) for complexes with cyclometalated ADC ligands,27 with (CIEx, CIEy) = (0.16, 0.21) and (0.17, 0.28) for blue-emitting compounds with “Chugaev-type” bis(ADC) ancillary ligands.26,28 The CIEy coordinates in particular are larger than the ideal values (<0.1) for high-purity blue emission; the most widely used industry standards are the International Electrotechnical Commission (IEC) sRGB standard, (CIEx, CIEy) = (0.15, 0.06), and the primary blue standards proscribed by National Television System Committee (NTSC) (CIEx, CIEy) = (0.14, 0.08) and Society of Motion Picture and Television Engineers (SMPTE-C) (CIEx, CIEy) = (0.155, 0.07). In addition, the blue-emitting ADC-containing complexes we prepared all use the C^N cyclometalating ligand 2-(2,4-difluorophenyl)pyridine (F2ppy), which is known to degrade in OLED devices via ligand defluorination.11 In the present work, we address these two limitations and improve upon our previous designs, introducing three new compounds that are devoid of sp2 C–F bonds and give rise to high-purity blue photoluminescence. These mixed-carbene compounds have the general formula Ir(C^C:NHC)2(C^C:ADC), where C^C:NHC is an N-heterocyclic carbene (NHC) derived cyclometalating ligand and C^C:ADC an ADC-containing cyclometalating ligand. These compounds expand the synthetic chemistry of heteroleptic NHC-derived cyclometalated iridium complexes and show that ADC ligands and NHC ligands can be partnered on the same complex. These compounds exhibit very promising photoluminescence attributes, in particular CIE coordinates that are very close to NTSC and IEC standards for color displays, making these compounds excellent candidates for incorporation into OLEDs.
Results and discussion
Synthesis and characterization
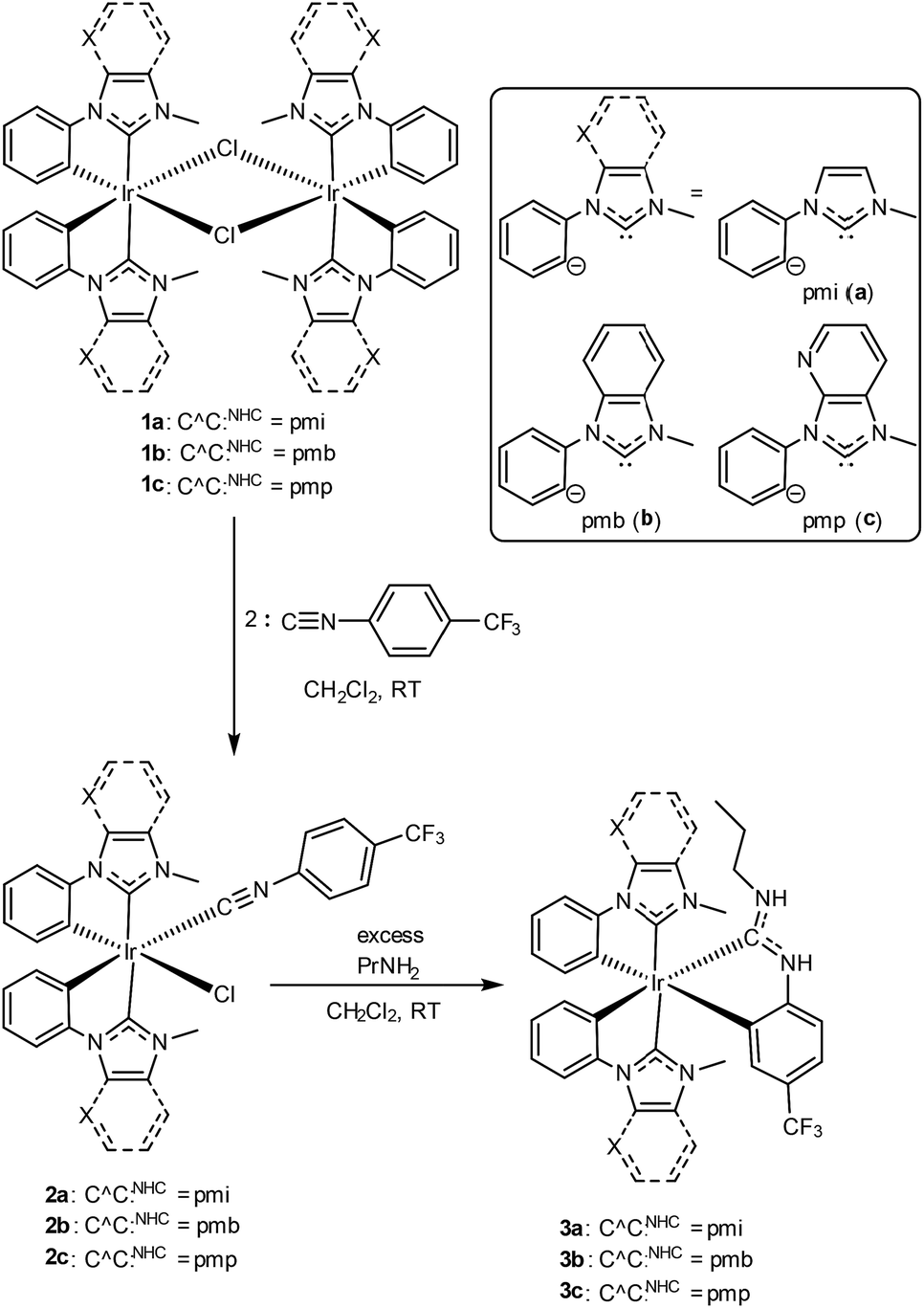
Scheme 1 outlines the synthesis of the compounds described in this work. Preparation of the mixed-carbene complexes begins with precursors of the type [Ir(C^C:NHC)2(μ-Cl)]2 (1a–c); two of these (1a and 1b) have been previously described.16 Treatment of dimers 1a–c with 4-trifluoromethylphenyl isocyanide forms mononuclear complexes of the type Ir(C^C:NHC)2(CN-p-C6H4CF3)(Cl) (2a–c), which then are treated with excess propylamine to form the final ADC-containing complexes 3a–c. Complexes 3a–c form via a cascade reaction involving metal-mediated nucleophilic addition of the amine to the coordinated isocyanide, followed by base-assisted cyclometalation to form the final neutral, tris-chelated heteroleptic products. This unconventional synthetic approach, which we introduced recently in a set of C^N-cyclometalated complexes,27 allows us to generate ADC-containing products that are not accessible using traditional ligand substitution or transmetalation approaches. The identity and purity of the final products were ascertained by 1H, 19F, and 13C{1H} NMR spectroscopy. Chemical inequivalency of the NHC's CH3 resonances (1H and 13C{1H}) and two sets of aromatic 1H signals for the two C^C:NHC ligands evince C1 symmetry, and characteristic resonances of the ADC, especially the propyl group and the two inequivalent N–H protons in the 1H spectrum and a downfield 13C{1H} resonance ∼200 ppm, are easily located in each case.
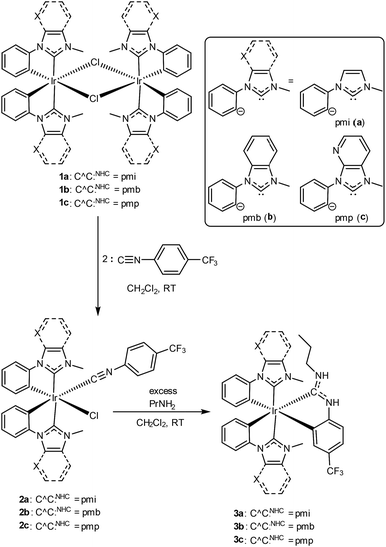 |
| | Scheme 1 Synthetic procedure to prepare blue-emitting complexes 3a–c. | |
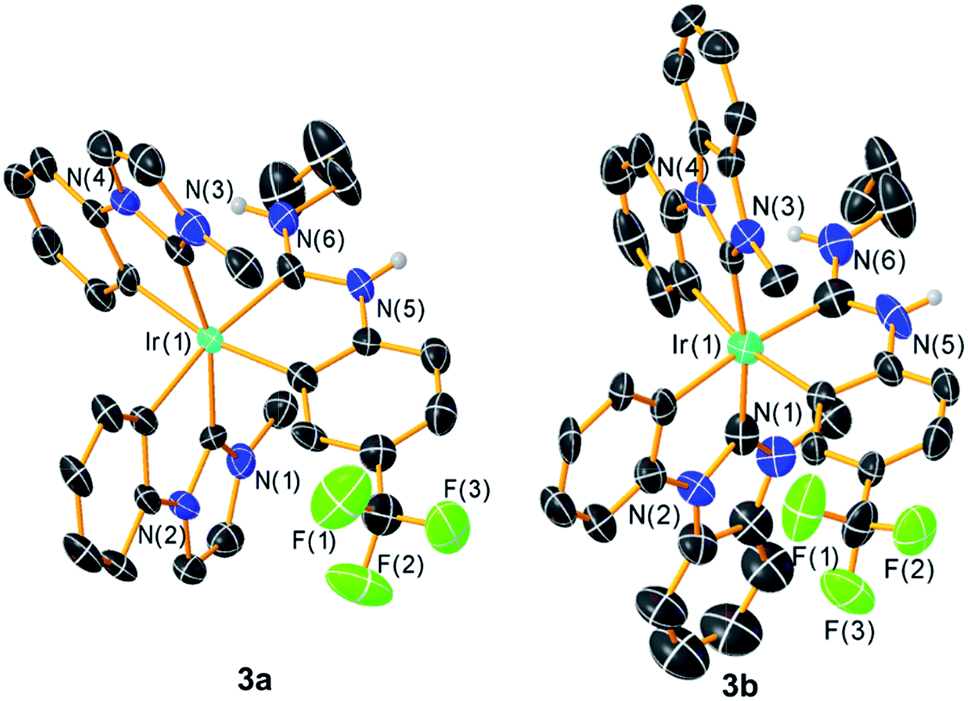
Further confirmation of the structures of 3a–c comes from single-crystal X-ray diffraction studies. Whereas the structure of 3c could be solved but refined poorly, structures of 3a‡ and 3b§ refined reasonably well and are included here in Fig. 2. X-ray crystallography confirms the pseudo-octahedral coordination geometry and trans arrangement of the NHC ligands and also verifies the chelated nature of the ADC ancillary ligand. Bond metrics of the ADC ligand, in particular the N–C bond distances and the carbene N–C–N bond angles, are very similar to the bond metrics observed in previously described complexes from our group with the same ancillary ligand partnered with pyridine- or thiazole-based C^N cyclometalating ligands.27 There are also not any substantial differences between Ir–C(NHC) and Ir–C(ADC) distances in these structures, with the latter distances falling in the range spanned by the Ir–C(NHC) distances.
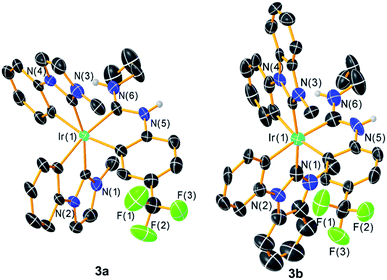 |
| | Fig. 2 Crystal structures of complexes 3a and 3b, with ellipsoids shown at the 50% probability level. Carbon atoms are shown in black and unlabeled, and carbon-bound hydrogen atoms are omitted for clarity. | |
Electrochemistry
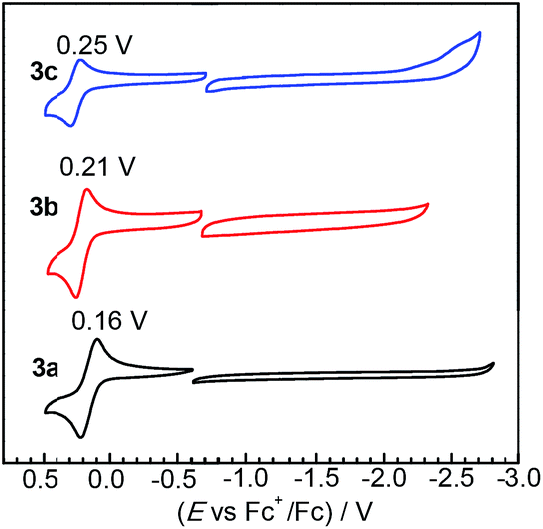
The redox properties of complexes 3a–c were investigated by cyclic voltammetry, which reveal that substituting NHC ligands with ADCs results in very small perturbations of frontier orbital energies. Fig. 3 overlays the cyclic voltammograms of 3a–c, recorded in MeCN with 0.1 M NBu4PF6 electrolyte. All three complexes exhibit reversible one-electron IrIV/IrIII couples upon sweeping anodically. Sweeping in the negative direction, complexes 3a and 3b do not show any discernible reduction waves within the solvent window, whereas 3c has an irreversible reduction at ca. −2.60 V vs. the Fc+/Fc couple. Compared to our previously reported complexes Ir(C^N)2(C^C:ADC) with nitrogen-containing cyclometalating ligands,27 complexes 3a–c are easier to oxidize by at least 160 mV, signifying a destabilized HOMO in these complexes. To contextualize these results further we can compare the potentials to homoleptic mer-Ir(C^C:NHC)3 complexes, which likewise place the two NHCs trans to one another but have another cyclometalated NHC occupying the remaining coordination sites. Replacing one NHC cyclometalating ligand with the ADC in 3a–c has a very small impact on the electrochemical potentials. Complex 3a is oxidized at 0.16 V, which is almost identical to the complex mer-Ir(pmi)3 (E1/2 = 0.14 V).13 Similarly, the oxidation potentials for the pairs 3b (E1/2 = 0.21 V) and mer-Ir(pmb)3 (E1/2 = 0.31 V)13 and 3c (E1/2 = 0.25 V) and mer-Ir(pmp)3 (E1/2 = 0.23 V)14 are within 100 mV of one another. The homoleptic mer-Ir(C^C:NHC)3 complexes and new complexes 3a–c are all very difficult to reduce, signifying very high-energy LUMOs that are responsible for the large HOMO–LUMO gaps in these C^C: cyclometalated compounds.
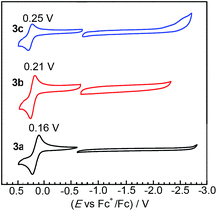 |
| | Fig. 3 Overlaid cyclic voltammograms of 3a–c, recorded in acetonitrile with 0.1 M NBu4PF6 electrolyte. A glassy carbon working electrode, platinum wire counter electrode, and silver wire pseudoreference electrode were used during the measurements, and potentials are referenced to an internal standard of ferrocene. Separate anodic (positive) and cathodic (negative) scans were recorded. Half-wave potentials (E1/2) are indicated above each oxidation wave, and currents are normalized to bring the plots onto the same scale. | |
Photophysical properties
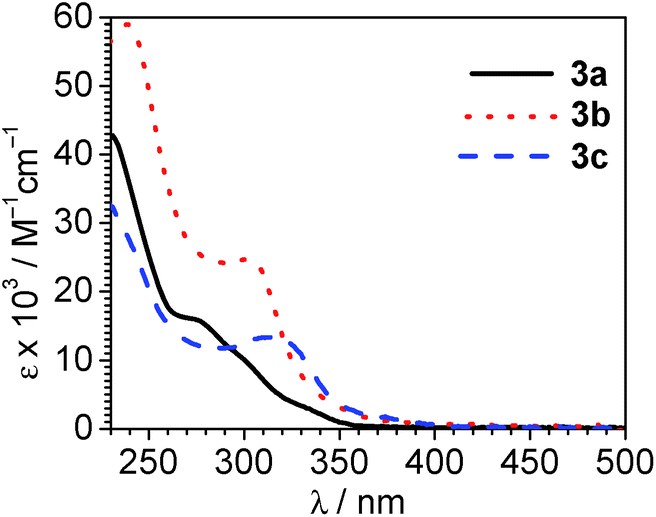
UV-vis absorption spectra, recorded at room temperature in CH2Cl2, are collected in Fig. 4. Mixed-carbene complexes 3a–3c are all colorless and only absorb in the UV region of the spectrum. All show intense absorption in the region of λ < 300 nm, consistent with ligand-centered π → π* transitions involving the NHC and/or ADC ligand. Consistent with the increased π conjugation of the benzo-fused pmb ligand, these UV absorption bands are more intense and less deep in the UV for complex 3b (C^C:NHC = pmb) compared to complex 3a (C^C:NHC = pmi). A weaker set of overlapping absorption bands is also observed, tailing to the low-energy reaches of the absorption profile. The overlapping bands include distinct maxima in 3b (λ = 302 nm) and 3c (λ = 315 nm), with multiple shoulders observed in each case. These absorption features are assigned to a combination of spin-allowed and spin-forbidden 1MLCT and 3MLCT transitions, as typically observed in cyclometalated iridium complexes. However, the destabilized LUMOs in these compounds, apparent from the cyclic voltammetry discussed above, result in these MLCT transitions occurring a higher energy than is typically observed in cyclometalated iridium complexes with C^N cyclometalating ligands.3,30 Cyclic voltammetry (Fig. 3) suggests that the LUMO in pyridyl-substituted complex 3c is lower energy than those of 3a and 3b, and in line with that observation we see that the MLCT bands in 3c occur at lower energy (longer wavelength) than the others.
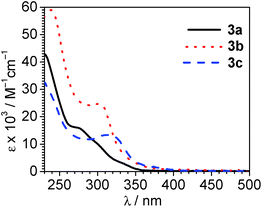 |
| | Fig. 4 Overlaid UV-vis absorption spectra of 3a–3c, recorded at room temperature in CH2Cl2. | |
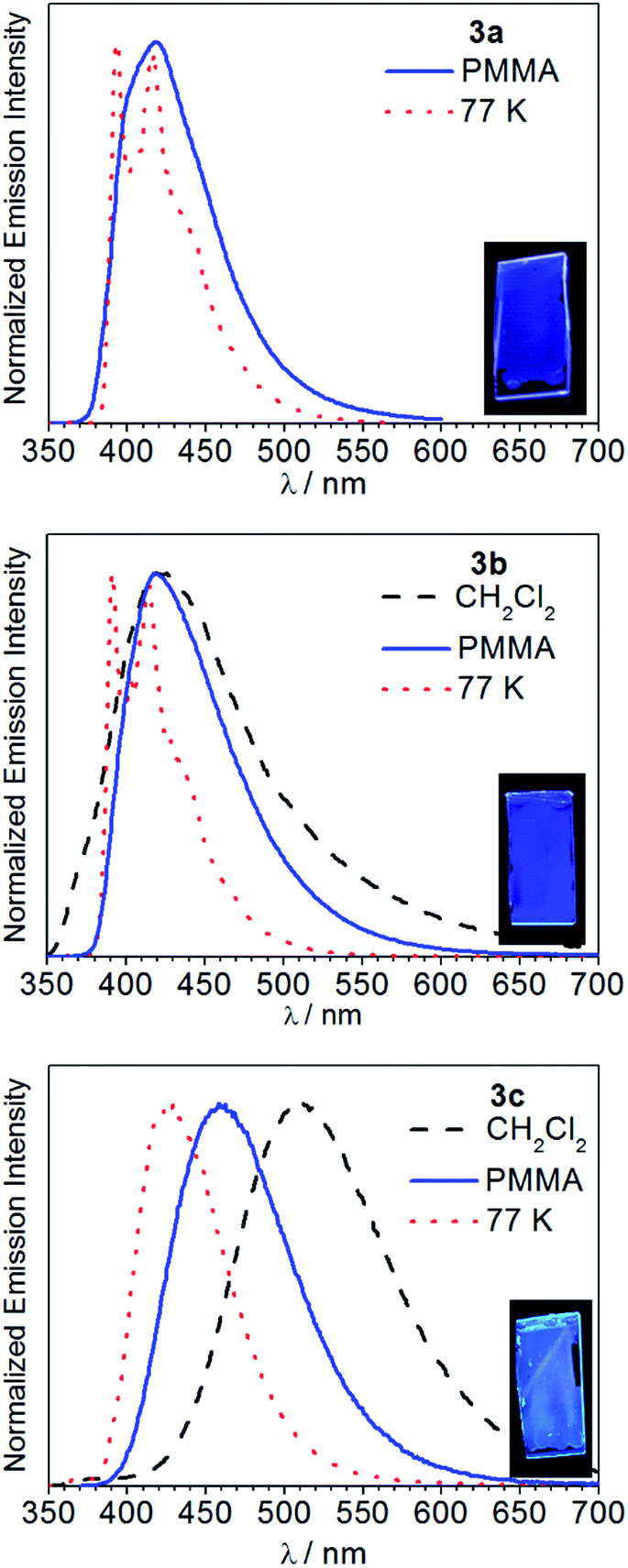
Fig. 5 overlays the room-temperature photoluminescence spectra of complexes 3a–c, whereas Table 1 summarizes the data. The complex Ir(pmi)2(C^C:ADC) (3a) is negligibly fluorescent in fluid solution, but when immobilized in poly(methyl methacrylate) (PMMA) at 2 wt% deep blue luminescence is observed, with a wavelength of maximum emission of 418 nm and a photoluminescence quantum yield (ΦPL) of 0.13. Replacing pmi with pmb in complex 3b has no impact on the observed wavelength of emission, but in this case the quantum efficiency is much higher. In CH2Cl2 solution complex 3b weakly emits, with a quantum yield of 0.013, and in PMMA the spectrum narrows and the quantum yield increases dramatically to 0.31. Finally, in complex 3c, where the pyridyl-substituted cyclometalating ligand pmp is used, the photoluminescence bathochromically shifts and is environmentally dependent. In solution complex 3c emits in the green region of the spectrum, with λ = 511 nm and a quantum yield of 0.39. The photoluminescence blue shifts significantly in PMMA film, with λ = 459 nm, and the quantum yield increases to 0.48. The photoluminescence lifetimes also vary across the series. When measured in PMMA film, the lifetimes decrease in the order 3a (τ = 6.1 μs), 3b (τ = 1.8 μs), and 3c (τ = 0.85 μs). These differences in lifetimes and quantum yields are primarily driven by differences in kr, which increase in the order 3a < 3b < 3c. Emission spectra were also recorded at 77 K in rigid solvent glass, and under these conditions the luminescence maximum displays a significant rigidochromic blue-shift in each case, signifying substantial charge-transfer. The spectra of 3a and 3b are much sharper with pronounced vibronic structure at 77 K, consistent with significant ligand-centered 3ππ* excited-state character as well, whereas the spectrum of 3c is devoid of vibronic structure even at 77 K, indicating a T1 state that is mainly a metal-to-ligand charge transfer (3MLCT) state.
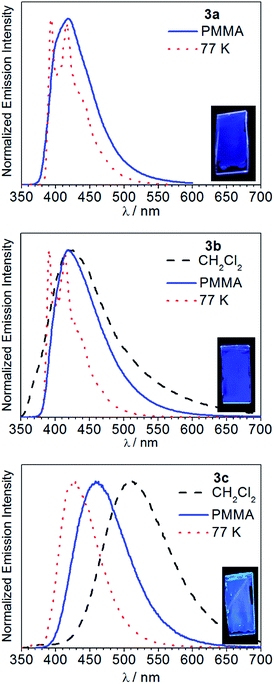 |
| | Fig. 5 Photoluminescence spectra of 3a–c recorded in CH2Cl2 at room temperature (black trace, dashed line), PMMA film at room temperature (blue trace, solid line), and in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 CH2Cl2/toluene glass at 77 K (red trace, dotted line). Samples were excited at λ = 310 nm. The insets show photographs of the PMMA films under UV illumination. :3 CH2Cl2/toluene glass at 77 K (red trace, dotted line). Samples were excited at λ = 310 nm. The insets show photographs of the PMMA films under UV illumination. | |
Table 1 Summary of photoluminescence data for 3a–ca
|
|
CH2Cl2, RT |
77c K |
PMMA, RTd |
|
λ/nm |
Φ
PL
|
λ/nm |
λ/nm |
Φ
PL
|
τ/μs |
k
r, knr × 10−5/s−1 |
(CIEx, CIEy)f |
|
For steady-state measurements, including quantum yields, λexc = 310 nm. For lifetime measurements, λexc = 330 nm.
Not luminescent in solution at RT.
Recorded in 1:3 CH2Cl2/toluene glass.
2 wt% complex.
Average of four or five independent trials, with standard deviation of the last digit shown in parentheses.
From PMMA film data.
|
|
3a
|
N.A.b |
N.A.b |
393, 417 |
418 |
0.13(1) |
6.1 |
0.21, 1.4 |
(0.16, 0.07) |
|
3b
|
420 |
0.013 |
391, 415 |
418 |
0.31(1) |
1.8 |
1.7, 3.8 |
(0.16, 0.10) |
|
3c
|
511 |
0.39 |
430 |
459 |
0.48(7) |
0.85 |
5.6, 6.1 |
(0.16, 0.18) |
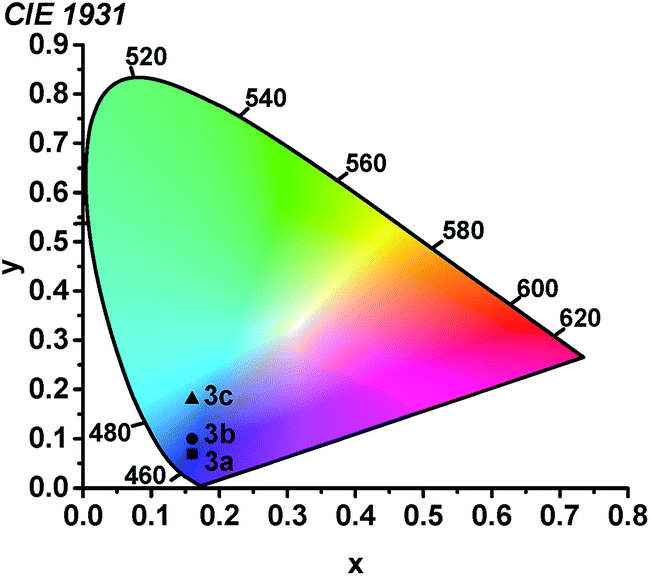
Using the thin-film photoluminescence data, CIE coordinates29 were determined and are summarized in Fig. 6. All three compounds fall in the blue region of the spectrum. The red-shifted emission in complex 3c engendered by the pyridyl-substituted NHC results in significant sky-blue coloration with (CIEx, CIEy) coordinates of (0.16, 0.18). In contrast, the color profiles for 3a and 3b represent pure blue luminescence, with CIE coordinates of (0.16, 0.10) for 3b and (0.16, 0.07) for 3a. The color profiles of 3a–c represent substantial improvements over our first two classes of cyclometalated iridium ADC complexes, all of which exhibited luminescence with significant sky blue to blue-green coloration rendering them unsuitable for device applications.26,27
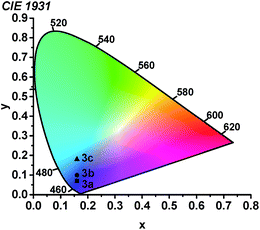 |
| | Fig. 6 Chromaticity diagram showing the (CIEx, CIEy) coordinates of 3a–c. | |
DFT calculations
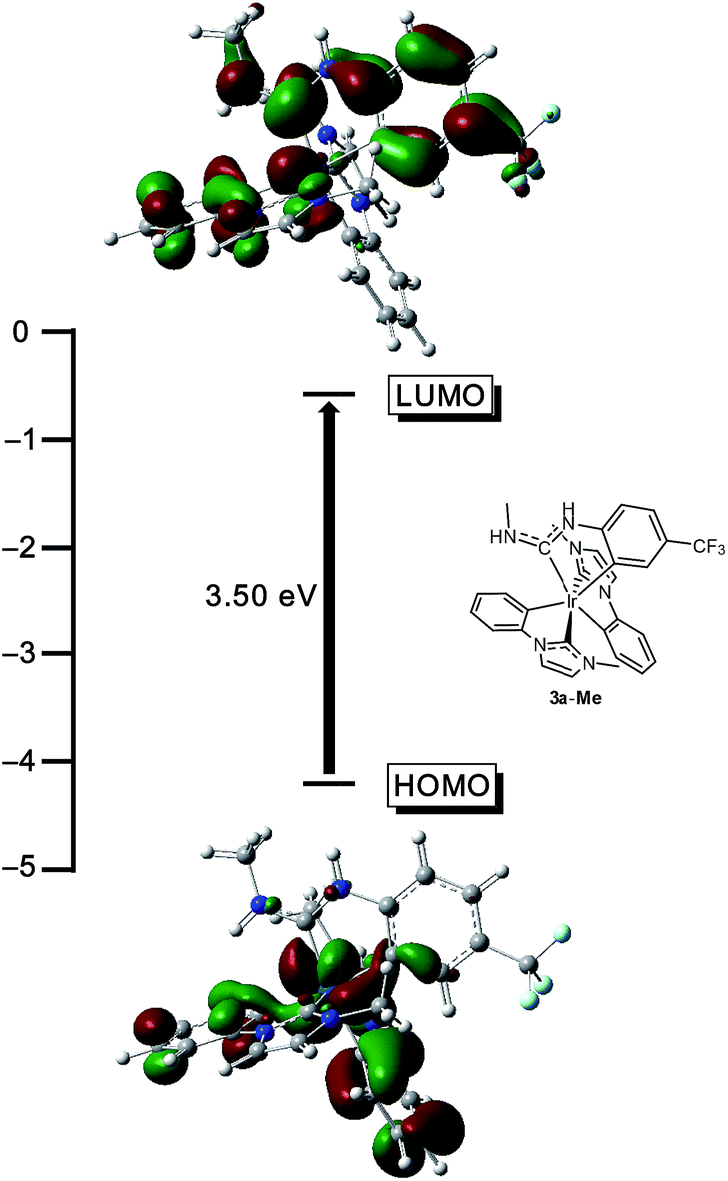
To validate the conclusions drawn from the experimental data presented above, and to determine the role of the ADC ligand in the frontier orbitals and triplet state, DFT calculations were performed on the model complex 3a-Me, where the N-propyl group on the ADC is truncated to a methyl group. All calculations were performed in the gas phase at MN15L/6-311G* for nonmetal atoms, with the SDD basis set used for Ir. The optimized geometry is C1-symmetric with structural metrics very similar to the crystal structure (Fig. 2). In particular, the ADC N–C–N bond angle, which optimized to 116.75°, is intermediate between the angles of 116.1(15)° and 117.5(14)°, observed for the two crystallographically independent molecules of 3a. Fig. 7 shows the computed HOMO and LUMO orbitals for 3a-Me. The HOMO is a mixed metal–ligand orbital consisting primarily of contributions from Ir d orbitals and the phenyl rings of the cyclometalated NHC ligands, as is typical for carbene-based iridium cyclometalates.13,15 In contrast, the LUMO is almost exclusively ligand-centered, though it does involve contributions from one of the cyclometalated NHC ligands and the cyclometalated ADC ligand. A HOMO–LUMO gap of 3.50 eV was computed, consistent with the large electrochemical HOMO–LUMO gap (>3 eV) observed via cyclic voltammetry (Fig. 3).
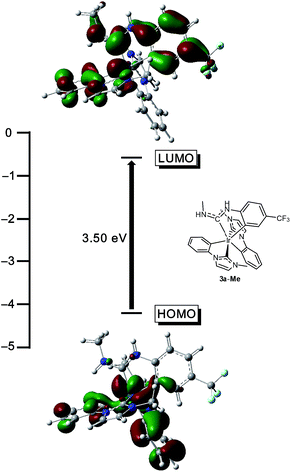 |
| | Fig. 7 DFT-computed HOMO and LUMO energy values and plots for the model complex 3a-Me (isovalue = 0.03 a.u.). Calculations were performed in the gas phase at MN15L/6-311G* for nonmetal atoms, with the SDD basis set used for Ir. The structure of 3a-Me is also shown, in the approximate orientation used for the orbital depictions. | |
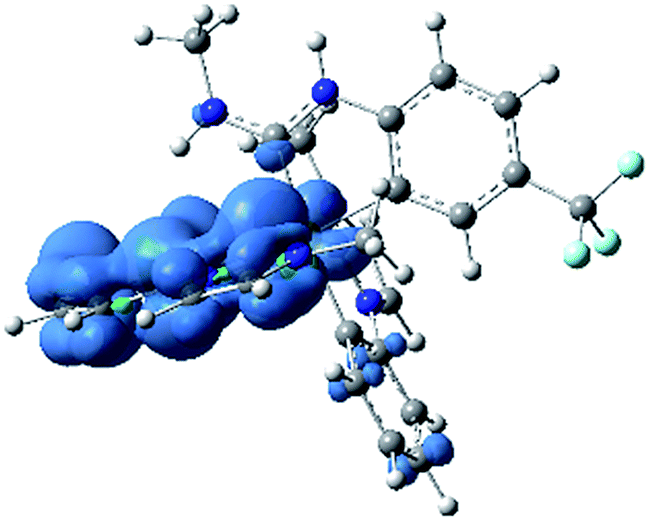
In addition to ground-state calculations in the singlet spin state, the lowest-energy triplet state was computed using unrestricted DFT. There are no major geometric differences between the singlet ground state and the triplet excited state, and the computed singlet–triplet gap of 3.20 eV is a very good match for the E0–0 value of 3.15 eV, estimated from the first maximum in the 77 K photoluminescence spectrum of 3a (Fig. 5). Fig. 8 shows the computed triplet spin density of 3a-Me, with the unpaired spin density found to reside exclusively on the Ir center and one of the pmi ligands, consistent with the formulation of the T1 state as a mixed 3LC/3MLCT state. Notably, there is almost no spin density on the ADC ligand, even though the ADC is involved substantially in the LUMO. The nature of this triplet state is very similar to that of homoleptic Ir(C^C:NHC) complexes, consistent with the observation that the photoluminescence spectra of 3a–c are quite similar to their homoleptic analogues (see below).
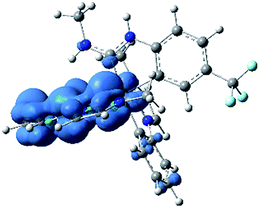 |
| | Fig. 8 UDFT calculated triplet spin density of complex 3a-Me. Contours are shown at an isovalue of 0.003 a.u. (blue indicates positive, green indicates negative). | |
Comparison of mixed-carbene and homoleptic NHC complexes
To further contextualize the photoluminescence properties and the CIE coordinates, we can compare complexes 3a–c to homoleptic Ir(C^C:NHC)3 complexes which use the same three NHC ligands. Although Ir(C^C:NHC)3 complexes are available in both fac and mer isomers, with the stereochemistry not having a large influence on photoluminescence attributes,13,14 we focus here on comparisons to the mer isomer, since this isomer and 3a–c include a trans arrangement of two of the NHC ligands. Immobilized in a thin film, compound 3c exhibits nearly identical photoluminescence to that of mer-Ir(pmp)3,14 with the same featureless profile and nearly identical maximum wavelengths and CIE coordinates ((CIEx, CIEy) = (0.16, 0.16) for mer-Ir(pmp)3). The photoluminescence attributes of 3a and 3b are also quite similar to their respective mer-Ir(C^C:NHC)3 (C^C:NHC = pmi, pmb) analogues. These compounds also emit with maxima near 400 nm, and although precise quantum yield values in polymer film were not reported, they were estimated to be greater than 0.1–0.2.13 The photoluminescence quantum yields of 3a (Φ = 0.13) and 3b (Φ = 0.31) are in the same range or greater, suggesting the photoluminescence efficiencies of 3a and 3b are very similar to or better than the homoleptic analogues. In addition, CIE coordinates for electroluminescent devices made from mer-Ir(pmb)3 were found to be (0.17, 0.06) and (0.17, 0.08),12 very similar to the color profile observed for the photoluminescence of 3a and 3b. One other point of comparison are homoleptic mer-Ir(C^C:NHC)3 complexes with CF3-substituted pmi ligands.15 The photoluminescence quantum yields of these compounds were measured in PMMA films, with one isomer having ΦPL = 0.14 and another with ΦPL = 0.47, similar values to those of 3a and 3b. CIE coordinates for devices made from these CF3-substituted Ir(pmi)3 analogues are also nearly identical to those observed for 3a and 3b. Taken together, these comparisons indicate that installing cyclometalated ADC ancillary ligands preserves the favorable photoluminescence characteristics of the carbene-based iridium cyclometalates that have been used in some of the top-performing blue devices.12,14,15 The subtle influence that the ADC has on the photoluminescence color when compared to homoleptic Ir(C^C:NHC)3 complexes suggests that the ADC-centered orbitals do not contribute substantially to the T1 state, which is born out in the DFT calculations described here.
Conclusions
In this work we address the long-standing challenge of designing effective blue phosphors for OLED applications. The compounds we describe are heteroleptic tris-cyclometalated iridium complexes with mixed-carbene ligation, prepared from isocyanide precursors by a nucleophilic addition/cyclometalation cascade sequence. The photoluminescence attributes of these compounds are admirable. In PMMA thin films, deep blue luminescence is observed for two members of the series, with CIE coordinates that match well with industry standards for blue phosphors. The photoluminescence quantum yields are good and the lifetimes are in the microsecond range, also important criteria for device applications. In short, the pure blue photoluminescence of the mixed-carbene compounds describe here and the potential advantages offered by the ADC moiety motivate continued evaluation of 3a–c and related analogues as dopants for blue OLEDs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
T. S. T. acknowledges support from the National Science Foundation (NSF) (CHE-1846831) and the Welch Foundation (grant no. E-1887) for funding this research. J. I. W. thanks the NSF for support (CHE-1751370), as well as computational resources provided by the uHPC cluster, managed by the University of Houston and acquired through support from the NSF (MRI-1531814).
Notes and references
-
Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, John Wiley & Sons, Inc, Chichester, West Sussex, 2017 Search PubMed.
-
Highly efficient OLEDs with phosphorescent materials, ed. H. Yersin, WILEY-VCH, Weinheim, 2008 Search PubMed.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef CAS PubMed.
- C.-H. Fan, P. Sun, T.-H. Su and C.-H. Cheng, Adv. Mater., 2011, 23, 2981–2985 CrossRef CAS PubMed.
- D.-L. Ma, V. P.-Y. Ma, D. S.-H. Chan, K.-H. Leung, H.-Z. He and C.-H. Leung, Coord. Chem. Rev., 2012, 256, 3087–3113 CrossRef CAS.
- T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., Int. Ed., 2012, 51, 4148–4151 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- M. S. Lowry and S. Bernhard, Chem.–Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed.
- Y. Tanabe and S. Sugano, J. Phys. Soc. Jpn., 1954, 9, 753–766 CrossRef CAS.
- S. Scholz, D. Kondakov, B. Lüssem and K. Leo, Chem. Rev., 2015, 115, 8449–8503 CrossRef CAS PubMed.
- R. J. Holmes, S. R. Forrest, T. Sajoto, A. Tamayo, P. I. Djurovich, M. E. Thompson, J. Brooks, Y.-J. Tung, B. W. D'Andrade, M. S. Weaver, R. C. Kwong and J. J. Brown, Appl. Phys. Lett., 2005, 87, 243507 CrossRef.
- T. Sajoto, P. I. Djurovich, A. Tamayo, M. Yousufuddin, R. Bau, M. E. Thompson, R. J. Holmes and S. R. Forrest, Inorg. Chem., 2005, 44, 7992–8003 CrossRef CAS PubMed.
- J. Lee, H.-F. Chen, T. Batagoda, C. Coburn, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Nat. Mater., 2015, 15, 92–98 CrossRef PubMed.
- A. K. Pal, S. Krotkus, M. Fontani, C. F. R. Mackenzie, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, Adv. Mater., 2018, 30, 1804231 CrossRef PubMed.
- C.-H. Hsieh, F.-I. Wu, C.-H. Fan, M.-J. Huang, K.-Y. Lu, P.-Y. Chou, Y.-H. O. Yang, S.-H. Wu, I.-C. Chen, S.-H. Chou, K.-T. Wong and C.-H. Cheng, Chem.–Eur. J., 2011, 17, 9180–9187 CrossRef CAS PubMed.
- Z. Chen, L. Wang, S. Su, X. Zheng, N. Zhu, C.-L. Ho, S. Chen and W.-Y. Wong, ACS Appl. Mater. Interfaces, 2017, 9, 40497–40502 CrossRef CAS PubMed.
- C.-F. Chang, Y.-M. Cheng, Y. Chi, Y.-C. Chiu, C.-C. Lin, G.-H. Lee, P.-T. Chou, C.-C. Chen, C.-H. Chang and C.-C. Wu, Angew. Chem., Int. Ed., 2008, 47, 4542–4545 CrossRef CAS PubMed.
- H. Sasabe, J. Takamatsu, T. Motoyama, S. Watanabe, G. Wagenblast, N. Langer, O. Molt, E. Fuchs, C. Lennartz and J. Kido, Adv. Mater., 2010, 22, 5003–5007 CrossRef CAS PubMed.
- T.-Y. Li, X. Liang, L. Zhou, C. Wu, S. Zhang, X. Liu, G.-Z. Lu, L.-S. Xue, Y.-X. Zheng and J.-L. Zuo, Inorg. Chem., 2015, 54, 161–173 CrossRef CAS PubMed.
- B. Wiegmann, P. G. Jones, G. Wagenblast, C. Lennartz, I. Münster, S. Metz, W. Kowalsky and H.-H. Johannes, Organometallics, 2012, 31, 5223–5226 CrossRef CAS.
- R. W. Alder, P. R. Allen, M. Murray and A. G. Orpen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1121–1123 CrossRef CAS.
- W. A. Herrmann, K. Öfele, D. v. Preysing and E. Herdtweck, J. Organomet. Chem., 2003, 684, 235–248 CrossRef CAS.
- P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef.
- V. P. Boyarskiy, K. V. Luzyanin and V. Yu. Kukushkin, Coord. Chem. Rev., 2012, 256, 2029–2056 CrossRef CAS.
- H. Na, A. Maity, R. Morshed and T. S. Teets, Organometallics, 2017, 36, 2965–2972 CrossRef CAS.
- H. Na and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 6353–6360 CrossRef CAS PubMed.
- H. Na, P. Lai, L. M. Cañada and T. S. Teets, Organometallics, 2018, 37, 3269–3277 CrossRef CAS.
- T. Smith and J. Guild, Trans. Opt. Soc., 1931, 33, 73–134 CrossRef.
- Y. K. Radwan, A. Maity and T. S. Teets, Inorg. Chem., 2015, 54, 7122–7131 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray crystallography summary table, UV-vis excitation spectra, and NMR spectra. CCDC 1879106 and 1879107. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01386e |
‡ 3a: CCDC 1879106, C31H30F3IrN6, M = 735.81, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 14.228(5) Å, b = 15.407(6) Å, c = 16.166(6) Å; α = 86.526(4)°, β = 65.199(4)°, γ = 85.672(4)°, Z = 4, 36765 tot. refln., 12993 ind. refln., Rint = 0.058, R1 = 0.098, wR2 = 0.300. , a = 14.228(5) Å, b = 15.407(6) Å, c = 16.166(6) Å; α = 86.526(4)°, β = 65.199(4)°, γ = 85.672(4)°, Z = 4, 36765 tot. refln., 12993 ind. refln., Rint = 0.058, R1 = 0.098, wR2 = 0.300. |
| § 3b: CCDC 1879107, C39H34F3IrN6, M = 835.92, monoclinic, C2/c, a = 27.270(8) Å, b = 20.718(6) Å, c = 14.832(4) Å; β = 116.007(3)°, Z = 8, 31630 tot. refln., 7309 ind. refln., Rint = 0.047, R1 = 0.088, wR2 = 0.211. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Zhili
Wen
,
Judy
I-Chia Wu
,
Zhili
Wen
,
Judy
I-Chia Wu