
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical and slow-chemistry radical transformations: a case of diorganozinc compounds and TEMPO†
Krzysztof
Budny-Godlewski
 a,
Iwona
Justyniak
b,
Michał K.
Leszczyński
b and
Janusz
Lewiński
*ab
a,
Iwona
Justyniak
b,
Michał K.
Leszczyński
b and
Janusz
Lewiński
*ab
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: lewin@ch.pw.edu.pl; Fax: +48 22 6282741; Tel: +48 22 2347315
bInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
First published on 17th June 2019
Abstract
From the green chemistry perspective, molecular solid-state transformations conducted under mild conditions are of great interest and desirability. However, research in this area lacked popularity in the previous century, and thus progressed slowly. In particular, the application of radical reactions in solid-state chemistry has been hampered by several long-standing challenges that are intrinsically associated with the apparent unpredictable nature of radical chemistry. We present a comparative study of model mechanochemical, slow-chemistry and solution radical reactions between TEMPO and homoleptic organozinc compounds (i.e., di-tert-butylzinc and diphenylzinc). In the case of the tBu2Zn/TEMPO reaction system only a dimeric diamagnetic complex [tBuZn(μ-TEMPO*)]2 is obtained in yields slightly varying with the method chosen. In contrast, when TEMPO is mixed with diphenylzinc in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio a novel paramagnetic Lewis acid–base adduct [[Ph2Zn(η1-TEMPO)]·TEMPO] is isolated in high yields regardless of the applied methodology. This adduct is also formed in the slow-chemistry process when TEMPO is gently mixed with Ph2Zn in a 1:1 molar ratio and left for two weeks at ambient temperature. Within the next week the reaction mixture gives in high yield a diamagnetic dinuclear compound [PhZn(μ-TEMPO*)][PhZn(μ2-η1:η1-TEMPO*)] and biphenyl. The analogous reaction conducted in toluene results in a much lower conversion rate. The reported results open up a new horizon in molecular solid-state radical transformations.
:1 molar ratio a novel paramagnetic Lewis acid–base adduct [[Ph2Zn(η1-TEMPO)]·TEMPO] is isolated in high yields regardless of the applied methodology. This adduct is also formed in the slow-chemistry process when TEMPO is gently mixed with Ph2Zn in a 1:1 molar ratio and left for two weeks at ambient temperature. Within the next week the reaction mixture gives in high yield a diamagnetic dinuclear compound [PhZn(μ-TEMPO*)][PhZn(μ2-η1:η1-TEMPO*)] and biphenyl. The analogous reaction conducted in toluene results in a much lower conversion rate. The reported results open up a new horizon in molecular solid-state radical transformations.
Introduction
The rational development of solid-state reactions is one of the objectives of the green chemistry movement. Notably, in the previous century research on molecular solid-state reactions mediated by mechanical forces progressed slowly which was aptly annotated by Braga and Grepioni: “Although research work in this area has never stopped, it has also lacked popularity and has remained confined to a few connoisseurs”.1 Only the last decade has witnessed a true renaissance of mechanochemistry2–5 applications of which include organic6–10 and organometallic synthesis,11–14 polymer chemistry,15,16 and materials science (e.g. in the preparation of MOFs,17,18 perovskites for solar cells19–22 or nanomaterials).3,23–26 In some cases mechanical stress can support electron transfer27 or cause homolytic bond cleavage and in consequence the formation of radicals. A textbook example of a radical solid-state chemistry phenomenon, which was already noted in ancient Greece, is the attraction between rubbed amber and some small objects.28 Only in 1967 Urbański revealed the formation of radicals on the surface of ground amber.29 The formation of radicals has also been observed upon mechanically induced reactions in polymeric materials.30,31 Grzybowski and co-workers demonstrated that these mechanoradicals can be responsible for static charge stabilization, and thus, polymers intentionally doped with radical scavengers (inter alia free stable radicals) are less vulnerable to static charging.32–34 Despite these seminal pioneering discoveries solid-state radical reactions have remained an underdeveloped area of research.Even though mechanochemistry is slowly becoming a routine solid-state synthesis tool, knowledge on the intimate mechanistic details is very limited.26,35,36 Remarkably, an emerging field of slow-chemistry (mild condition reactions occurring in the solid-state without an external stimuli)37 or accelerated aging reactions (i.e., solid-state transformations where reaction rates are enhanced by the presence of solvent vapour)38 nicely complements and enriches solid-state processes, and may turn out to be an area to help better understand mechanochemical reactions. Slow-chemistry reactions are widely observed in nature, e.g. biomineralization, pearl formation or mineral neogenesis, and only recently have these natural processes been mimicked in synthetic procedures.39–42
Herein we present unprecedented solvent-free slow-chemistry and mechanical force triggered radical transformations involving solid homoleptic organozinc compounds and an intentionally added free stable nitroxyl radical, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO). Very recently, in contrast to common beliefs,43 our group and independently Stephan's group demonstrated that in solution TEMPO is non-innocent towards Et2Zn44,45 and (η5-C5Me5)2Zn,46 respectively, and smoothly induces radical transformations triggered by single-electron transfer (SET)47 from the Zn–C bond to TEMPO, to afford organozinc complexes incorporating TEMPO anions. We were wondering whether TEMPO will also remain non-innocent towards homoleptic diorganozinc compounds in the solid-state and to this end we chose di-tert-butylzinc (tBu2Zn) and diphenylzinc (Ph2Zn) solid homoleptic organometallics with versatile properties and reactivity.48–50 The solid-state reactions were commenced with comparative solution phase investigations. Remarkably, while in the case of the tBu2Zn/TEMPO system either wet chemistry or mechanochemical and slow-chemistry transformations provided essentially the same outcome, the character of the products of the Ph2Zn/TEMPO system depends significantly on the stoichiometry of the reaction and the selected synthetic methodology.
Results and discussion
Comparative investigations in solution
Initially, when a toluene solution of tBu2Zn was treated with an orange toluene solution of TEMPO (2 equiv.) at −78 °C, an instantaneous decolourization took place. Standard work-up of the post-reaction mixture afforded colourless crystals of a diamagnetic dimeric complex [tBuZn(μ-TEMPO*)]2 (12) (Fig. 1a, TEMPO* stands for the TEMPO anion) and TEMPO–tBu as the primary products. The formation of 12 is likely triggered by an incipient tBu2Zn/TEMPO adduct followed by an intramolecular SET from the Zn–tBu bond to TEMPO, and TEMPO–tBu results from the subsequent release of a tBu˙ radical and its recombination with the second TEMPO molecule.51 In this case we were not able to isolate the anticipated paramagnetic Lewis acid–base R2Zn(TEMPO) adduct due to the high reactivity of homoleptic zinc alkyls towards TEMPO. Remarkably, the respective paramagnetic adduct was isolated from a similar reaction with Ph2Zn. Clearly, Ph2Zn is less prone to react as a SET reagent, and moreover the Ph2Zn/TEMPO system appeared also sensitive to the molar ratio of the reagents and reaction time. A 1:1 mixture of the starting materials was dissolved in toluene and stirred at room temperature for an hour and then cooled to −26 °C. Within 24 hours yellow crystals of a supramolecular complex [[Ph2Zn(η1-TEMPO)]·TEMPO] (2·TEMPO) deposited in moderate yield. Compound 2·TEMPO is composed of a paramagnetic Lewis acid–base adduct [Ph2Zn(η1-TEMPO)] (2) with a hydrogen-bonded TEMPO molecule (Fig. 1b). Interestingly, a prolonged (two weeks) reaction between Ph2Zn and 1 equiv. of TEMPO conducted at ambient temperature in toluene generated a new diamagnetic dinuclear complex [PhZn(μ-TEMPO*)][PhZn(μ2-η1:η1-TEMPO*)] (3, Fig. 1c) (yield ca. 35%) as well as biphenyl (PhPh) resulting from homocoupling of the Ph˙ radicals. Remarkably, the Ph˙ radicals do not recombine with TEMPO molecules on this occasion as we did not observe the signals characteristic of TEMPO–Ph in the 1H NMR spectrum of the post-reaction mixture.
 | ||
| Fig. 1 Unprecedented variety of outcomes of the solid-state and wet reactions of TEMPO with tBu2Zn and Ph2Zn: (a) [tBuZn(μ-TEMPO*)]2 (12), (b) [[Ph2Zn(η1-TEMPO)]·TEMPO] (2·TEMPO), and (c) [PhZn(μ-TEMPO*)][PhZn(μ2-η1:η1-TEMPO*)] (3). | ||
To clarify the factors determining the formation of the paramagnetic complex 2·TEMPO and the diamagnetic complex 3, we performed the reaction of Ph2Zn with 2 equiv. of TEMPO in a manner analogous to that of the equimolar system (for details, see the Experimental procedures). In this case the SET process was suppressed and 2·TEMPO was isolated essentially quantitatively. Undoubtedly, a key aspect of the Ph2Zn/TEMPO reaction system is the reduction of TEMPO to its anionic form by Ph2Zn (with the subsequent coupling of the formed Ph˙ radicals to yield PhPh) only for the 1:1 mixture while a higher amount of TEMPO inhibits the SET step (for details see Fig. S22 in the ESI†).
Crystal structure analysis
Compounds 12, 2·TEMPO and 3 were characterized by NMR spectroscopy and single-crystal X-ray diffraction, and additionally the paramagnetic character of 2·TEMPO was confirmed by EPR spectroscopy. Compounds 12 and 3 show sharp 1H NMR resonances characteristic of the zinc bonded organic groups and the TEMPO* anion, while the NMR spectrum of 2·TEMPO confirms its paramagnetic character (Fig. S1–S8 in the ESI†). The diffusion ordered NMR spectroscopy (DOSY) experiments show that 12 and 3 most likely exist as monomers in solution (for details, see Table S1 in the ESI†). Remarkably, the solution and solid-state EPR spectra of 2·TEMPO show one set of signals analogous to that observed for the free TEMPO radical (see Fig. S13 and S14†).X-ray crystal structure analysis showed that the molecular structure of the tert-butyl–zinc derivative 12 (Fig. 1a) is essentially analogous to the previously reported diamagnetic ethylzinc derivative [EtZn(μ-TEMPO*)]2,46 and comprises bridging μ-TEMPO* ligands and a planar Zn2O2 ring (Zn1–O1 = 1.939(3) Å and Zn1–O1′ = 1.964(3) Å). In turn, the relevant phenyl–zinc derivative 3 crystallizes as an asymmetric dimer that contains a rarely encountered mix of coordination μ-O- and μ-η1:η1-NO-modes of the anionic TEMPO* ligands (Fig. 1c).52 Two three-coordinate zinc atoms are bridged by μ-O-TEMPO* and μ-η1:η1-NO-TEMPO* units, thus forming a slightly bent five-membered [ZnOZnNO] ring (Zn1–O1 = 1.916(3) Å, O1–Zn2 = 1.943(3) Å, Zn2–N2 = 2.078(3) Å, N2–O2 = 1.441(4) Å, and O2–Zn1 = 1.898(2) Å).
In the paramagnetic adduct 2·TEMPO, one TEMPO radical coordinates to the zinc centre in an η1-mode (Zn1–O1 = 2.0841(18) Å, O1–N1 = 1.294(3) Å, and Zn1–O1–N1 = 143.50(17)°) (Fig. 1b). The second TEMPO molecule interacts by hydrogen bonding involving the oxygen atom and the C–H bond of one Zn-bonded Ph group (H5–O2 = 2.323(9) Å, O2–N2 = 1.288(3) Å, and H5–O2–N2 = 86.517(13)°) and the observed hydrogen-bonding interaction torsion angle N2–O2⋯H5–C5 = 45.227(20)° is similar to the calculated N–O⋯H–C torsion angle (45°) for the CH3CN/TEMPO system.53 The zinc atom occupies a site within a trigonal-planar arrangement of the oxygen and carbon atoms (C1–Zn1–C7 = 145.61(12), C1–Zn1–O1 = 112.8(1)°, and O1–Zn1–O7 = 101.4(1)) with the Zn1–O1 bond length markedly longer than that observed for the Zn–O bond in zinc complexes incorporating an external anionic η1-TEMPO* ligand (cf. the Zn–O of 1.854(6) in [Zn(TEMPO*)2]2).46 To the best of our knowledge, the adduct 2 is the first isolated and structurally characterized simple Lewis acid adduct of TEMPO with an organometallic molecule.54 The revealed hydrogen-bond interaction between the free TEMPO molecule and the Zn-bonded Ph group as well as a vast number of other non-covalent interactions observed in the solid-state structure of 2·TEMPO (see Fig. S27†) are likely responsible for an increased stability of 2·TEMPO in the solid-state (note that 2·TEMPO does not transform into 3 in the solid-state, vide infra).
Reactions in the solid-state
Equipped with the above knowledge about the reactivity of TEMPO towards tBu2Zn and Ph2Zn in solution, we performed analogous reactions with these reagents in the solid-state. The progress of both slow-chemistry and mechanochemical reactions was monitored using PXRD analysis (for extended diffractograms see Fig. S9–S12†) and additionally confirmed by 1H NMR spectroscopy; all operations were conducted under an argon inert gas atmosphere.We revealed that both slow- and mechanochemical reactions take place in the case of the tBu2Zn/TEMPO system, and selectively afford 12 and TEMPO–tBu in high yield (Fig. 2). The mechanochemical reaction involving grinding tBu2Zn with 2 equiv. of TEMPO with a glass rod at 0 °C occurs within circa 20 minutes (Fig. 2b). In the slow-chemistry reaction, tBu2Zn was gently mixed with 2 equiv. of TEMPO and then stored at 5 °C under an inert gas atmosphere and in this case the reaction required circa 24 hours for completion (Fig. 2c).
 | ||
| Fig. 2 The PXRD patterns for the reactions of TEMPO with tBu2Zn: (a) the simulated PXRD spectrum of 12, (b) the mixture of tBu2Zn and 2 equiv. of TEMPO ground with a glass rod for 20 minutes at 0 °C and (c) the mixture of tBu2Zn and 2 equiv. of TEMPO gently mixed and stored at 5 °C for 24 h. | ||
Strikingly, transformations involving Ph2Zn and TEMPO proceed entirely differently than in the case of the tBu2Zn/TEMPO system (Fig. 3). Grinding Ph2Zn with 1 equiv. of TEMPO at ambient temperature for over 15 minutes gives a mixture of the paramagnetic adduct 2·TEMPO and unreacted Ph2Zn as judged by the PXRD analysis (Fig. 3b). However, when Ph2Zn is ground with 2 equiv. of TEMPO, then 2·TEMPO is obtained selectively as the only product (Fig. 3c). Regardless of the grinding conditions (molar ratio of substrates, prolonged time, and higher frequency of milling), no trace of the 3 phase was observed in the PXRD pattern of the postreaction powder.‡ What is more, the pure compound 2·TEMPO does not transform into the diamagnetic compound 3 even after over 6 months of aging (20 °C and inert gas atmosphere).
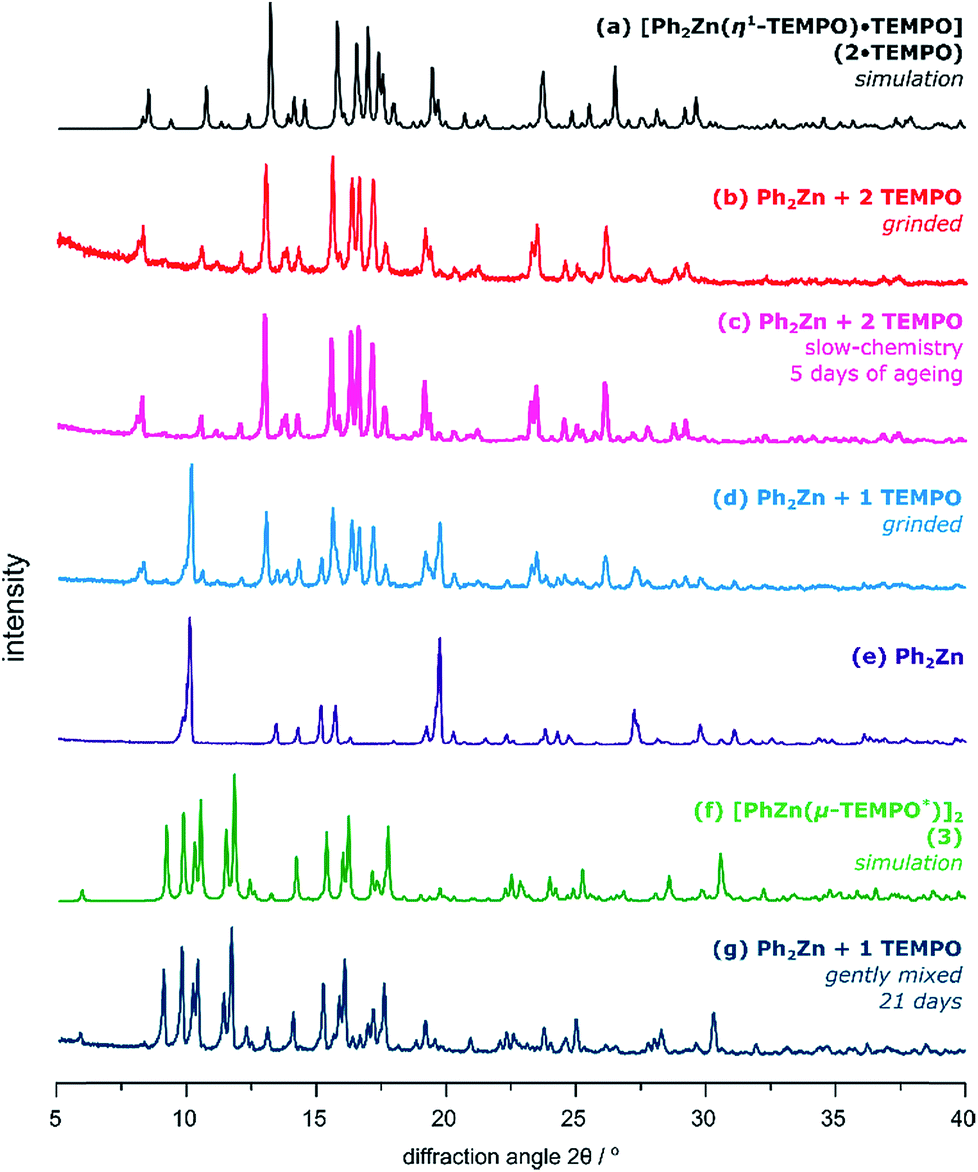 | ||
| Fig. 3 The PXRD patterns of various reaction systems involving TEMPO and Ph2Zn: (a) the simulated PXRD spectrum of 2·TEMPO, (b) Ph2Zn ground with 2 equiv. of TEMPO, (c) Ph2Zn gently mixed with 2 equiv. of TEMPO and stored under an inert gas atmosphere at 20 °C for 5 days, (d) Ph2Zn ground with 1 equiv. of TEMPO, and (e) Ph2Zn. (f) The simulated PXRD spectrum of 3 and (g) Ph2Zn gently mixed with 1 equiv. of TEMPO stored at 20 °C for 21 days. | ||
The slow chemistry reactions involving Ph2Zn and TEMPO in various molar ratios are more subtle. The paramagnetic adduct 2·TEMPO is obtained selectively within ca. 5 days of aging (20 °C) of a mixture consisting of Ph2Zn gently mixed with 2 equiv. of TEMPO (Fig. 3d). No further transformation of 2·TEMPO was observed in the solid state even after over 6 months of aging. In the case of the equimolar mixture of Ph2Zn and TEMPO the slow-chemistry reaction proceeds differently and consists of two distinguishable steps. In the first step, the reaction yields an intermediate mixture of 2·TEMPO and Ph2Zn, which in the second step quantitatively transforms into an equimolar mixture of 3 and PhPh (Fig. 4). Moreover, the evolution of PXRD diffractograms of the equimolar mixture of Ph2Zn and TEMPO over time depends on the way the substrates were initially mixed (ground or gently mixed). The PXRD analysis of the initially ground substrate mixture reveals patterns characteristic of 2·TEMPO and Ph2Zn. Then after 21 days of aging the PXRD patterns evolve into that characteristic of 3 (patterns characteristic of PhPh are not observed but its formation was confirmed by 1H NMR spectroscopy). In contrast, the gently mixed substrate mixture affords an intermediate mixture of 2·TEMPO and Ph2Zn within 7 days of aging and then within the next 14 days transforms into 3 and PhPh.
 | ||
| Fig. 4 Proposed mechanism of the reaction of TEMPO with Ph2Zn (1:1 molar ratio) in the solid-state. | ||
The above results indicate that the solid-state equimolar mixture of Ph2Zn and TEMPO initially smoothly transforms into a 2·TEMPO supramolecular complex while half the amount of Ph2Zn remains intact. Subsequently, the mixture of 2·TEMPO and Ph2Zn transforms into 3 in the slow-chemistry process. Remarkably, this transformation is not observed for the pure phase 2·TEMPO complex under either mechanochemical or slow-chemistry conditions. Thus, at this stage of studies it is reasonable to propose that 2·TEMPO and Ph2Zn equilibrate in the solid state with the simple Lewis acid–base adduct 2 that is active in the SET process leading to 3 (Fig. 4).
Conclusions
Investigations of solid-state chemistry reactions under mild conditions have remained both one of the greatest mysteries and one of the most challenging tasks for chemists and materials scientists. We demonstrated that mechanochemical and slow-chemistry processes can be used as efficient tools to control the outcome of organometallic radical reactions. Moreover, in our opinion, the reported studies are a significant advance in molecular solid-state chemistry and should aid in unlocking the potential of radical slow-chemistry reactions as a novel tool for the preparation of desired products. At this stage with little knowledge of the mechanisms of the titular solid-state transformations, the best approach to extending this idea would seemingly be through trial and error. Further work in this regard is currently underway in our laboratory.Experimental
tBu2Zn was prepared according to literature procedures48 under an argon atmosphere using standard Schlenk techniques. Ph2Zn and all other reagents were of commercial grade obtained from Sigma-Aldrich Co. The solvents were carefully dried and distilled under a nitrogen atmosphere prior to use. TEMPO was sublimed and stored under an argon atmosphere.[tBuZn(μ-TEMPO*)]2 (12)
[Ph2Zn(η1-TEMPO)·TEMPO] (2·TEMPO)
:1 molar ratio (ca. 39%). The paramagnetic nature of the product was confirmed by EPR spectroscopy. Pure compound 2·TEMPO left for over 6 months at 20 °C under an inert gas atmosphere does not transform into the diamagnetic compound 3. C,H,N analysis (%) calcd for C30H46N2O2Zn1: C 67.72, H 8.71, N 5.26; found: C 67.75, H 8.68, N 5.24.
[PhZn(μ2-TEMPO*)][PhZn(μ2-η1:η1-TEMPO*)] (3)
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support of the Foundation for Polish Science TEAM Program co-financed by the European Union under the European Regional Development Fund TEAM/2016-2/14 and the support of the National Science Centre 2015/19/N/ST5/02194. We are grateful to Dr Krzysztof Kruczała and Prof. Zbigniew Sojka for the EPR measurements and Prof. Zbigniew Ochal for GC measurements.Notes and references
- D. Braga and F. Grepioni, Angew. Chem., Int. Ed., 2004, 43, 4002–4011 CrossRef CAS PubMed.
- V. Boldyrev and K. Tkáčová, J. Mater. Synth. Process., 2000, 8, 121–132 CrossRef CAS.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2011, 41, 413–447 RSC.
- Themed Issue: Mechanochemistry, Chem. Soc. Rev., 2013, 42, 7487–7740 Search PubMed.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317–2329 RSC.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- D. Tan and T. Friščić, Eur. J. Org. Chem., 2018, 2018, 18–33 CrossRef CAS.
- J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC.
- D. Tan and F. García, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC.
- N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352–2362 RSC.
- J. Lewiński, M. Dutkiewicz, M. Lesiuk, W. Śliwiński, K. Zelga, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2010, 49, 8266–8269 CrossRef PubMed.
- J. G. Hernández, I. S. Butler and T. Friščić, Chem. Sci., 2014, 5, 3576–3582 RSC.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- N. Willis-Fox, E. Rognin, T. A. Aljohani and R. Daly, Chem, 2018, 4, 2499–2537 CAS.
- D. Prochowicz, K. Sokołowski, I. Justyniak, A. Kornowicz, D. Fairen-Jimenez, T. Friščić and J. Lewiński, Chem. Commun., 2015, 51, 4032–4035 RSC.
- K. Užarević, T. C. Wang, S.-Y. Moon, A. M. Fidelli, J. T. Hupp, O. K. Farha and T. Friščić, Chem. Commun., 2016, 52, 2133–2136 RSC.
- D. Prochowicz, M. Franckevičius, A. M. Cieślak, S. M. Zakeeruddin, M. Grätzel and J. Lewiński, J. Mater. Chem. A, 2015, 3, 20772–20777 RSC.
- P. Pal, S. Saha, A. Banik, A. Sarkar and K. Biswas, Chem.–Eur. J., 2018, 24, 1811–1815 CrossRef CAS PubMed.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, M. Saski, P. Yadav, D. Bi, N. Pellet, J. Lewiński, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2018, 140, 3345–3351 CrossRef CAS PubMed.
- M. Saski, D. Prochowicz, W. Marynowski and J. Lewiński, Eur. J. Inorg. Chem., 2019, 2680–2684 CrossRef CAS.
- M. J. Rak, T. Friščić and A. Moores, Faraday Discuss., 2014, 170, 155–167 RSC.
- M. Y. Malca, H. Bao, T. Bastaille, N. K. Saadé, J. M. Kinsella, T. Friščić and A. Moores, Chem. Mater., 2017, 29, 7766–7773 CrossRef CAS.
- A. Moores, Current Opinion in Green and Sustainable Chemistry, 2018, 12, 33–37 CrossRef.
- P. Krupiński, A. Kornowicz, K. Sokołowski, A. M. Cieślak and J. Lewiński, Chem.–Eur. J., 2016, 22, 7817–7823 CrossRef PubMed.
- Y. Li, N. L. Haworth, L. Xiang, S. Ciampi, M. L. Coote and N. Tao, J. Am. Chem. Soc., 2017, 139, 14699–14706 CrossRef CAS PubMed.
- P. Iversen and D. J. Lacks, J. Electrost., 2012, 70, 309–311 CrossRef.
- T. Urbański, Nature, 1967, 216, 577–578 CrossRef.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- B. Baytekin, H. T. Baytekin and B. A. Grzybowski, Angew. Chem., Int. Ed., 2014, 53, 6946–6950 CrossRef CAS PubMed.
- H. T. Baytekin, B. Baytekin and B. A. Grzybowski, Angew. Chem., Int. Ed., 2012, 51, 3596–3600 CrossRef CAS PubMed.
- H. T. Baytekin, B. Baytekin, T. M. Hermans, B. Kowalczyk and B. A. Grzybowski, Science, 2013, 341, 1368–1371 CrossRef CAS PubMed.
- A. M. Belenguer, G. I. Lampronti, D. J. Wales and J. K. M. Sanders, J. Am. Chem. Soc., 2014, 136, 16156–16166 CrossRef CAS PubMed.
- K. Užarević, I. Halasz and T. Friščić, J. Phys. Chem. Lett., 2015, 6, 4129–4140 CrossRef PubMed.
- X. Lim, Nature, 2015, 524, 20–21 CrossRef CAS PubMed.
- M. J. Cliffe, C. Mottillo, R. S. Stein, D.-K. Bučar and T. Friščić, Chem. Sci., 2012, 3, 2495–2500 RSC.
- F. Nudelman and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2012, 51, 6582–6596 CrossRef CAS PubMed.
- C. Mottillo, Y. Lu, M.-H. Pham, M. J. Cliffe, T.-O. Do and T. Friščić, Green Chem., 2013, 15, 2121–2131 RSC.
- F. Qi, R. S. Stein and T. Friščić, Green Chem., 2014, 16, 121–132 RSC.
- C. Mottillo and T. Friščić, Molecules, 2017, 22, 144–181 CrossRef PubMed.
- A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, E. Alvarez, E. Gutierrez-Puebla, A. Monge, A. Galindo, D. del Río and R. A. Andersen, J. Am. Chem. Soc., 2007, 129, 693–703 CrossRef CAS PubMed.
- K. Budny-Godlewski, D. Kubicki, I. Justyniak and J. Lewiński, Organometallics, 2014, 33, 5093–5096 CrossRef CAS.
- M. Kubisiak, K. Zelga, W. Bury, I. Justyniak, K. Budny-Godlewski, Z. Ochal and J. Lewiński, Chem. Sci., 2015, 6, 3102–3108 RSC.
- P. Jochmann and D. W. Stephan, Chem. Commun., 2014, 50, 8395–8397 RSC.
- For selected examples of the role of SET in organozinc chemistry, see: (a) E. Wissing, S. van der Linden, E. Rijnberg, J. Boersma, W. J. J. Smeets, A. L. Spek and G. van Koten, Organometallics, 1994, 13, 2602–2608 CrossRef CAS; (b) J. Lewiński, W. Śliwiński, M. Dranka, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2006, 45, 4826–4829 CrossRef PubMed; (c) J. Lewiński, K. Suwała, M. Kubisiak, Z. Ochal, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2008, 47, 7888–7891 CrossRef PubMed; (d) I. Dranka, M. Kubisiak, I. Justyniak, M. Lesiuk, D. Kubicki and J. Lewiński, Chem.–Eur. J., 2011, 17, 12713–12721 CrossRef CAS PubMed; (e) E. Folkertsma, S. H. Benthem, J. T. B. H. Jastrzebski, M. Lutz, M.-E. Moret and R. J. M. Klein Gebbink, Eur. J. Inorg. Chem., 2018, 1167–1175 CrossRef CAS PubMed.
- J. Lewiński, M. Dranka, W. Bury, W. Śliwiński, I. Justyniak and J. Lipkowski, J. Am. Chem. Soc., 2007, 129, 3096–3098 CrossRef PubMed.
- G. Wittig, F. J. Meyer and G. Lange, Justus Liebigs Ann. Chem., 1951, 571, 167–201 CrossRef CAS.
- P. R. Markies, G. Schat, O. S. Akkerman, F. Bickelhaupt, W. J. J. Smeets and A. L. Spek, Organometallics, 1990, 9, 2243–2247 CrossRef CAS.
- D. R. Armstrong, L. Balloch, J. J. Crawford, B. J. Fleming, L. M. Hogg, A. R. Kennedy, J. Klett, R. E. Mulvey, C. T. O'Hara, S. A. Orr and S. D. Robertson, Chem. Commun., 2012, 48, 1541–1543 RSC.
- For selected examples of TEMPO* anion coordination modes in organozinc chemistry, see: ref. 44 and 46, and (a) G. C. Forbes, A. R. Kennedy, R. E. Mulvey and P. J. A. Rodger, Chem. Commun., 2001, 1400–1401 RSC; (b) Z. Zhu, J. C. Fettinger, M. M. Olmstead and P. P. Power, Organometallics, 2009, 28, 2091–2095 CrossRef CAS; (c) J. Francos, A. R. Kennedy and C. T. O'Hara, Dalton Trans., 2016, 45, 6222–6233 RSC.
- J. L. Russ, J. Gu, K.-H. Tsai, T. Glass, J. C. Duchamp and H. C. Dorn, J. Am. Chem. Soc., 2007, 129, 7018–7027 CrossRef CAS PubMed.
- M. R. Haneline and F. P. Gabbaï, Inorg. Chem., 2005, 44, 6248–6255 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization data of new products: the NMR, EPR, IR spectra, extended PXRD diffractograms and crystallographic data. CCDC 1550886–1550888. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01396b |
| ‡ Bearing in mind that TEMPO melts at approximately 40 °C, we conducted an experiment between an equimolar mixture of Ph2Zn and TEMPO at 40 °C. According to PXRD analysis, the reaction gives a mixture of 2·TEMPO and Ph2Zn within 30 minutes, which then after 4 days at 40 °C gives 3 and PhPh in excellent yield (for details see Fig. S24). We assume that prolonged (days) grinding will lead to the same results as the simple thermal experiment described above, although as a reaction in the melted phase it should no longer be considered as mechanochemistry. |
| This journal is © The Royal Society of Chemistry 2019 |