
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTumour suppression by targeted intravenous non-viral CRISPRa using dendritic polymers†
Jessica A.
Kretzmann
 ab,
Cameron W.
Evans
a,
Colette
Moses
bc,
Anabel
Sorolla
b,
Amy L.
Kretzmann
a,
Edina
Wang
b,
Diwei
Ho
a,
Mark J.
Hackett
d,
Benjamin F.
Dessauvagie
ef,
Nicole M.
Smith
a,
Andrew D.
Redfern
f,
Charlene
Waryah
b,
Marck
Norret
a,
K. Swaminathan
Iyer
*a and
Pilar
Blancafort
*b
ab,
Cameron W.
Evans
a,
Colette
Moses
bc,
Anabel
Sorolla
b,
Amy L.
Kretzmann
a,
Edina
Wang
b,
Diwei
Ho
a,
Mark J.
Hackett
d,
Benjamin F.
Dessauvagie
ef,
Nicole M.
Smith
a,
Andrew D.
Redfern
f,
Charlene
Waryah
b,
Marck
Norret
a,
K. Swaminathan
Iyer
*a and
Pilar
Blancafort
*b
aSchool of Molecular Sciences, The University of Western Australia, 35 Stirling Hwy, Crawley, WA 6009, Australia. E-mail: swaminatha.iyer@uwa.edu.au
bHarry Perkins Institute of Medical Research, 6 Verdun St, Nedlands, WA 6009, Australia. E-mail: pilar.blancafort@uwa.edu.au
cSchool of Human Sciences, The University of Western Australia, 35 Stirling Hwy, Crawley, WA 6009, Australia
dCurtin Institute for Functional Molecules and Interfaces, Curtin Health Innovation Research Institute, Department of Chemistry, Curtin University, Bentley, WA 6845, Australia
eAnatomical Pathology, PathWest Laboratory Medicine, Fiona Stanley Hospital, Murdoch, WA, Australia
fSchool of Medicine, The University of Western Australia, Crawley, WA, Australia
First published on 27th June 2019
Abstract
Aberrant gene expression is a hallmark of cancer. Although transcription is traditionally considered ‘undruggable’, the development of CRISPR-associated protein 9 (Cas9) systems offers enormous potential to rectify cancer-associated transcriptional abnormalities in malignant cells. However delivery of this technology presents a critical challenge to overcome in order to realize clinical translation for cancer therapy. In this article we demonstrate for the first time, a fully synthetic strategy to enable CRISPR-mediated activation (CRISPRa) of tumour suppressor genes in vivo using a targeted intravenous approach. We show this via highly efficient transcriptional activation of two model tumour suppressor genes, Mammary Serine Protease Inhibitor (MASPIN, SERPINB5) and cysteine-rich 61/connective tissue growth factor/nephroblastoma-overexpressed 6 (CCN6, WISP3), in a mouse model of breast cancer. In particular, we demonstrate that targeted intravenous delivery of can be achieved using a novel nanoscale dendritic macromolecular delivery agent, with negligible toxicity and long lasting therapeutic effects, outlining a targeted effective formulation with potential to treat aggressive malignancies.
Introduction
Malignant transformations in various cancers may be induced by inappropriate expression of genes as a consequence of mutation or epigenetic alterations. The resulting complex and dynamic transcriptional dysregulation of multiple genes is central to cancer progression.1,2 There is mounting evidence that artificial transcription factors (ATFs) capable of targeted regulation of genes can constitute a therapeutic strategy to reverse these aberrant malignant transformations. Programmable DNA-binding proteins have recently emerged as an exciting platform for engineering synthetic transcription factors to enable precise transactivation of dormant tumour suppressor genes, and offer major advantages such as flexibility and ease of programming for multiple target genes, and a wider range of potential target genes, compared to delivery of traditional expression cassettes simply encoding the gene of interest.3–6 Among such established custom DNA-binding domains, zinc finger (ZF) proteins and transcription activator-like effectors (TALE) have led to the rational design of DNA binding domains but these ATFs have issues such as limited binding specificity, transcriptional activation of target genes, and still require complete re-design with each new target gene.7 Currently the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) system have emerged as the most readily scalable to facilitate transcriptional control due to its simplicity of programming. Specifically, the CRISPR-mediated gene activation, termed CRISPRa, using deactivated Cas9 (dCas9) fusion proteins to recruit transcriptional activators have enabled multiple fold enhanced activation of endogenous genes in vitro.3,8,9Initial artificial transcription factor design constituted only one effector domain such as the herpes simplex activation domain (VP16), a tetrameric repeat of VP16's minimal activation domain (VP64) or VP160 fused with dCas9.8,10 However second-generation systems such as dCas9-SunTag, VP64-p65-Rta (VPR)11 and synergistic activation mediators (SAM)12 systems have more recently been employed for more potent gene activation. These systems function effectively by recruiting multiple, mechanistically distinct transactivators at dCas9-binding sites. A major translational bottleneck in rendering the technology therapeutically relevant is the lack of an efficient delivery methodology. Indeed efficient delivery of the multiple constituents of these artificial transcription factors in primary cells has represented a major challenge both in vitro and in vivo. As evident in the case of the SAM system, concurrent delivery of three components; dCas9 fused with VP64 (dCas9-VP64), sgRNA containing two MS2 RNA aptamers (sgRNAMS2), and chimeric activator fused with MS2 coat protein (MS2-p65-HSF1), is pivotal to achieve significant activation of silenced tumour suppressor genes. To date, CRISPR/Cas9 technology has been delivered in vivo by direct injection into the treatment site,13 intraperitoneal injections,14 intratumoral injections,15 or co-delivered with viral systems,16 all of which have limited clinical applicability. Therapeutic in vivo delivery via lentiviral and adeno-associated viral (AAV) vectors is intrinsically limited by off-target toxicity concerns and limited packaging capacity.14 Although physical and mechanical means of delivery such as electroporation and membrane deformation have shown promising results in in vitro and ex vivo, there are substantial barriers to their implementation in whole animals.14,17 While significant effort has been made to develop non-viral liposome- and polymer-based delivery agents, these strategies remain hindered by low delivery efficiency.18 We recently reported a novel design strategy in which a linear copolymer backbone was used to anchor dendrons of varying generations (G1–G5) using copper-catalysed click chemistry to generate a library of transfection agents that outperformed commercially available regents in terms of biocompatibility, non-toxicity and high transfection efficiencies.19 Importantly we demonstrated that significant gene reactivation of Mammary serine Protease inhibitor (MASPIN), a silenced tumour suppressor gene, can be achieved following dendrimer-based in vitro to deliver CRISPR/dCas9 fused with VP64 transcriptional activator in MCF-7 cells.19 Since the polymeric delivery platform is amenable to multimodal functionalisation to incorporate whole body imaging agents and targeting moieties, in the current study we have endeavoured to test the hypothesis that targeted CRISPRa via intravenous delivery will result in optimal activation of tumour suppressor genes in an in vivo mouse model of breast cancer. It is important to note that both efficient gene reactivation and functional tumour cell normalization have not previously been realised in the tumour tissue in a mouse model. In particular we investigated two model tumour suppressor genes – Mammary Serine Protease Inhibitor (MASPIN, SERPINB5) and Cysteine-rich 61/Connective tissue growth factor/Nephroblastoma-overexpressed 6 (CCN6, WISP3), which are often silenced by DNA methylation and by histone methylation (H3K9me) in mesenchymal and highly metastatic cells.20,21 Downregulation of MASPIN expression has been observed in clinical studies of breast, ovarian and lung cancers, and is associated with tumour progression and poor prognosis.20,22,23CCN6 is a secreted tumour suppressor gene commonly downregulated in highly aggressive, mesenchymally enriched breast cancers, including triple negative breast cancers (TNBC) and metaplastic carcinomas.24–26
Results and discussion
In vitro activation of tumour suppressor genes MASPIN and CCN6 causes subsequent loss of tumorigenic properties
The macromolecular delivery agent composed of a linear random copolymer backbone of a hydroxyethyl methacrylate (HEMA, 1) and glycidyl methacrylate (GMA, 2), with polyamido amine (PAMAM) fifth-generation dendrons attached along the backbone via a copper-catalyzed click reaction (Schemes S1 and S2†). The agent was then reacted with heptafluorobutyric anhydride to further enhance transfection efficiency (Scheme S3†). The generation and substitution density of PAMAM dendrons were chosen from the previously reported in vitro screen to achieve highest transfection efficiency (∼80% and ∼50% in MCF-7 cells when delivering a 5.3 kb and 10.3 kb EGFP plasmid respectively, compared to Lipofectamine 2000 achieving ∼80% and ∼30% in MCF-7 cells when delivering the 5.3 kb and 10.3 kb EGFP plasmids respectively).19 Firstly, we reconfirmed the expression of MASPIN and CCN6 in normal human breast tissue. MASPIN was expressed and quantified in normal myoepithelial cells, while CCN6 was expressed by normal breast luminal epithelial cells, and expression quantified in luminal epithelial cells (Fig. S1†), both consistent with previous literature.27,28 Next, the efficacy of the polymeric platform to enable non-viral activation of two tumour suppressor genes MASPIN or CCN6 via transfection with a range of CRISPRa systems was investigated in human breast adenocarcinoma cells (MCF-7) and human non-small lung carcinoma cells (H157), where both MASPIN and CCN6 are significantly downregulated (Fig. 1 and S4†). To achieve maximum upregulation, sgRNAs targeting the proximal promoter of either MASPIN or CCN6 were delivered along with dCas9 fused to either VP64 or VPR activation domains, with and without the SAM complex (Fig. 1b and d for MASPIN and CCN6 respectively). Significant upregulation of MASPIN (∼67-fold) and CCN6 (∼19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500-fold) was observed in vitro 72 h post-transfection in MCF-7 cells with the combination of CRISPR/dCas9-VPR/SAM and CRISPR/dCas9-VPR, respectively. Off-target effects of CRISPR-based technology resulting from sgRNA binding at non-cognate sites have posed concerns for its clinical application.11 For each gene, we selected 20 predicted off-target sites with the fewest sequence mismatches to the cognate recognition sequence and closest proximity to a promoter or enhancer region with regulatory potential, and assessed mRNA expression levels by qRT-PCR (Fig. 1e and S5†). We found no significant regulation of the potential off-target genes at 48 h post-transfection.
500-fold) was observed in vitro 72 h post-transfection in MCF-7 cells with the combination of CRISPR/dCas9-VPR/SAM and CRISPR/dCas9-VPR, respectively. Off-target effects of CRISPR-based technology resulting from sgRNA binding at non-cognate sites have posed concerns for its clinical application.11 For each gene, we selected 20 predicted off-target sites with the fewest sequence mismatches to the cognate recognition sequence and closest proximity to a promoter or enhancer region with regulatory potential, and assessed mRNA expression levels by qRT-PCR (Fig. 1e and S5†). We found no significant regulation of the potential off-target genes at 48 h post-transfection.
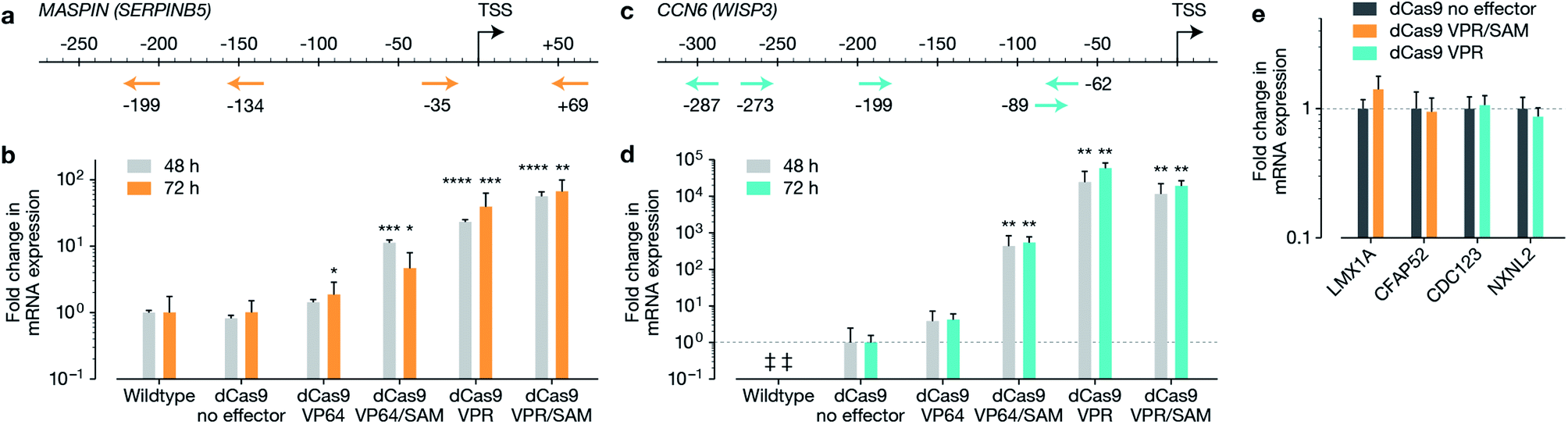 | ||
| Fig. 1 Activation of tumour suppressor genes, MASPIN and CCN6 in MCF-7 cells. Guide design for each of the target genes is demonstrated in (a) and (c) for MASPIN and CCN6, respectively. Activation of MASPIN and CCN6 n MCF-7 cells demonstrated in (b) and (d) respectively, using various combinations of effectors fused to CRISPR/dCas9. Analysis of potential off-target sites (e) demonstrated no significant alteration in expression at 48 h. Transfection was completed using non-targeted polymer formulation 7F. The symbol ‘‡’ is used when signal could not be detected by qRT-PCR. Student's t-test was performed against control for (b), (d) and (e). All data in graphs are represented as mean ± SEM (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001). | ||
We next interrogated whether target gene re-expression was associated with phenotypic tumour cell reprograming, including a decrease in cell migration, colony formation and proliferation after activation of both MASPIN and CCN6 (Fig. 2). Since silencing of MASPIN and CCN6 in cancer cell lines has previously been associated with increased cellular migration, we first investigated the impact of increased expression of each of these genes on the in vitro migration potential of MCF-7 cells using a Boyden chamber with serum as the chemoattractant, and found a significant decrease in the number of migrating cells when CCN6 was reactivated (Fig. 2a). To assess tumour formation potential, cells transfected for the reactivation of MASPIN and CCN6 were tested in a soft agar assay, which measures anchorage-independent growth, and is a standard assay for detecting malignant transformation of cells in in vitro conditions.29 After 3 weeks of colony growth, MASPIN and CCN6 activation conditions resulted in significantly smaller colonies (Fig. 2b and e) and fewer colonies (Fig. 2c and f) than wild-type MCF-7 cells or cells transfected with control plasmid CRSIPR/dCas9 no effector (representative images in Fig. S6†). Additionally, cell viability and proliferation were monitored 48–120 h post transfection. At 48 h there were significantly fewer cells in conditions expressing high levels of both MASPIN and CCN6 (Fig. 2d and g for MASPIN and CCN6 respectively).
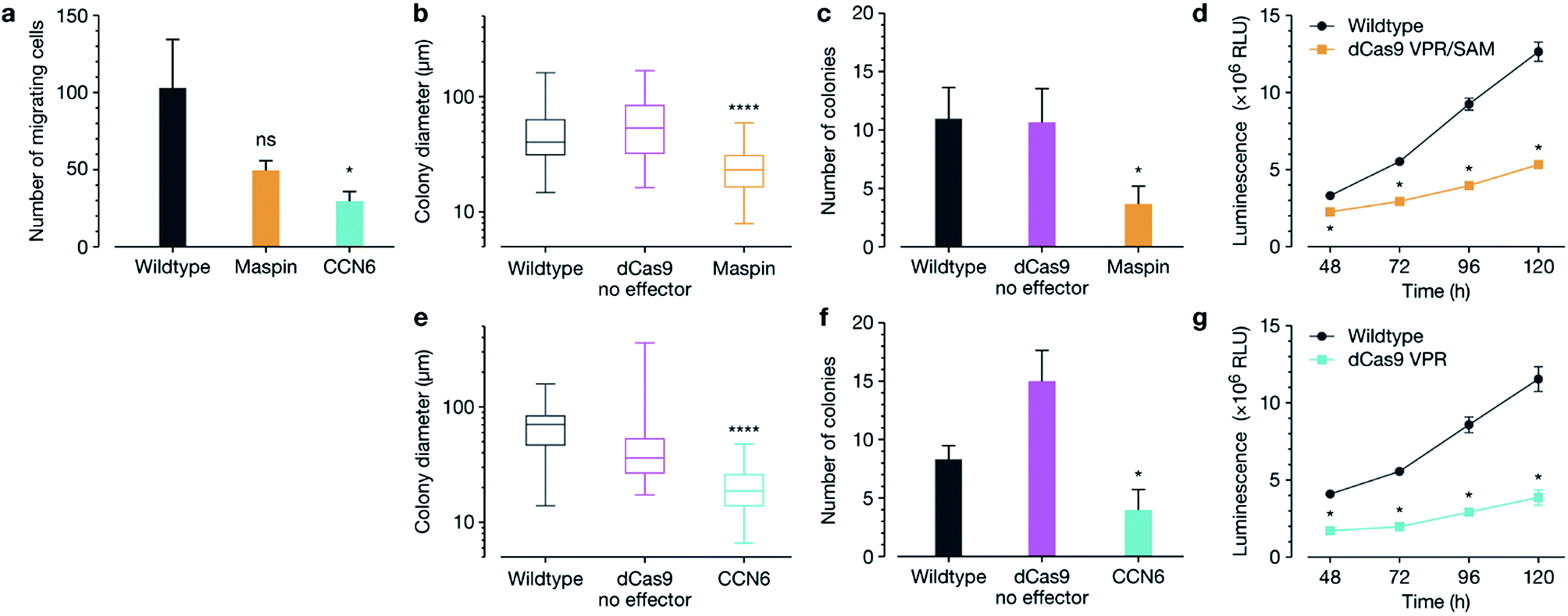 | ||
| Fig. 2 Loss of migratory and proliferation properties in MCF-7 cells after re-activation of MASPIN and CCN6. (a) Significantly decreased migratory potential of MCF-7 cells 48 h after transfection for MASPIN and CCN6 activation, demonstrated using a transwell migration assay. Wildtype and treated MCF-7 cells were seeded for soft agar assays and colony diameter and number assessed after 3 weeks of growth, as shown for upregulation of MASPIN (b and c) and CCN6 (e and f). Activation of both MASPIN and CCN6 resulted in significantly fewer and smaller colonies. Cell proliferation was significantly lower in MCF-7 cells after activation of MASPIN (d) and CCN6 (g) 48–120 h post-transfection. Transfection was completed using non-targeted polymer formulation 7F. Statistical analysis was completed against wildtype cells as a control in all cases, where Student's t-test was performed against control for (a–c, e and f). In (d and g) multiple t-tests were performed against control at the appropriate time point. Box plots show center line as median, box limits as upper and lower quartiles, and whiskers given minimum and maximum values (*p ≤ 0.05, ****p ≤ 0.0001, ns p > 0.05). | ||
Cyclic RGD functionalised polymer enables targeted in vivo delivery of plasmid DNA
Following the in vitro characterisation, polymer formulations were next modified for targeted in vivo delivery by the attachment of polyethylene glycol (PEG) and either cyanine-7 (Cy7) fluorescent dye (non-targeted, Fig. 3b, 12), or cyclic RGD peptide with Cy7 attached (cRDG-Cy7, targeted, Fig. 3c, 16). Reaction schematics can be found in Schemes S4–S6.† PEG decoration confers ‘stealth’ properties to the resulting polyplex by shielding the majority of the positive charge, allowing for longer circulation time and minimizing non-specific uptake.30 A cyclic RGD peptide was incorporated to enable homing of the polyplexes to the tumour through binding with αvβ-family integrins that are overexpressed on the surface of many cancer cells, including MCF-7, and on the endothelial cells of the tumour vasculature.31–34 While RGD is typically used to target overexpression of αvβ3 integrins, MCF-7 cells express ανβ5 and ανβ6 integrins, which are further overexpressed in the stimulated tumour microenvironment.32,35 Polyplex size and charge before and after PEG-cRGD-Cy7 attachment was assessed using dynamic light scattering and zeta potential measurements in phosphate buffered saline solution (PBS). Effective binding of pDNA was maintained, with an accompanying increase in polyplex size and decrease in charge with attachment of PEG-cRGD-Cy7, as expected (Fig. S7†), however in vitro transfection efficiency decreased significantly with the introduction of PEG-cRGD (Fig. S8†), a common phenomenon associated with PEG.36 Fluorescent spectra of the Cy7-modified polymers was confirmed in Fig. S9.† | ||
| Fig. 3 Design and in vivo validation of cRGD-targeted polymeric design for increased MCF-7 tumour uptake. (a) Whole animal imaging showing distribution of non-targeted (b) and targeted (c) Cy7 labelled polyplexes 24 h after i.v. injection, tumour is outlined in yellow. Polymer surface was decorated with PEG12 chains and either Cy7 ((b), non-targeted formulation, 12) or cRGD-Cy7 ((c), targeted formulation, 16). (d) cRGD-Cy7 targeted polymer formulation 16 demonstrated increased tumour uptake 72 h after i.v. injection (N = 3), measured by normalized photon flux of tumours ex vivo. cRGD-Cy7 targeted formulation 16 demonstrated significant kidney clearance from 48 h to 72 h as measured by ex vivo measurements ((e), total N = 7). (f) Brain and tumour tissue sections labelled with Hoechst (blue) and anti-mCherry (red). mCherry plasmid was delivered and successfully expressed in tumour tissue by cRGD-targeted polymer 16. (g) Flow cytometry of harvested tissue demonstrating Cy7+ and mCherry+ cell populations 72 h after single i.v. injection delivered using cRGD-targeted formulation 16. mCherry expression was localized to the tumour, with low expression in off-target tissue. Data was analyzed using Student t-test comparing targeted formulation against the non-targeted as a control in the appropriate tissue (*p < 0.05, **p < 0.01). | ||
To confirm targeting, in vivo BALB/c nude mice implanted with a MCF-7 xenograft stably expressing luciferase in the right flank received a single intravenous injection of either the non-targeted or targeted formulation bound with mCherry-encoding plasmid. Distribution of each formulation was monitored over 72 h using whole body live animal imaging, which indicated faster clearance of the targeted formulation through the kidney, and preferential homing in the tumour (Fig. 3a, d and S10–S13†). Mice were sacrificed at 72 h, with additional animals sacrificed at 24 and 48 h post-injection, and both Cy7 and mCherry fluorescence was quantified using ex vivo imaging (Fig. 3d, S11 and S12†), confocal microscopy (Fig. 3f) and flow cytometry (Fig. 3g and S13†). Overall the targeted formulation 16 demonstrated significant kidney clearance over 72 h (Fig. 3e), high in vivo transfection efficiency of ∼20% in the tumour, and low plasmid expression in off-target tissues, as shown by confocal imaging (Fig. 3f) and flow cytometry (Fig. 3g). FTIR analysis of the kidneys (Fig. S14†) demonstrated nucleic acid content increasing over time following injection, with a maximum at 48 h, before decreasing at 72 h. The FTIR spectra taken at the edge of the nucleic acid influx showed a large reduction in phosphate content compared to the ribose content, suggestive of DNA degradation.
In vivo re-activation of tumour suppressor genes suppresses tumour growth in MCF-7 xenograft in BALB/c mice
Following confirmation of tumour targeting and uptake in the BALB/c MCF-7 luciferase xenograft model with the targeted polymer formulation 15 (without Cy7 fluorescent tag), we proceeded to deliver CRISPR-based transactivation plasmids for MASPIN and CCN6, and investigate the functional outcome in that model. The experimental timeline is illustrated in Fig. 4a. Mice received a total of five intravenous injections (one injection every 72 h), from days 7 to 19, with each dosage approximately 0.3 mg kg−1 of plasmid. This dosage is significantly lower to that reported for similar studies which have used 0.75 mg kg−1 (ref. 37) and 6 mg kg−1.38 Mice received polymer delivering either non-coding pcDNA, CRISPR/dCas9 no effector, CRISPR/dCas9-VPR/SAM targeting MASPIN activation or CRISPR/dCas9-VPR targeting CCN6 activation (N = 11 per group). For all treatment groups, mice (N = 3 per group, randomly selected) were humanely sacrificed 72 h after the last injection (day 23) to assess tissue for tumour histology and toxicity with haematoxylin and eosin staining. Kidneys demonstrated normal histological characteristics across all treatment groups. Livers demonstrated mild non-specific inflammation in 2/3 MASPIN and CCN6 treatment mice and 3/3 CRISPR control mice but not pcDNA animals. Similarly, mild non-specific lung inflammation was seen in 1/3 MASPIN and CCN6 treatment mice and 3/3 CRISPR control mice, but not in pcDNA animals (Table S7, Fig. S18 and S19†). One CCN6-treated animal demonstrated an acute lung injury with capillaritis. However, as no other mice had exhibited this effect it is uncertain whether this was a result of the treatment (Fig. S20†). To further assess histology and toxicity in liver and lungs, additional mice (N = 3 per group, randomly selected) were humanely sacrificed at day 39, nearly 3 weeks after treatment had ceased (Table S7†). Livers demonstrating mild non-specific inflammation decreased to 1/3 animals for pcDNA, MASPIN and CRISPR control mice and 3/3 CCN6-treated mice. Lungs demonstrated mild non-significant inflammation for 3/3 pcDNA mice and 2/3 CCN6 mice, while CRISPR control and MASPIN-treated mice demonstrated normal histological characteristics (Table S8†). Mouse body weight and tumour volume were measured every 2 days and are shown in Fig. S15.† No significant difference in animal body weight was observed. By day 46, median tumour volumes were significantly lower for MASPIN and CCN6 CRISPRa treatment groups (155 ± 72 and 79.9 ± 10 mm3 respectively) than for the no effector control mice (320 ± 93 mm3). Animals were imaged on days 25, 32, 39 and 46 and tumour size assessed by bioluminescent intensity (Fig. 4b, S16 and S17†). Tumour progression was monitored and normalised to the animals for pcDNA, MASPIN and CRISPR control mice, and 3/3 CCN6 treatment mice. Lungs demonstrated mild non-significant inflammation for 3/3 pcDNA mice, and 2/3 CCN6 mice, while CRISPR control and MASPIN bioluminescent reading taken on day 25, where tumour size was not significantly different across groups (Fig. 4d–f and S16†). At day 32, mice treated with activation of CCN6 had tumour growth arrest (Fig. 4d), followed by tumour regression first evident by day 39 in some mice, and more prominent by day 46 (Fig. 4e and f respectively). Mice treated with MASPIN activation demonstrated tumour growth arrest by day 39 (Fig. 4e), with mixed effects evident by day 46 ranging from regression (0.44-fold change in size), to growth (2.8-fold change in size) over the course of the week. In the case of MASPIN the overall tumour suppression is associated not only with the level of expression but also but also the level of subcellular distribution in cancer cells (cytoplasmic, nuclear or both cytoplasmic-nuclear expression).39 This would imply that in order to have a therapeutic effect upon activation in the overall tumour the majority of the cancer cells have to not only have increased expression but concomitant subcellular localisation profiles. Importantly our result is similar to previous studies where in vivo MASPIN activation with zinc fingers ATFs demonstrated tumour growth arrest, however in that case the expression of zinc finger proteins was induced in genetically engineered cells using viral vectors.40 Whilst in the case of CCN6 which is a secreted extracellular matrix protein,41,42 the overall tumour suppression is achievable even if only select regions of the tumour received the treatment for CCN6 activation: the effect can spread throughout the whole tumour making CCN6 the more potent tumour suppressor target. It is noteworthy that the timelines for anti-tumorigenic onset observed in the case of CCN6 activation is in accordance with previously reported studies. This is associated with the activation of downstream cell signalling pathways implicated in the regulation of nuclear cycling and/or apoptotic functions, angiogenesis and possible tumour suppression by binding IGF-like ligands.43 In the present case all remaining mice were sacrificed at day 46 for histological assessment of tumours (photographed Fig. 4c and histology Fig. 4g). Histology of all tumours demonstrated grade 3 MCF-7 tumours with no change of tumour grade (tubule formation, nuclear pleomorphism, mitotic rate) over the course of the treatment. However, CCN6-induced tumours also exhibited a large areas of tumour regression with malignant tissue replaced by benign sclerosis (Fig. 4g and S21†). | ||
| Fig. 4 Activation of tumour suppressor genes in vivo by targeted polymeric delivery of CRISPR/dCas9 variants. (a) MCF-7 luciferase tumours were inoculated at day 0 by subcutaneous injection into the right flank of BALB/c nude mice (N = 11 per group). Mice received a treatment regime of one i.v. injection every 72 h, for a total of 5 injections between day 7 and day 19. Mice were euthanized at day 23 for histological assessment (N = 3 per group), and remaining mice were imaged on days 25, 32 and 39 for tumour burden assessment (N = 8 per group), more mice were sacrificed for histological assessment on day 39 after imaging (N = 3 per group). Final imaging for tumour burden was conducted on day 46, and all remaining mice were sacrificed for histological assessment. (b) Bioluminescence images of mice treated with either dCas9 no effector (control) or dCas9-VPR for the activation of CCN6 from days 25 to 46. (c) Representative ex vivo MCF-7 luciferase tumours excised at day 46 of experiment. (d–f) Tumour growth by luciferase bioluminescence shown as a fold change in size comparing days 25 vs. 32 (d), 25 vs. 39 (e) and 25 vs. 46 (f) respectively. At day 39 MASPIN- and CCN6-treated tumours demonstrated significant tumour growth arrest compared to dCas9 no effector control. At day 46 significant tumour repression was observed in CCN6 tumours. Each point represents an individual animal, while colored points represent animals which remained for the entire experiment. (g) Hematoxylin and eosin stained tumour tissues demonstrating grade 3 MCF-7 tumours for dCas9 no effector and MASPIN, and regressed tumour area for CCN6. Data was analyzed with a Mann–Whitney U test, against the control (*p ≤ 0.05, **p ≤ 0.01, ns p > 0.05). | ||
Animals were sacrificed at days 23, 39, and 46 for immunofluorescence (IF) assessment of processes occurring in the tissue at different stages. At day 23 animals treated with the activation of MASPIN and CCN6 demonstrated expression of the CRISPR/dCas9-VPR protein in ∼25% of cells (Fig. 5a–d). Tissue sections obtained from animals treated for the activation of MASPIN and CCN6 demonstrated significantly greater presence of the CRISPR/dCas9 protein than tissue obtained from control pcDNA animals. MASPIN and CCN6 treatment groups demonstrated similar transfection efficiencies, which lead to significantly higher MASPIN and CCN6 protein expression in the tumours of the treated animals compared to the control tissue (Fig. 5m–p). Expression of the CRISPR/dCas9 plasmid was further monitored in off-target tissues that demonstrated relatively high uptake in the biodistribution study, including lungs, liver and kidneys. No significant expression was observed (Fig. S22†). It is important to note that this does not necessarily equate to no off-target gene regulation. In this study it is not possible to quantify changes in gene expression in off-target tissue due insufficient homology between the human sgRNAs designed for CCN6 and MASPIN and the mouse genome to achieve effective binding and transcriptional regulation, with the vast majority of sgRNAs having more than 3 mismatches (Tables S8, S9 and Fig. S24†). Future studies will involve the implementation of syngeneic mouse models to fully address this aspect. Cell proliferation and apoptosis was monitored at day 39 (Fig. S23†). Both MASPIN and CCN6-treated tumours showed a significant decrease in proliferation compared to the no effector control group. At day 46, tumours in the no effector control group were still proliferating at a significantly higher rate than that in treated tissue, as assessed by IF staining for the Ki67 proliferation marker (Fig. 5e–h). In addition to inhibition of cell proliferation, tumours treated by activation of CCN6 also displayed apoptosis in approximately 8% of cells at day 46 compared to 1% in control tissue (Fig. 5i–k), as assessed by IF. This observation is consistent with previous in vitro and in vivo studies, which have demonstrated a significant increase in apoptosis with increased CCN6 expression.42 Therefore, the activation of CCN6 resulted in long-term tumour suppression, with effects ongoing four weeks after treatment had ceased.
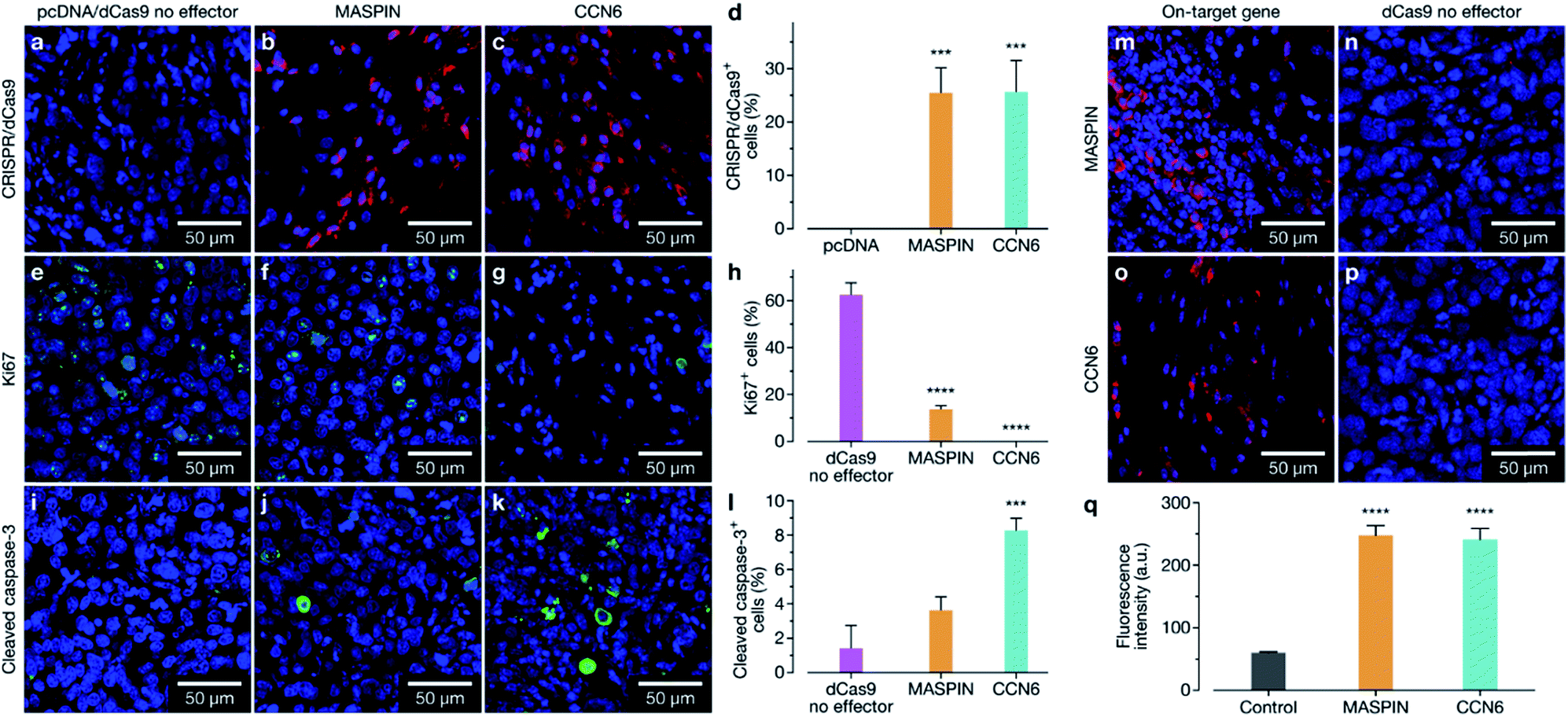 | ||
| Fig. 5 Immunofluorescence staining of MCF-7 luciferase tumour sections. (a–c) Tumour sections taken from day 23 were stained with an anti-CRISPR/Cas9 antibody, to assess expression of the CRISPR/dCas9 within the cells. (d) Quantification of CRISPR/dCas9-positive cells; tumours which received CRISPR/dCas9-VPR/SAM and CRISPR/dCas9-VPR targeting MASPIN and CCN6 respectively demonstrated a significant increase in the number of CRISPR/dCas9-positive cells compared to the pcDNA control. Tumour sections taken from day 46 were stained for proliferation marker Ki67 (e–g) and apoptosis marker cleaved caspase-3 (i–k). MASPIN and CCN6 upregulation in tumours caused significant decreases in proliferation (h), while upregulation of CCN6 also demonstrated significant levels of apoptosis (l) when compared to the dCas9 no effector control. Tumour sections from day 23 were stained using anti-MASPIN (m) and anti-CCN6 (o) antibodies, and demonstrated significant upregulation of the target protein (q) when compared to tissue which received the dCas9 no effector control (n and p). Data was analyzed using a standard one-way ANOVA against the control, with multiple comparisons, (**p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, ns p > 0.05). | ||
Conclusions
In summary, we have demonstrated for the first time that targeted intravenous CRISPRa using complete synthetic delivery strategy is indeed achievable in a mouse model of breast cancer. In particular we have shown that targeted intravenous delivery of CRISPR/dCas9 platforms for the activation of silenced tumour suppressor genes can be achieved efficiently, with no significant observable toxicity from both the synthetic non-viral vector formulation, and the delivery of the CRISPR plasmid DNA. Non-viral methods for the delivery of CRISPR systems, as demonstrated, hold promise for intravenous therapy, with the benefit of engineering multimodality into the delivery platform to enable both targeted delivery and imaging, and at the same time overcoming the bottlenecks associated with viral delivery. We believe this study provides a strong platform for further research involving the development of medically translatable CRISPR-based technology, allowing the field to expand through the use of fully non-viral delivery tools. Future work will focus on developing a deeper understanding of the potential of this synthetic approach in the delivery to other cancer models, delivery of alternative cargos such as RNA or protein, gene regulation in models of other diseases and most importantly to fully unravel the longevity of treatment using CRISPR based therapeutic technology.Experimental
Materials
All chemicals were purchased from Sigma Aldrich (Australia) and used without further purification unless otherwise stated. Cyanine-7 NHS ester (Cy7-NHS) was purchased from Lumiprobe. Peptides (H-DfC(1230)RG-cyclic, 99% purity 14 and H-DfC(51)RG-cyclic, 97% purity 13) were custom designed and synthesized by Mimotopes (Melbourne, Australia). SM(PEG)12 (Thermo Scientific, 100 mg) was diluted by adding 360 μL of dry DMSO, as per manufacturer's protocol, and stored under argon at −20 °C.Polymer synthesis
Detailed polymer synthesis and characterisation can be found in ‘Polymer Synthesis and Characterisation’ in ESI.† P(HEMA0.84-ran-GMA0.16) copolymer was synthesized as previously described, given in ESI Scheme S1.†19 Briefly, inhibitors for HEMA 1 and GMA 2 were removed and the monomers were reacted via an atom-transfer radical polymerization method to afford the final copolymer 3. The copolymer was then functionalized with azido groups as described previously, given in ESI Scheme S2,† to afford product 4.19Propargyl functionalized polyamido amine (PAMAM) dendrons were synthesized via a method adapted by Lee et al.44 and Lin et al.,45 to afford 4.5 generation dendron 5.
Azido-functionalized polymer 4 and propargyl functionalized PAMAM dendron 5 were then reacted via an azide–alkyne copper catalyzed click reaction and the generation was finalized to generation 5 with ethylene diamine to give the final product 7. Method was adapted from those previously described, and is given in ESI Scheme S2.†19,46
Partial fluorination of surface primary amines was achieved as described by Wang et al.,47 by reaction with heptafluorobutyric anhydride, as depicted in ESI Scheme S3,† giving product 7F. 1H NMR and elemental analysis can be found in Fig. S3a and Table S5†.
Decoration of surface amines for in vivo studies for non-targeted and targeted formulations, in addition to custom peptides, are given in ESI Schemes S4–S6.† Briefly, functionalisation was achieved through reaction of a bifunctional SM(PEG)12 linker, where the NHS ester reacted with the surface amines of the polymer, and the malemide functionality was used to react with the thiol moiety on either the Cy7 (product 12), or RGD peptide (product 15 and product 16, with and without Cy7 respectively), for non-targeted and targeted formulations respectively. 1H NMR demonstrating PEG decoration can be found in Fig. S3b.†
Cell culture and transfection
Detailed cell culture and transfection protocols can be found in ‘In vitro experiments’ in ESI,† including plasmid details and sgRNA sequences (Table S1†).MCF-7 and MCF-7 luciferase (human breast adenocarcinoma cell line, ATCC, and MCF-7 modified in-house to express luciferase) were cultured in Minimum Essential Medium α (MEM α, Gibco) supplemented with 0.15% sodium bicarbonate, 10% FBS and 1× GlutaMAX. H157 (human lung adenocarcinoma cell, ATCC) were cultured in RPMI1640 supplemented with 10% FBS and 1× GlutaMAX. All cell lines were grown in a humidified incubator at 37 °C with 5% CO2. Cells were seeded at appropriate densities 16–24 h prior to transfection.
To perform transfections, polymer solution and pDNA were diluted to appropriate concentrations in Opti-MEM, at a final N/P ratio of 5:1 for a total of 1 μg of pDNA in a 12-well standard plate. Solutions were mixed thoroughly and left to incubate at r.t. for 30 min prior to addition to cultured cells. Cell media was removed, cells were washed with PBS, and Opti-MEM was transferred to each well. Transfections were performed in non-serum conditions, for 48 h or 72 h for RNA extraction. Cells were harvested at 48 h for downstream functional assays.
RNA extraction and PCR
Detailed RNA extraction and PCR protocols, including all primer and data analysis details, can be found in ‘In vitro experiments’ in ESI.†Total RNA was extracted using Trizol reagent (Invitrogen), as per the manufacturer's protocol. Extracted total RNA was converted to cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). RT-qPCR was carried out using TaqMan Fast Universal PCR Master Mix (Applied Biosystems), which GAPDH as housekeeping control. Additional off-target analysis was achieved using RT2 SYBR Green ROX qPCR Mastermix. Primer details can be found in ESI Tables S3 and S2.†
Functional assays
Detailed protocols for cell viability, migration and soft agar assays can be found in ‘In vitro experiments’ in ESI.†Cells were transfected 48 h prior to being collected and counted for seeding for the following assays:
Cell viability assay was carried out using CellTiter-Glo reagent (Promega) according to the manufacturer's protocol. Cells were seeded at 2.5 × 104 cells per well in an opaque 96-well flat bottom tissue culture plate.
Comparative migration assay was performed as per manufacturer's protocol (CytoSelect Migration Assay, 8 μm, colorimetric format, Cell Biolabs, Inc.). Cells were seeded at 0.75 × 106 cells in 500 μL in upper chamber without serum. Full growth media was added to the chamber below to encourage cell migration through the polycarbonate membrane.
Soft agar assays were performed by seeding 5000 cells per well on a 6 well plate with growth medium, on top of a solidified layer of 0.5% agarose in growth medium, and before a 0.35% agarose/growth medium mixture. Cells were fed every 3 days with growth medium.
Ethics statements
All experiments were conducted in accordance with the Code of Practice for the Care and Use of Animals and were approved by the University of Western Australia Animal Ethics Committee (RA/3/100/1336).Normal human breast tissues were taken from the de-identified control tissue archive routinely used by PathWest to provide validatory controls as part of routine clinical practice.
In vivo experiments
Detailed protocols for in vivo experiments, including full tissue collection, staining and preparation, and microscopy and imaging protocols can be found in ‘BALB/c nude mice in vivo experiments’ in ESI.† Primary antibody details and dilution protocols can be found in ESI Table S5.†MCF-7 luciferase (MCF-7-luci) cells were cultured routinely as described above. For tumour inoculation, cells were collected and resuspended in 50:50 mixture of MEM α and Matrigel (Corning BD Bioscience), with 1 μg estrogen (β-estradiol 17-valerate, Sigma Aldrich). Animals were injected in right flank with cell suspension (5 × 106 cells per animal, 60 μL). Tumours were maintained by subcutaneous injections of estrogen (20 μL, 50 ng μL−1 in peanut oil) located near the periphery of the visible tumour every 48 h.
For biodistribution and functional in vivo experiments, MCF-7-luci tumours were inoculated and maintained as above. 7 days after tumour inoculation, mice were randomly divided into groups and injected.
For biodistribution experiment, animals were injected by a single intravenous injection containing either the targeted 16 or non-targeted 12 formulations and mCherry plasmid (10 μg plasmid/injection, N/P = 5 for each polymer formulation). Animals (N = 3 per group) were anesthetized (isoflurane) and imaged using CRi Maestro 2 (wavelengths, conditions) at 24, 48 and 72 h timepoints, and culled at 72 h for ex vivo imaging of tissue. Additional animals (N = 2 per group, per timepoint) were culled at 24 and 48 h timepoints. Tissue was analysed ex vivo by Cri Maestro 2 imaging, flow cytometry using BD FACS CantoII flow cytometer, or snap frozen in OCT for cryosectioning and confocal imaging, and FTIR analysis.
For functional in vivo experiment, intravenous injections were given to mice (N = 11 per group, except for CCN6 which had N = 9) every 72 h, starting 7 days after tumour inoculation. Mice were injected with targeted polymer formulation (14, 56.4 μg) and plasmid DNA (5 μg). Mice were treated with either pcDNA, dCas9 no effector, dCas9-VPR/SAM (MASPIN) or dCas9-VPR (CCN6) formulations. Mice received a total of 5 injections. Tumour progression was followed by caliper measurements every 48 h, and bioluminescence imaging using Caliper IVIS Lumina II imaging system, on days 25, 32, 39 and 46 of the study. Mice were sacrificed at day 23, 39 and 46 (N = 3 per timepoint) for tissue collection and analysis. Tissue was collected and preserved in 4% paraformaldehyde in PBS, and later embedded in wax. Sections were cut at 5 μm and underwent standard hematoxylin and eosin staining, or were stained for immunofluorescence analysis.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was funded by the Australian Research Council (ARC), the National Health and Medical Research Council (NHMRC) of Australia, the Cancer Council of Western Australia, and the National Institutes of Health. The authors would like to acknowledge the facilities and the technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments. JAK acknowledges Cancer Council Western Australia for a PhD Top Up Scholarship. AS is supported by a postdoctoral fellowship from the National Breast Cancer Foundation (PF-15-001). We would like to acknowledge Dr Janice H. C. Plani-Lam for the production of the MCF-7 luciferase cell line.References
- F. J. Sánchez-Rivera and T. Jacks, Nat. Rev. Cancer, 2015, 15, 387–395 CrossRef PubMed.
- S. Stolzenburg, A. S. Beltran, T. Swift-Scanlan, A. G. Rivenbark, R. Rashwan and P. Blancafort, Oncogene, 2015, 34, 5427–5435 CrossRef CAS PubMed.
- B. Garcia-Bloj, C. Moses, A. Sgro, J. Plani-Lam, M. Arooj, C. Duffy, S. Thiruvengadam, A. Sorolla, R. Rashwan, R. L. Mancera, A. Leisewitz, T. Swift-Scanlan, A. H. Corvalan and P. Blancafort, Oncotarget, 2016, 7, 60535–60554 CrossRef PubMed.
- R. Barrangou and J. A. Doudna, Nat. Biotechnol., 2016, 34, 933–941 CrossRef CAS PubMed.
- T. Gaj, C. A. Gersbach and C. F. Barbas III, Trends Biotechnol., 2013, 31, 397–405 CrossRef CAS PubMed.
- E. D. Papadakis, S. A. Nicklin, A. H. Baker and S. J. White, Curr. Gene Ther., 2004, 4, 89–113 CrossRef CAS PubMed.
- F. Farzadfard, S. D. Perli and T. K. Lu, ACS Synth. Biol., 2013, 2, 604–613 CrossRef CAS PubMed.
- P. Perez-Pinera, D. D. Kocak, C. M. Vockley, A. F. Adler, A. M. Kabadi, L. R. Polstein, P. I. Thakore, K. A. Glass, D. G. Ousterout, K. W. Leong, F. Guilak, G. E. Crawford, T. E. Reddy and C. A. Gersbach, Nat. Methods, 2013, 10, 973–976 CrossRef CAS.
- M. F. L. Russa and L. S. Qi, Mol. Cell. Biol., 2015, 35, 3800–3809 CrossRef.
- C. A. Giménez, M. Ielpi, A. Mutto, L. Grosembacher, P. Argibay and F. Pereyra-Bonnet, Gene Ther., 2016, 23, 543–547 CrossRef PubMed.
- A. Chavez, J. Scheiman, S. Vora, B. W. Pruitt, M. Tuttle, E. P. R. Iyer, S. Lin, S. Kiani, C. D. Guzman, D. J. Wiegand, D. Ter-Ovanesyan, J. L. Braff, N. Davidsohn, B. E. Housden, N. Perrimon, R. Weiss, J. Aach, J. J. Collins and G. M. Church, Nat. Methods, 2015, 12, 326–328 CrossRef CAS PubMed.
- H. Zhang, Y. Ma, Y. Xie, Y. An, Y. Huang, Z. Zhu and C. J. Yang, Sci. Rep., 2015, 5, 10099 CrossRef CAS PubMed.
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z.-Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS PubMed.
- L. Li, L. Song, X. Liu, X. Yang, X. Li, T. He, N. Wang, S. Yang, C. Yu, T. Yin, Y. Wen, Z. He, X. Wei, W. Su, Q. Wu, S. Yao, C. Gong and Y. Wei, ACS Nano, 2017, 11, 95–111 CrossRef CAS PubMed.
- H.-X. Wang, Z. Song, Y.-H. Lao, X. Xu, J. Gong, D. Cheng, S. Chakraborty, J. S. Park, M. Li, D. Huang, L. Yin, J. Cheng and K. W. Leong, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4903–4908 CrossRef CAS PubMed.
- H. Yin, C.-Q. Song, J. R. Dorkin, L. J. Zhu, Y. Li, Q. Wu, A. Park, J. Yang, S. Suresh, A. Bizhanova, A. Gupta, M. F. Bolukbasi, S. Walsh, R. L. Bogorad, G. Gao, Z. Weng, Y. Dong, V. Koteliansky, S. A. Wolfe, R. Langer, W. Xue and D. G. Anderson, Nat. Biotechnol., 2016, 34, 328–333 CrossRef CAS PubMed.
- X. Han, Z. Liu, M. chan Jo, K. Zhang, Y. Li, Z. Zeng, N. Li, Y. Zu and L. Qin, Sci. Adv., 2015, 1, e1500454 CrossRef PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- J. A. Kretzmann, D. Ho, C. W. Evans, J. H. C. Plani-Lam, B. Garcia-Bloj, A. E. Mohamed, M. L. O'Mara, E. Ford, D. E. K. Tan, R. Lister, P. Blancafort, M. Norret and K. S. Iyer, Chem. Sci., 2017, 8, 2923–2930 RSC.
- N. Maass, M. Teffner, F. Rösel, R. Pawaresch, W. Jonat, K. Nagasaki and P. Rudolph, J. Pathol., 2001, 195, 321–326 CrossRef CAS PubMed.
- R. Kikuchi, H. Tsuda, Y. Kanai, T. Kasamatsu, K. Sengoku, S. Hirohashi, J. Inazawa and I. Imoto, Cancer Res., 2007, 67, 7095–7105 CrossRef CAS PubMed.
- N. Maass, T. Hojo, F. Rösel, T. Ikeda, W. Jonat and K. Nagasaki, Clin. Biochem., 2001, 34, 303–307 CrossRef CAS PubMed.
- M. Nakagawa, H. Katakura, M. Adachi, K. Takenaka, K. Yanagihara, Y. Otake, H. Wada and F. Tanaka, Annals of Surgical Oncology, 2006, 13, 1517–1523 CrossRef PubMed.
- Y. Zhang, K. A. Toy and C. G. Kleer, Mod. Pathol., 2012, 25, 178–184 CrossRef CAS PubMed.
- J.-I. Jun and L. F. Lau, Nat. Rev. Drug Discovery, 2011, 10, 945–963 CrossRef CAS PubMed.
- E. E. Martin, W. Huang, T. Anwar, C. Arellano-Garcia, B. Burman, J.-L. Guan, M. E. Gonzalez and C. G. Kleer, Oncogene, 2017, 36, 2275–2285 CrossRef CAS PubMed.
- J. S. Reis-Filho, F. Milanezi, P. Silva and F. C. Schmitt, Pathol., Res. Pract., 2001, 197, 817–821 CrossRef CAS PubMed.
- W. Huang, Y. Zhang, S. Varambally, A. M. Chinnaiyan, M. Banerjee, S. D. Merajver and C. G. Kleer, Am. J. Pathol., 2008, 172, 893–904 CrossRef CAS.
- S. Stolzenburg, M. G. Rots, A. S. Beltran, A. G. Rivenbark, X. Yuan, H. Qian, B. D. Strahl and P. Blancafort, Nucleic Acids Res., 2012, 40, 6725–6740 CrossRef CAS PubMed.
- B. Pelaz, P. del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernández, J. M. de la Fuente, G. U. Nienhaus and W. J. Parak, ACS Nano, 2015, 9, 6996–7008 CrossRef CAS PubMed.
- J. A. Menendez, L. Vellon, I. Mehmi, P. K. Teng, D. W. Griggs and R. Lupu, Oncogene, 2004, 24, 761–779 CrossRef PubMed.
- A. Taherian, X. Li, Y. Liu and T. A. Haas, BMC Cancer, 2011, 11, 293 CrossRef CAS PubMed.
- E. Ruoslahti, Annu. Rev. Cell Dev. Biol., 1996, 12, 697–715 CrossRef CAS PubMed.
- C. Rüegg, A. Yilmaz, G. Bieler, J. Bamat, P. Chaubert and F. J. Lejeune, J. Nat. Med., 1998, 4, 408, DOI:10.1038/nm0498-408.
- D. Katoh, K. Nagaharu, N. Shimojo, N. Hanamura, M. Yamashita, Y. Kozuka, K. Imanaka-Yoshida and T. Yoshida, Oncogenesis, 2013, 2, e65 CrossRef CAS PubMed.
- L. Y. Song, Q. F. Ahkong, Q. Rong, Z. Wang, S. Ansell, M. J. Hope and B. Mui, Biochim. Biophys. Acta, Biomembr., 2002, 1558, 1–13 CrossRef CAS.
- C. Liang, F. Li, L. Wang, Z.-K. Zhang, C. Wang, B. He, J. Li, Z. Chen, A. B. Shaikh, J. Liu, X. Wu, S. Peng, L. Dang, B. Guo, X. He, D. W. T. Au, C. Lu, H. Zhu, B.-T. Zhang, A. Lu and G. Zhang, Biomaterials, 2017, 147, 68–85 CrossRef CAS PubMed.
- K. Lee, M. Conboy, H. M. Park, F. Jiang, H. J. Kim, M. A. Dewitt, V. A. Mackley, K. Chang, A. Rao, C. Skinner, T. Shobha, M. Mehdipour, H. Liu, W. Huang, F. Lan, N. L. Bray, S. Li, J. E. Corn, K. Kataoka, J. A. Doudna, I. Conboy and N. Murthy, Nat. Biomed. Eng., 2017, 1, 889–901 CrossRef CAS.
- S. H. Dzinic, A. Kaplun, X. Li, M. Bernardo, Y. Meng, I. Dean, D. Krass, P. Stemmer, N. Shin, F. Lonardo and S. Sheng, PLoS One, 2013, 8, e74502 CrossRef.
- A. S. Beltran and P. Blancafort, Epigenetics, 2011, 6, 224–235 CrossRef CAS PubMed.
- C. G. Kleer, Y. Zhang, Q. Pan and S. D. Merajver, Neoplasia, 2004, 6, 179–185 CrossRef CAS PubMed.
- C. G. Kleer, Y. Zhang, Q. Pan, K. L. van Golen, Z.-F. Wu, D. Livant and S. D. Merajver, Oncogene, 2002, 21, 3172–3180 CrossRef CAS PubMed.
- G. Lorenzatti, W. Huang, A. Pal, A. M. Cabanillas and C. G. Kleer, J. Cell Sci., 2011, 124, 1752–1758 CrossRef CAS PubMed.
- J. W. Lee, B.-K. Kim, H. J. Kim, S. C. Han, W. S. Shin and S.-H. Jin, Macromolecules, 2006, 39, 2418–2422 CrossRef CAS.
- Y.-J. Lin, B.-K. Tsai, C.-J. Tu, J. Jeng and C.-C. Chu, Tetrahedron, 2013, 69, 1801–1807 CrossRef CAS.
- P. Zhao, Y. Yan, X. Feng, L. Liu, C. Wang and Y. Chen, Polymer, 2012, 53, 1992–2000 CrossRef CAS.
- M. Wang, H. Liu, L. Li and Y. Cheng, Nat. Commun., 2014, 5, 3053 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01432b |
| This journal is © The Royal Society of Chemistry 2019 |