DOI:
10.1039/C9SC01460H
(Edge Article)
Chem. Sci., 2019,
10, 5258-5266
Sequence information transfer using covalent template-directed synthesis†
Received
25th March 2019
, Accepted 22nd April 2019
First published on 26th April 2019
Abstract
Template-directed synthesis is the biological method for the assembly of oligomers of defined sequence, providing the molecular basis for replication and the process of evolution. To apply analogous processes to synthetic oligomeric molecules, methods are required for the transfer of sequence information from a template to a daughter strand. We show that covalent template-directed synthesis is a promising approach for the molecular replication of sequence information in synthetic oligomers. Two monomer building blocks were synthesized: a phenol monomer and a benzoic acid monomer, each bearing an alkyne and an azide for oligomerization via copper catalyzed azide alkyne cycloaddition (CuAAC) reactions. Stepwise synthesis was used to prepare oligomers, where information was encoded as the sequence of phenol (P) and benzoic acid (A) units. Ester base-pairing was used to attach monomers to a mixed sequence template, and CuAAC was used to zip up the backbone. Hydrolysis of the ester base-pairs gave back the starting template and the sequence complementary copy. When the AAP trimer was used as the template, the complementary sequence PPA was obtained as the major product, with a small amount of scrambling resulting in PAP as a side-product. This covalent base-pairing strategy represents a general approach that can be implemented in different formats for the replication of sequence information in synthetic oligomers.
Introduction
Template-directed synthesis is the method used in biology for production of oligomers with defined sequence. The information encoded in DNA templates is copied into DNA duplicates, RNA transcripts and translated proteins, and these information transfer processes are the molecular basis for the evolution of living systems.1–3 The development of template-directed methods for synthesising non-natural oligomers of defined sequence would open the way for exploitation of directed evolution to identify synthetic oligomer sequences with useful properties, Orgel's Holy Grail.4 Nucleic acids are the only materials currently capable of sequence information transfer, so the use of evolution to explore chemical space is limited to systems that interface with DNA.5–8 Here we describe a template-directed approach to synthetic oligomers that uses covalent base-pairing as the basis for sequence information transfer between parent and daughter strands. This chemical templating process represents a first step towards the application of evolution to tackle the vast chemical space constituted by synthetic oligomer sequences.
The information transfer that takes place on nucleic acid templates is based on selective H-bonding interactions that hold the correct monomer unit in place for attachment to a growing oligomer chain. Non-covalent templating is well-established in the field of supramolecular chemistry and has been widely applied in the synthesis of macrocycles9–12 and mechanically interlocked molecules.13–18 Although non-covalent templates have been used for homo-oligomerisation reactions,19–24 template-directed synthesis of mixed sequence oligomers remains exclusively the domain of nucleic acids.25–27 Some of the reasons are outlined in Fig. 1. Ideally, the monomers would bind to the template, and then covalent bond formation in the ZIP step would give the complementary sequence as the product. However, when the base-pairing interactions are reversible, there are additional equilibria that compete with assembly of the key pre-ZIP intermediate: incomplete binding of monomers to the template, intramolecular folding of the template, and formation of a stable duplex that limits the availability of template due to product inhibition. Non-enzymatic experiments using nucleic acid templates show that even using DNA, it is difficult to achieve the process shown in Fig. 1.28–30 In biology, all of the competing equilibria are prevented by complex enzymatic machinery that controls each step of the process to attach the correct monomers one by one.31–33
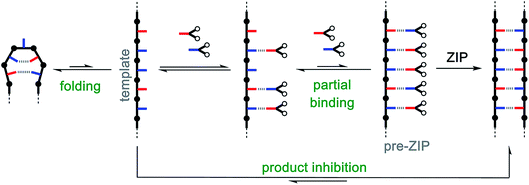 |
| | Fig. 1 Non-covalent template-directed synthesis of mixed sequence oligomers. The red and blue bars represent two different bases that form a base-pair via non-covalent interactions. Reversible binding of monomer building blocks to the template followed by a zipping up process covalently couples adjacent monomers to yield a duplex with a product oligomer that is a complementary copy of the template. Competing equilibria that interfere with formation of the key pre-ZIP complex are highlighted in green. | |
Approach
Fig. 2 illustrates an alternative chemical approach to assembly of the pre-ZIP intermediate that avoids the competing equilibria shown in Fig. 1. In this case, the monomers are covalently attached to the template using kinetically stable bonds that can be cleaved again after the ZIP step, regenerating the starting template along with the templated product. The key to the success of the ZIP step in Fig. 2 is that the reactions on the template are intramolecular and hence more favourable than competing intermolecular reactions, which can be suppressed by operating at high dilution. Covalent template-directed synthesis is relatively unexplored compared with non-covalent approaches. There are some reports on the synthesis of macrocycles and mechanically interlocked molecules by covalent templating.34–41 A cored dendrimer has been prepared by using a porphyrin as a covalent template for cross-linking the external arms of the dendrimer.42,43 Covalent templating has also been used for the synthesis of polydisperse homo-polymers suggesting that the key ZIP step in Fig. 2 is applicable to the synthesis of macromolecules.44–46 However, implementation of the scheme in Fig. 2 requires efficient orthogonal chemistry for selective attachment of two different types of monomer, zipping up the backbone, and cleavage of the product duplex. Here we describe one promising solution and demonstrate the utility by using a mixed sequence trimer as a template for synthesis of the complementary sequence.
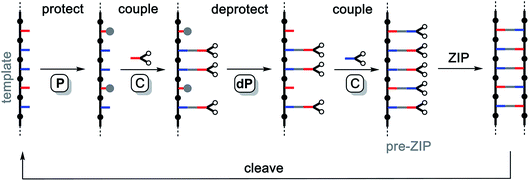 |
| | Fig. 2 Covalent template-directed synthesis of mixed sequence oligomers. The red and blue bars represent bases that pair via a kinetically inert covalent bond. The pre-ZIP intermediate is assembled using a sequence of irreversible chemical reactions. Selective protection of the red bases (with grey balls, P), followed by covalent coupling of the blue bases with red monomers (C), deprotection of the red bases (dP), and covalent coupling of the red bases with blue monomers (C) yields the key intermediate for the ZIP step with no competing processes. The product oligomer and starting template are recovered by cleaving the covalent bonds connecting the base-pairs in the duplex. | |
Results
Covalent base-pairing chemistry
Fig. 3 shows how a covalent base-pairing system can be implemented based on formation of an ester between a phenol (red base) and a benzoic acid (blue base). The base-pair attachment sequence shown in Fig. 2 can be achieved by selective protection of the phenol bases (P), ester coupling of the benzoic acid bases with phenol monomers (C), deprotection of the phenol bases (dP) and coupling of the phenol bases with benzoic acid monomers (C). Hydrolysis of the esters recovers the starting template and a complementary copy.
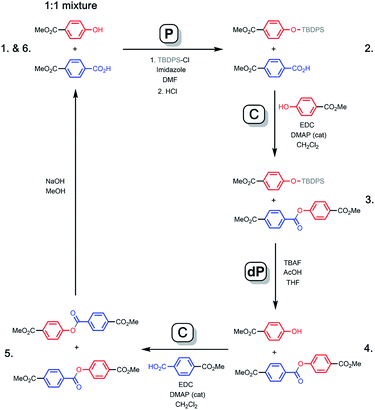 |
| | Fig. 3 Formation of an ester between a phenol (red) and benzoic acid (blue) gives a covalent base-pair. Starting from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two template bases, sequential formation of two base-pairs via the P–C–dP–C sequence is illustrated. Cleavage by hydrolysis gives the two template bases and two product bases, which are identical to the original template bases. The 1–6 numbering corresponds to the 1H NMR spectra shown in Fig. 4. :1 mixture of two template bases, sequential formation of two base-pairs via the P–C–dP–C sequence is illustrated. Cleavage by hydrolysis gives the two template bases and two product bases, which are identical to the original template bases. The 1–6 numbering corresponds to the 1H NMR spectra shown in Fig. 4. | |
In order to demonstrate the viability of this covalent base-pair attachment and cleavage chemistry, we carried out the complete reaction cycle on the model system shown in Fig. 3. In this case, the two different bases that represent the template (a phenol and a benzoic acid) are present as a mixture of two separate molecules rather than connected as part of an oligomer. Fig. 4 shows 1H NMR spectra of the crude reaction mixtures obtained from the sequence of reactions in Fig. 3. Selective formation and then cleavage of the esters of both bases were achieved quantitatively and the only purification required between steps was an aqueous work-up. Ester base-pairing chemistry therefore appears to be an ideal candidate for the development of robust quantitative covalent template-directed synthesis methodologies.
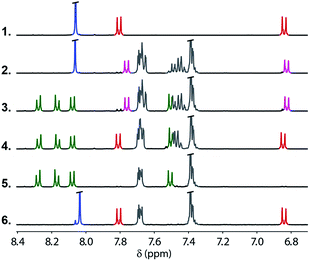 |
| | Fig. 4 Partial 400 MHz 1H NMR spectra in DMSO-d6 at 298 K of the crude reaction mixtures for the ester base-pair attachment and cleavage cycle shown in Fig. 1. Spectrum 1: the starting template phenol (red) and benzoic acid (blue). Spectrum 2: selective protection of the phenol (pink) in the presence of the benzoic acid (blue). Spectrum 3: formation of the first base-pair (green) in the presence of the protected phenol (pink). Spectrum 4: deprotection of the phenol (red) in the presence of the first base-pair (green). Spectrum 5: formation of the second base-pair (green). Spectrum 6: products of the cleavage reaction, the phenol (red) and benzoic acid (blue). There is a small blue signal due to partial hydrolysis of the spectator methyl esters, and the grey signals are TBDPS residues not removed by aqueous work-up. | |
Backbone chemistry
The copper catalysed azide–alkyne cycloaddition (CuAAC) reaction is high yielding and compatible with a wide range of chemical functionality,47,48 so this reaction was selected for the ZIP step in Fig. 2. Fig. 5(a) shows the structures of monomer building blocks, equipped with an azide, an alkyne and either a phenol (P) or a benzoic acid (A), and a trimer template (AAP) that can be assembled from these monomers through sequential CuAAC and TMS deprotection reactions. The product of the ZIP step in Fig. 2 is a duplex, and it is therefore essential that the conformational properties of the backbone formed in CuAAC oligomerisation reactions are compatible with this polymacrocyclic architecture. The molecular mechanics model of the AAP·APP duplex shown in Fig. 5(b) indicates that the ring strain is low: 18–35 kJ mol−1 per macrocyclisation, which is comparable to cyclopentane (26 kJ mol−1).49,50 The CuAAC backbone shown in Fig. 5 is directional (we write the sequence in the alkyne to azide direction), so two isomeric duplexes can be formed in the ZIP step, parallel and antiparallel. Fig. 5(b) shows the antiparallel AAP·APP duplex, which was calculated to be 5 kJ mol−1 more stable than the corresponding parallel duplex, so selectivity in the ZIP step can be anticipated (see ESI† for details).
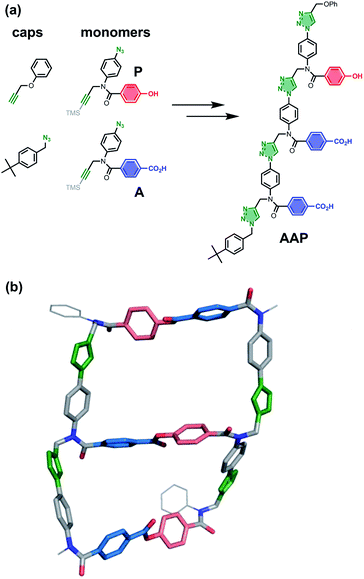 |
| | Fig. 5 (a) Monomer building blocks (A and P) and monofunctional caps for assembly of mixed sequence oligomers by sequential CuAAC coupling and TMS deprotection reactions: the AAP trimer is shown. (b) Molecular mechanics structure of the antiparallel AAP·APP duplex (lowest energy structure from conformational searches). | |
Synthesis of the monomers
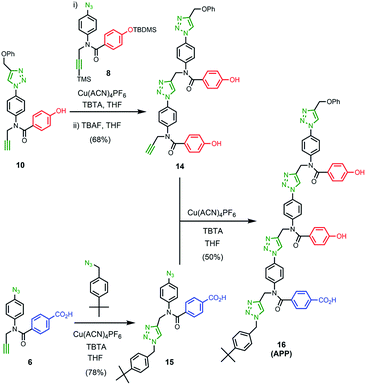
Scheme 1 shows the synthesis of the two monomer building blocks. p-Azidoaniline 2 was prepared according to a procedure described in the literature,51 and subsequent amide coupling with mono-methyl terephthalate gave 3 in excellent yield. Alkylation of 3 using TMS-protected propargyl bromide and sodium hydride gave a mixture of 4 and the TMS-deprotected alkyne 5. TBAF deprotection of 4 and hydrolysis of 5 using lithium hydroxide afforded the benzoic acid monomer 6 in excellent yield. For the phenol monomer, aniline 2 was coupled with TBDMS-protected 4-hydroxybenzoic acid to give the amide 7 in excellent yield. Subsequent alkylation with TMS-protected propargyl bromide provided 8, and TBAF-mediated deprotection of both silyl protecting groups gave phenol monomer 9 in excellent yield.
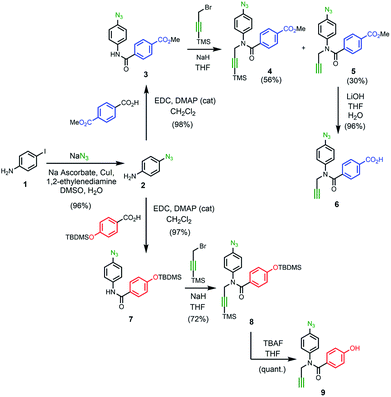 |
| | Scheme 1 | |
Synthesis of the template
The two monomer building blocks were used to prepare a template oligomer through sequential CuAAC and TMS deprotection reactions. Scheme 2 shows the synthesis of a mixed sequence trimer. In order to prevent reaction with the ends of the template chain in the ZIP step, monofunctional chain terminating groups (phenyl propargyl ether and p-tert-butylbenzylazide) were used to cap the terminal azide and alkyne. The protected phenol monomer 8 was first coupled with phenyl propargyl ether using a CuAAC reaction. TBAF-mediated deprotection of the product gave the capped phenol monomer 10 in good yield. CuAAC coupling of 10 with 4 gave access to mixed 2-mer 11 in excellent yield. CuAAC coupling of 11 with 4 afforded 3-mer 12. CuAAC coupling of 12 with p-tert-butylbenzylazide followed by LiOH-mediated basic hydrolysis yielded template 13 in good yield. We will use the shorthand AAP for this acid–acid-phenol template, writing the sequence in the alkyne to azide direction starting at the t-butyl benzyl terminus.
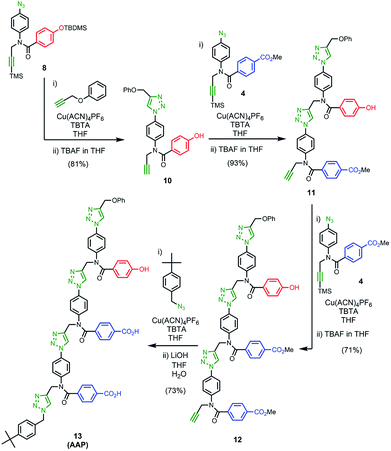 |
| | Scheme 2 | |
Oligomer synthesis on a trimer template
The AAP trimer was used as a template for the synthesis of a sequence-complementary copy using the ester base-pairing chemistry shown in Fig. 3 and CuAAC backbone chemistry for the ZIP step. The two different monomer units were loaded onto the template using the protection-coupling-deprotection-coupling reaction sequence shown in Fig. 6.
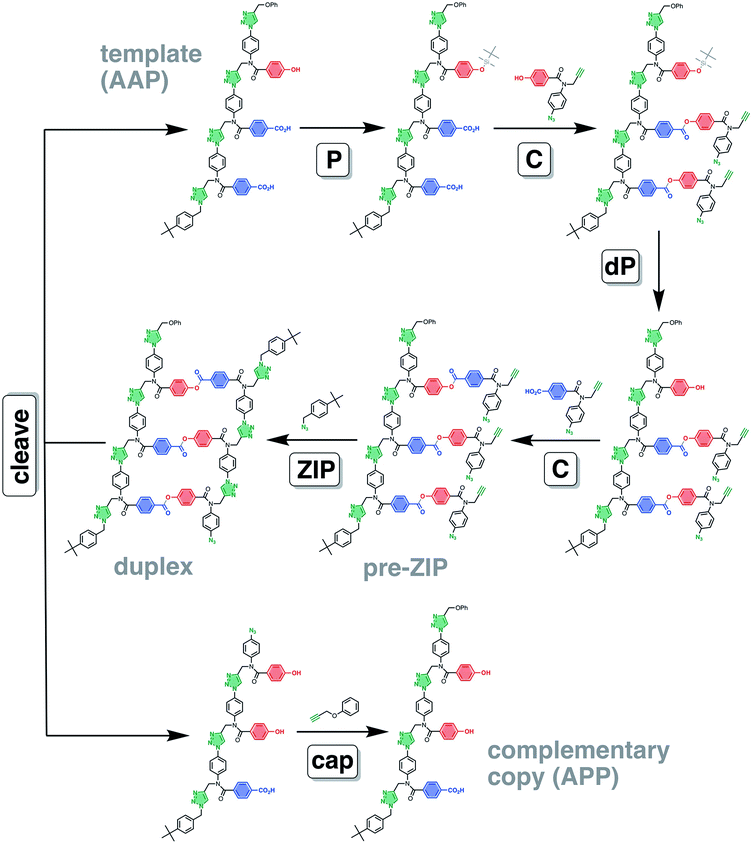 |
| | Fig. 6 Covalent template-directed synthesis of a mixed sequence trimer. Ester base-pair chemistry was used to load two different monomer units onto the AAP template with the P–C–dP–C reaction sequence. The resulting pre-ZIP intermediate was subjected to a CuAAC reaction to give the duplex. Hydrolysis of the ester base-pairs in the cleave step regenerated the template, and capping of the terminal azide of the other product provided the complementary copy APP. The antiparallel duplex is shown as the product of the ZIP step, but the parallel duplex leading to PPA as the complementary copy is also possible. Conditions: P TBDMS-Cl, imidazole, then HCl; C EDC, DMAP; dP TBAF; ZIP Cu(CH3CN)4PF6, TBTA; cleave LiOH; cap Cu(CH3CN)4PF6, TBTA. | |
Fig. 7 shows the corresponding UPLC traces of the crude reaction mixtures obtained in each step of the base-pair attachment sequence. In each case, quantitative conversion of starting material to product was achieved with aqueous work-up as the only purification required. Fig. 7(a) and (b) show that the TBDMS protection step cleanly converted the template into the protected phenol. Fig. 7(c) shows the crude reaction mixture from the EDC coupling of the exposed benzoic acids on the template with an excess of the phenol monomer. Base-pair formation proceeded quantitatively, and the only other species present was the excess of the phenol monomer. The phenol group on the template was deprotected cleanly using TBAF, and the excess of phenol monomer used in the coupling step was the only impurity carried through in this step (Fig. 7(d)). The second base-pair coupling reaction to load the benzoic acid monomer onto the template gave quantitative conversion to the pre-ZIP intermediate. The only other species present in the crude reaction mixture shown in Fig. 7(e) is the ester formed between benzoic acid monomer and the excess of the phenol monomer carried through from the previous steps. This side product was easily removed by chromatography to obtain the pre-ZIP intermediate in high purity and 63% isolated yield (Fig. 8(a)).
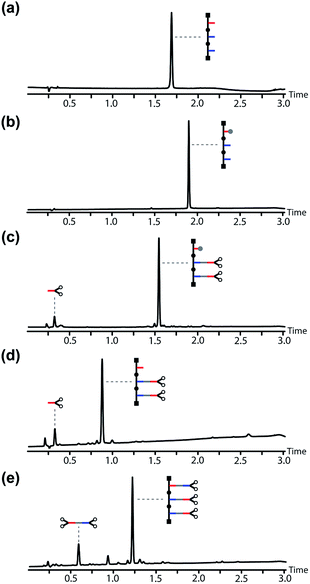 |
| | Fig. 7 UPLC traces of crude reaction mixtures obtained after aqueous work-up in the base-pair attachment sequence using the AAP template: (a) the starting template, and the crude products obtained after (b) TBDMS protection, P, (c) EDC coupling with the phenol monomer, C, (d) TBAF deprotection, dP, (e) EDC coupling with the benzoic monomer, C. Conditions: C18 column at 40 °C using a gradient of water/formic acid (0.1%) and CH3CN/formic acid (0.1%). The gradients used in (a) and (b) were different from those used in (c–e). | |
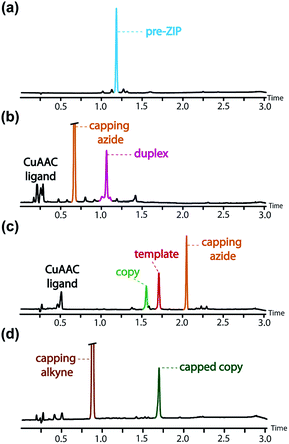 |
| | Fig. 8 UPLC traces of crude reaction mixtures obtained after aqueous work-up in conversion of the pre-ZIP intermediate to the templated product: (a) the starting pre-ZIP intermediate and the crude products obtained after (b) the CuAAC ZIP step, (c) LiOH base-pair cleavage, (d) CuAAC capping of the copy strand. Conditions: C18 column at 40 °C using a gradient of water/formic acid (0.1%) and CH3CN/formic acid (0.1%). The capping azide appears at different retention times in (b) and (c), because the gradient used in (a) and (b) was different from that used in (c) and (d). | |
The ZIP step to oligomerise the monomers on the template was carried out using copper(I) catalysis under dilute conditions to minimize competing intermolecular reactions. An azide chain terminator was also added to the reaction mixture to prevent oligomerisation of the product duplex through the unreacted terminal alkyne and azide groups. Fig. 8(b) shows the UPLC trace of the crude reaction mixture obtained after aqueous work-up, which apart from the reagents, contained a single major species with a molecular weight that corresponds to the capped AAP·APP duplex shown in Fig. 6. The crude reaction mixture was then subject to lithium hydroxide hydrolysis to cleave the ester base-pairs, which cleanly converted the duplex into the template and copy strands (Fig. 8(c)). The two products were separated by flash chromatography (see ESI†), and the terminal azide group in the copy strand was then capped with phenyl propargyl ether in a quantitative CuAAC reaction to give the final templated product (Fig. 8(d) shows the crude reaction mixture obtained from the final capping step). In summary, the complete reaction sequence shown in Fig. 6 can be achieved with very high efficiency and almost quantitative conversion in each of the seven steps.
Sequencing the products of templated oligomerisation
In order to demonstrate successful sequence information transfer in the covalent template-directed synthesis cycle shown in Fig. 6, a method for sequencing the products is required. The MS–MS methods used in peptide sequencing failed for these triazole oligomers, which did not fragment cleanly.52,53 Sequencing was achieved using a combination of mass spectrometry, NMR spectroscopy and synthesis. The mass spectra shown in Fig. 9(a) are consistent with recovery of the AAP template strand and the sequence-complementary copy APP shown in Fig. 6 as the only products. The two bases have different molecular weights, so the presence of sequences containing different numbers of A and P units can be ruled out. The 1H NMR spectrum of the recovered template is identical to the spectrum of the starting material, apart from traces of the CuAAC ligand, which were not removed in the purification step (Fig. 9(b)). The template strand is therefore stable to the conditions of the templating cycle and can be recovered to be re-used in subsequent cycles.
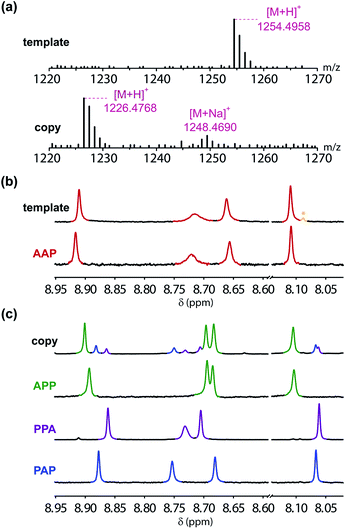 |
| | Fig. 9 Sequencing of the products. (a) HRMS of the recovered template and the templated copy. (b) Triazole region of 500 MHz 1H NMR spectra in DMSO-d6 at 298 K of the recovered template (traces of the CuAAC ligand are marked in yellow with an asterisk) and the starting AAP template. (c) Triazole region of 500 MHz 1H NMR spectra in DMSO-d6 at 298 K of the templated copy and samples of all possible sequences containing one A and two P units, which were each synthesized independently. | |
The 1H NMR spectrum of the copy strand is more complicated (Fig. 9(c)). Although there is one major product (green signals), there are traces of two other species (blue and purple signals). The mass spectrum of this sample indicates that all three species contain one A and two P units, so the products must be the three isomeric sequences: APP, PPA and PAP. Pure samples of these three oligomers were obtained by direct synthesis using a similar sequence of CuAAC and TMS deprotection reactions used to prepare the template oligomer AAP. The synthesis of APP and PAP exploited intermediates 10 and 11 used in the synthesis of the original template (Schemes 3 and 5). The synthesis of PPA used the phenol dimer 20, which was fortuitously obtained from spontaneous azide–alkyne cycloaddition of the monomer building block 9 upon storage (Scheme 4).
 |
| | Scheme 3 | |
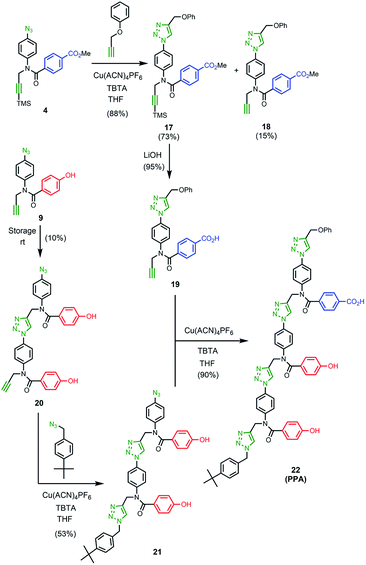 |
| | Scheme 4 | |
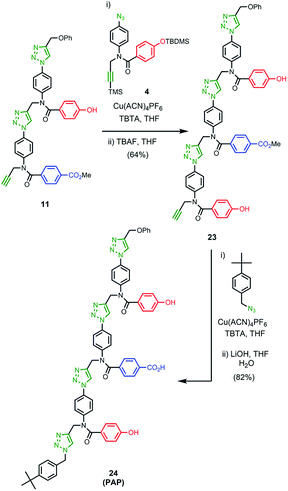 |
| | Scheme 5 | |
Fig. 9(c) compares the 1H NMR spectra of the independently synthesised samples of APP, PPA and PAP with the 1H NMR spectrum of the copy sample obtained from the template-directed synthesis reaction. The major product (72%) from the templating reaction is APP, the sequence-complementary copy of the AAP template resulting from the antiparallel duplex shown in Fig. 6. One of the minor products (11%) is PPA, the sequence-complementary copy of the AAP template that would result from formation of the parallel duplex in the ZIP step. The other minor species (17%) is the scrambled sequence PAP, which must result from intramolecular coupling between the two terminal monomer units on the template. We conclude that the templating cycle proceeds through the antiparallel duplex to give a single major product, which is the sequence complement of the template. The fidelity of the information transfer process is degraded somewhat by a side-reaction that involves long range coupling between monomer units that are not attached at adjacent sites on the template.
Conclusions
Biological synthesis of oligomers of defined sequence is based on non-covalent template-directed synthesis. We have developed an alternative chemical method based on covalent template-directed synthesis. The covalent base-pairing methodology described here provides a mechanism for quantitative attachment of monomers to a template without the competing equilibria that occur with reversible base-pairing interactions.54 The result is a robust method for the transfer of sequence information between parent and daughter strands. A mixed sequence trimer was successfully used to template the synthesis of a sequence-complementary copy. Minor quantities of a scrambled sequence were also observed, but rather than compromising the fidelity of the information transfer process, scrambled sequences could serve as a useful source of mutation, if these systems are developed for evolutionary searching of chemical structure space.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the ERC (ERC-2012-AdG 320539-duplex) and the Herchel Smith Fund for funding.
Notes and references
-
R. R. Sinden, DNA Structure and Function, Academic Press, 1994 Search PubMed.
-
A. J. F. Griffiths, W. M. Gelbart, J. H. Miller and R. C. Lewontin, Modern Genetic Analysis, Freeman, 1999 Search PubMed.
- K. Chen and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5618 CrossRef CAS.
- L. E. Orgel, Acc. Chem. Res., 1995, 28, 109 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818 CrossRef CAS PubMed.
- J. W. Chin, Nature, 2017, 550, 53 CrossRef CAS.
- D. L. Usanov, A. I. Chan, J. P. Maianti and D. R. Liu, Nat. Chem., 2018, 10, 704 CrossRef CAS PubMed.
- J. C. W. Willis and J. W. Chin, Nat. Chem., 2018, 10, 831 CrossRef CAS PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017 CrossRef CAS.
- S. Anderson, H. L. Anderson and J. K. M. Sanders, Acc. Chem. Res., 1993, 26, 469 CrossRef CAS.
- R. L. E. Furlan, S. Otto and J. K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4801 CrossRef CAS PubMed.
- P. S. Bols and H. L. Anderson, Acc. Chem. Res., 2018, 51, 2083 CrossRef CAS.
- C. O. Dietrich-Buchecker and J.-P. Sauvage, Tetrahedron Lett., 1983, 24, 5095 CrossRef CAS.
- P. R. Ashton, T. T. Goodnow, A. E. Kaifer, M. V. Reddington, M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vincent and D. J. Williams, Angew. Chem., Int. Ed., 1989, 28, 1396 CrossRef.
- C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303 CrossRef CAS.
- F. B. L. Cougnon and J. K. M. Sanders, Acc. Chem. Res., 2012, 45, 2211 CrossRef CAS.
- R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434 CrossRef CAS.
- J.-F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Chem. Soc. Rev., 2013, 42, 1700 RSC.
- X. Li, Z.-Y. J. Zhan, R. Knipe and D. G. Lynn, J. Am. Chem. Soc., 2002, 124, 746 CrossRef CAS PubMed.
- P. K. Lo and H. F. Sleiman, J. Am. Chem. Soc., 2009, 131, 4182 CrossRef CAS PubMed.
- S. Anderson, H. L. Anderson and J. K. M. Sanders, Angew. Chem., Int. Ed., 1992, 31, 907 CrossRef.
- S. Anderson, H. L. Anderson and J. K. M. Sanders, J. Chem. Soc., Perkin Trans. 1, 1995, 18, 2247 RSC.
- A. Cnossen, C. Roche and H. L. Anderson, Chem. Commun., 2017, 53, 10410 RSC.
- N. Kamonsutthipaijit and H. L. Anderson, Chem. Sci., 2017, 8, 2729 RSC.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef.
- D. K. O'Flaherty, N. P. Kamat, F. N. Mirza, L. Li, N. Prywes and J. W. Szostak, J. Am. Chem. Soc., 2018, 140, 5171 CrossRef PubMed.
- Y. Ura, J. M. Beierle, L. J. Leman, L. E. Orgel and M. R. Ghadiri, Science, 2009, 325, 73 CrossRef CAS PubMed.
- S. S. Mansy, J. P. Schrum, M. Krishnamurthy, S. Tobé, D. A. Treco and J. W. Szostak, Nature, 2008, 454, 122 CrossRef CAS PubMed.
- S. Zhang, J. C. Blain, D. Zielinska, S. M. Gryaznov and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17732 CrossRef CAS PubMed.
- B. D. Heuberger, A. Pal, F. Del Frate, V. V. Topkar and J. W. Szostak, J. Am. Chem. Soc., 2015, 137, 2769 CrossRef CAS PubMed.
- C. M. Joyce and S. J. Benkovic, Biochemistry, 2004, 43, 14317 CrossRef CAS PubMed.
- A. J. Berdis, Chem. Rev., 2009, 109, 2862 CrossRef CAS PubMed.
- E. Nudler, Annu. Rev. Biochem., 2009, 78, 335 CrossRef CAS.
- G. Schill and A. Luttringhaus, Angew. Chem., Int. Ed., 1964, 3, 546 CrossRef.
- W. Moneta, P. Baret and J. Pierre, J. Chem. Soc., Chem. Commun., 1985, 899 RSC.
- S. Höger, A.-D. Meckenstock and H. Pellen, J. Org. Chem., 1997, 62, 4556 CrossRef.
- S. Höger and A.-D. Meckenstock, Chem.–Eur. J., 1999, 5, 1686 CrossRef.
- K. Hiratani, M. Kaneyama, Y. Nagawa, E. Koyama and M. Kanesato, J. Am. Chem. Soc., 2004, 126, 13568 CrossRef CAS PubMed.
- H. Kawai, T. Umehara, K. Fujiwara, T. Tsuji and T. Suzuki, Angew. Chem., Int. Ed., 2006, 45, 4281 CrossRef CAS.
- C. Schweez, P. Shushkov, S. Grimme and S. Höger, Angew. Chem., Int. Ed., 2016, 55, 3328 CrossRef CAS.
- L. Steemers, M. J. Wanner, M. Lutz, H. Hiemstra and J. H. Van Maarseveen, Nat. Commun., 2017, 8, 15392 CrossRef.
- S. C. Zimmerman, M. S. Wendland, N. A. Rakow, I. Zharov and K. S. Suslick, Nature, 2002, 418, 399 CrossRef CAS.
- S. C. Zimmerman, I. Zharov, M. S. Wendland, N. A. Rakow and K. S. Suslick, J. Am. Chem. Soc., 2003, 125, 13504 CrossRef CAS PubMed.
- N.-T. Lin, S.-Y. Lin, S.-L. Lee, C.-h. Chen, C.-H. Hsu, L. P. Hwang, Z.-Y. Xie, C.-H. Chen, S.-L. Huang and T.-Y. Luh, Angew. Chem., Int. Ed., 2007, 46, 4481 CrossRef CAS PubMed.
- Y.-Z. Ke, S.-L. Lee, C.-h. Chen and T.-Y. Luh, Chem.–Asian J., 2011, 6, 1748 CrossRef CAS PubMed.
- Y.-Z. Ke, R.-J. Ji, T.-C. Wei, S.-L. Lee, S.-L. Huang, M.-J. Huang, C.-h. Chen and T.-Y. Luh, Macromolecules, 2013, 46, 6712 CrossRef CAS.
- M. Meldal, Macromol. Rapid Commun., 2008, 29, 1016 CrossRef CAS.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC.
-
Schrödinger Release 2016-4: MacroModel, Schrödinger, LLC, New York, 2016 Search PubMed.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 1998, 120, 4450 CrossRef CAS.
- W. Li, T. Tian, W. Zhu, J. Cui, Y. Ju and G. Li, Polym. Chem., 2013, 4, 3057 RSC.
- J.-A. Amalian, T. T. Trinh, J.-F. Lutz and L. Charles, Anal. Chem., 2016, 88, 3715 CrossRef CAS PubMed.
- J. S. Cottrell, J. Proteomics, 2011, 74, 1842 CrossRef CAS PubMed.
- Templated dimerization was recently reported using dynamic covalent base-pairing: K. R. Strom, J. W. Szostak and N. Prywes, J. Org. Chem., 2019, 84, 3754 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthetic procedures, full characterization of all compounds, UPLC traces for full replication cycle and molecular modelling are available. See DOI: 10.1039/c9sc01460h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Maria
Ciaccia
,
Giulia
Iadevaia
,
Maria
Ciaccia
,
Giulia
Iadevaia













