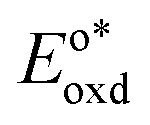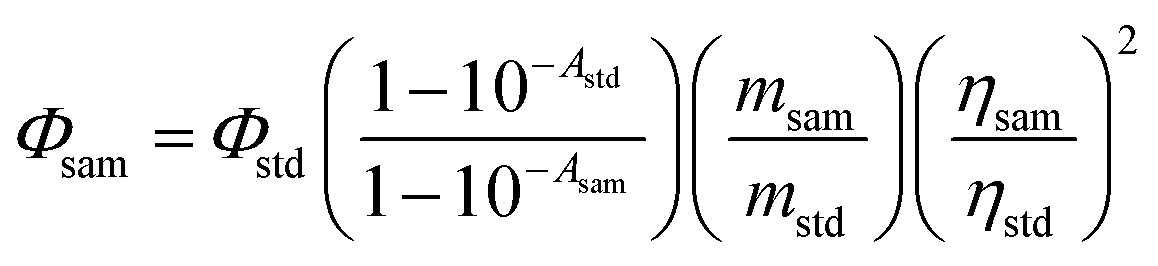DOI:
10.1039/C9SC01463B
(Edge Article)
Chem. Sci., 2019,
10, 7091-7103
Phosphorus corrole complexes: from property tuning to applications in photocatalysis and triplet–triplet annihilation upconversion†
Received
25th March 2019
, Accepted 12th June 2019
First published on 13th June 2019
Abstract
Efficient triplet photosensitizers are important for fundamental photochemical studies and applications such as triplet–triplet annihilation upconversion (TTA UC), photoredox catalytic organic reactions and photovoltaics. We now report a series of phosphorus corrole compounds as efficient visible light-harvesting metal-free triplet photosensitizers. While the heavy-atom-free phosphorus corroles show absorption in the visible spectral region (centered at 573 nm) and have a decent triplet state quantum yield (ΦΔ = 49%), iodo-substitution on the corrole core induces red-shifted absorption (589 nm) and improves intersystem crossing significantly (ΦΔ = 67%). Nanosecond transient absorption spectra confirm triplet state formation upon photoexcitation (τT = 312 μs) and the iodinated derivatives also display near IR phosphorescence in fluid solution at room temperature (λem = 796 nm, τp = 412 μs). Both singlet oxygen (1O2) and superoxide radical anions (O2−˙) may be produced with the phosphorus corroles, which are competent photocatalysts for the oxidative coupling of benzylamine (the Aza Henry reaction). Very efficient TTA UC was observed with the phosphorus corroles as triplet photosensitizers and perylene as the triplet acceptor, with upconversion quantum yields of up to ΦUC = 38.9% (a factor of 2 was used in the equation) and a very large anti-Stokes effect of 0.5 eV.
Introduction
Energy up-conversion is a highly desirable process, as incident photons of a given energy are converted into photons with higher energy.1 It is important for applications in many fields including photovoltaic devices,2,3 luminescent probes for bio-imaging,4–9 photocatalysis by low-energy photons,10–15 and photoinduced charge separation.16–19 When the efficiency of up-conversion is high enough, low-power up-conversion may be used to harness low-energy photons that, otherwise, would be lost in practical applications. For instance, it could be applied to solar cells to harvest the red and NIR part of the solar spectrum, which has energy lower than the band-gap of the employed photoactive material.20 Its applicability for bioimaging is even more intriguing, since excitation in the NIR region offers advantages of deeper depth penetration, suppressed background fluorescence and reduced phototoxicity.21
One possible strategy for photon upconversion comes from harnessing sensitized triplet–triplet annihilation (TTA) photochemistry. Higher energy photons are generated from the absorption of lower energy light by an efficient triplet sensitizer through sequential, highly allowed one-photon absorption, subsequent intersystem crossing (ISC) and intermolecular triplet–triplet energy transfer (TTET) to the energy acceptor, and the population of the S1 state of the triplet acceptor by the TTA process.1,2,22,23 In essence, the energy stored in two sensitized triplet molecules is combined to produce one higher energy singlet state and a corresponding ground-state species. TTA upconversion is particularly interesting for its advantages over conventional upconversion methods where two photons require excitation with coherent light sources (lasers) of very high-power density (e.g. nonlinear optical crystals). In contrast, TTA upconversion requires only noncoherent low-power excitation and is characterized by intense absorption of excitation light as well as high upconversion quantum yields.1,2,11 This phenomenon was first introduced by Parker and Hatchard in the 1960s, utilizing organic chromophores as triplet sensitizers, which however exhibited low intersystem crossing efficiencies thereby limiting upconversion.24,25 TTA upconversion has witnessed great development in recent years.26,27
One challenge for the development of TTA upconversion is the limited availability of appropriate triplet photosensitizers.11 Commonly investigated typical triplet photosensitizers used for TTA upconversion include porphyrin and phthalocyanine complexes,28 phosphorescent transition metal complexes with diimine ligands,29 bromo- and iodo-organic chromophores,30 and a few heavy-metal-free triplet photosensitizers.31 It is relatively easy to study the intermolecular triplet energy transfer of phosphorescent photosensitizers, however, when the triplet sensitizer is non-phosphorescent, quenching must be measured by the variation of the lifetime of the T1 excited state of the sensitizer with the aid of time-resolved transient absorption spectroscopy.11 During the last decade, we and other groups have studied corroles chelated by metal ions such as gold and iridium as triplet photosensitizers, by taking advantage of the heavy atom effect for inducing efficient singlet-triplet interconversion (ISC).32–37 In parallel, we have also introduced corroles chelating non-redox active and much more abundant elements, such as Ga, Al, and P, wherein the non-radiative decay of the S1 state can be inhibited. ISC enhancement was encouraged by bromides or iodides,38,39 which in the corrole case are present on the corrole skeleton and hence not released to the environment.40,41 Corroles with reasonable ISC probability have been introduced for photodynamic therapy,34,42,43 molecular spintronics,44 energy transfer studies,45 and oxygen sensing.35 But to the best of our knowledge, corroles were rarely used for TTA upconversion,32,46 despite the easy tuning of their redox and photophysical properties and the extremely high stability regarding demetallation.
Results and discussion
Rational design of the compounds
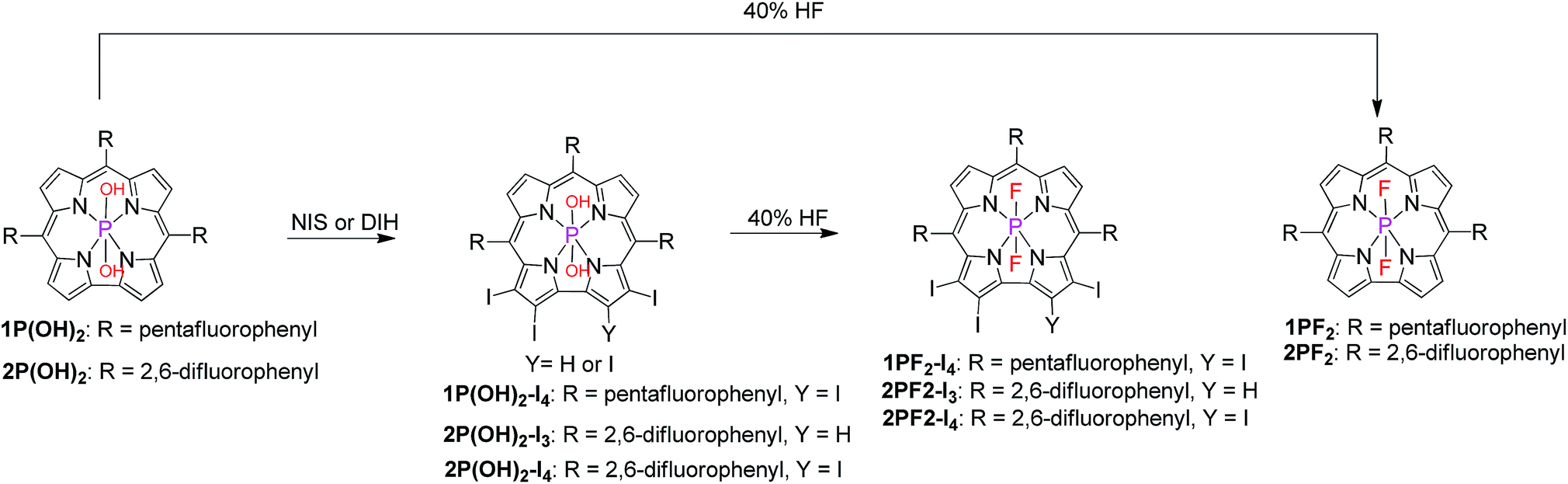
We decided to focus our investigations on compounds composed of non-toxic and non-precious C, H, F, I, and P elements only (Scheme 1), as opposed to the intensively used Ru/Pt/Ir photosensitizers. The most prominent feature of corroles, particularly meso-C6F6-substituted derivatives, is strong fluorescence. As this testifies for very little non-radiative loss of energy,47 the hypothesis was that halogenation of the corrole core will increase the ISC probability and consequently the yields of the triplet excited states. Another advantage is that iodination increases red shifts of the absorbance/emission maxima and also redox potentials. The latter aspect is also tunable by the axial ligands and the number of F atoms in the meso-aryl substituents. The novelty regarding the synthetic part was to move away from hydroxo- and oxo-phosphorus corroles, which are substitution labile (converted into each other and react with solvents),47,48 to a well-defined series of substitution-inert trans-difluorophosphorus corroles, so as to allow for the systematic research described in this paper.49
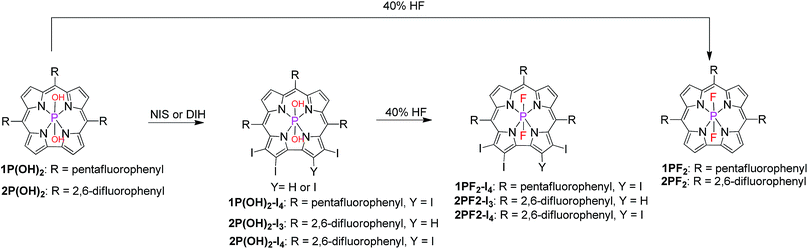 |
| | Scheme 1 Axial ligand exchange and macrocycle iodination of phosphorus corroles. | |
Treatment of phosphorus corroles with N-iodosuccinimide (NIS) or with 1,3-diiodo-5,5-dimethylhydantoin (DIH) led to selective iodination on the corrole macrocycle (Scheme 1).41 The axial hydroxyl ligands of these iodinated complexes were exchanged with fluorides after treatment with aqueous hydrofluoric acid (Scheme 1).41 This transformation resulted in very significant and enlightening changes in the NMR spectra (Fig. S1–S17†). The 1H resonances of hydroxyl groups disappear and the 19F signals of fluoride ligands appear, well separated from those of C6F5 and C6H3F2 aryl groups. Another prominent feature is the J ≅ 830 Hz mutual splitting coupling constant between the axial fluorides (appear as a doublet) and phosphorus (appears as a triplet).
X-ray crystallography
Being concerned about the effect of the rather large iodo-substituents on the planarity of the complexes, the X-ray structures of 1PF2-I4 and 2PF2-I4 were obtained. Fig. 1 clearly demonstrates that the deviations of the β-pyrrole substituted moieties of 1PF2-I4 and 2PF2-I4 relative to the average mean plane of all 23 carbon atoms are quite small, in the range of 0.032–0.287 Å for 2PF2-I4 and 0.000 Å for the extremely planar 1PF2-I4. The peripheral phenyl rings are not in coplanar geometry with the corrole core, and thus the π-conjugation framework of the substituted corroles is not heavily affected by the phenyl rings.
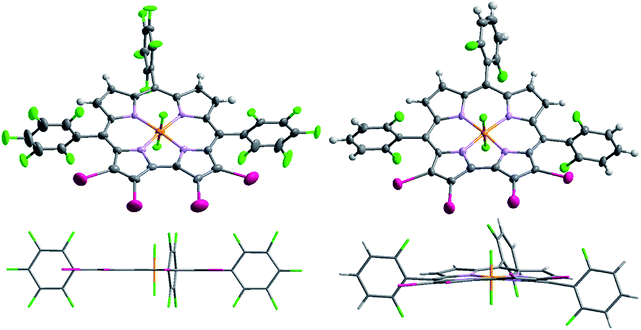 |
| | Fig. 1 Top and side views of the single-crystal molecular structure of 1PF2-I4 (left) and 2PF2-I4 (right). | |
UV-vis absorption and photoluminescence of the compounds
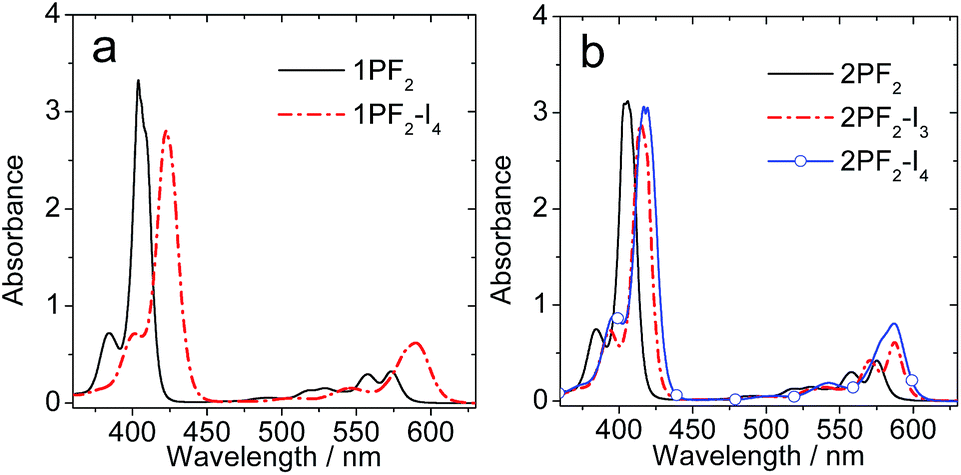
The UV-vis absorption spectrum of 1PF2 displays an intense absorption band at 404 nm and weaker ones in the range of 500–600 nm (Fig. 2a). The former is attributed to the higher-energy and more allowed Soret band and the latter to lower energy and symmetry-forbidden Q bands of the corrole core. Similar absorptions were observed for 1PF2-I4, but red-shifted by 19 nm for the Soret band and 16 nm for the Q bands, and the absorption of the lower energy band at 589 nm is intensified as compared to that of 1PF2. Intensified absorption in the red-shifted region is beneficial for the application of these heavy-metal-free photosensitizers. Similar absorption profiles were observed for 2PF2, 2PF2-I3 and 2PF2-I4 (Fig. 2b).
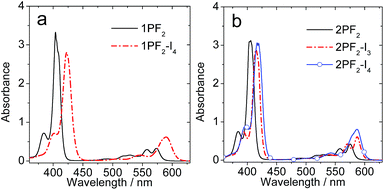 |
| | Fig. 2 UV-vis absorption spectra of (a) 1PF2 and 1PF2-I4; (b) 2PF2, 2PF2-I3 and 2PF2-I4. c = 1.0 × 10−5 M in toluene, 20 °C. | |
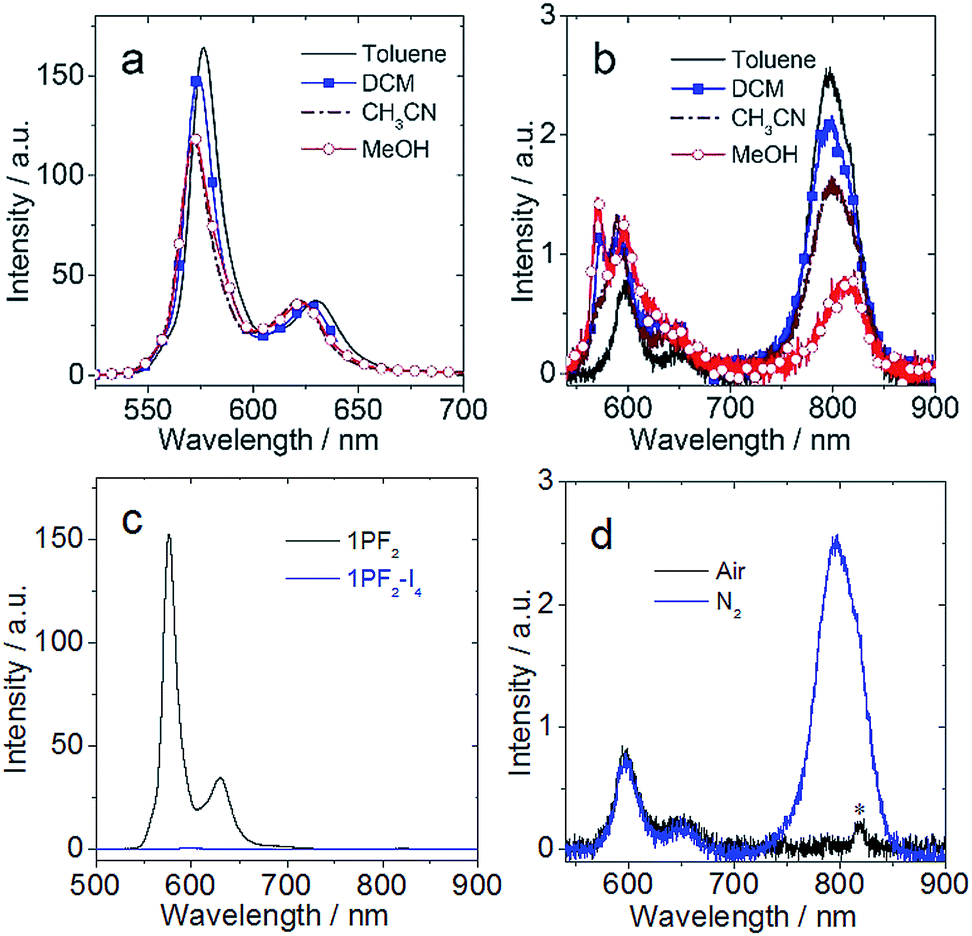
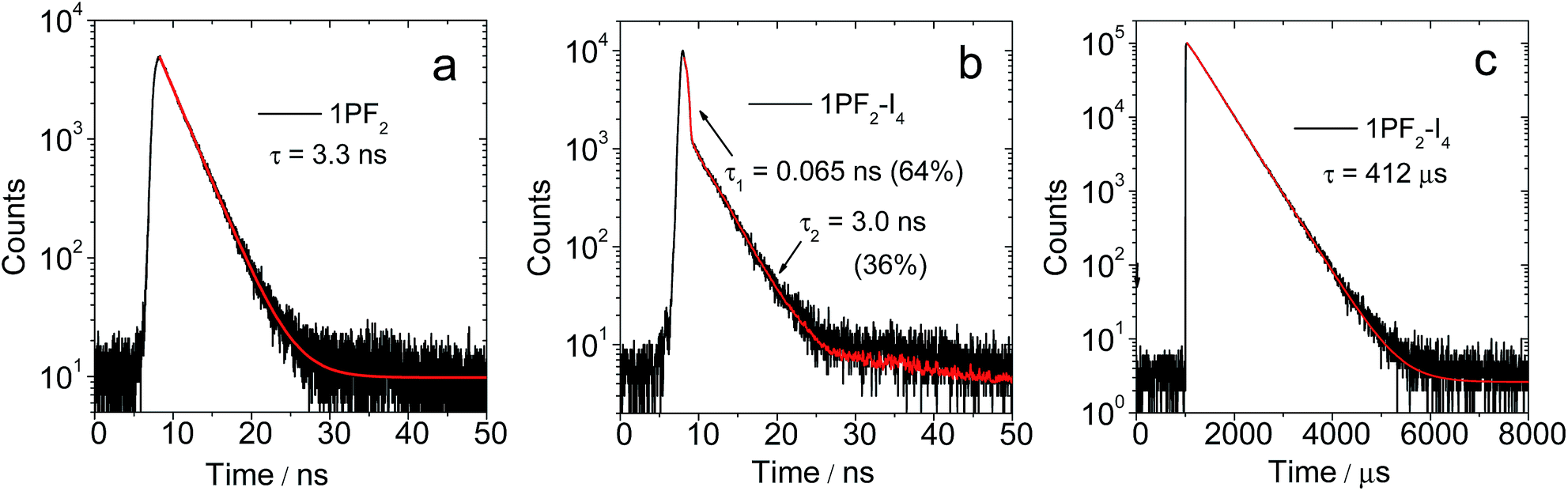
In terms of luminescence properties, there is a significant difference between 1PF2 and 1PF2-I4 (Fig. 3). 1PF2 displays emission bands in the range of 525–700 nm in different organic solvents (DCM, CH3CN, and MeOH), with significant vibrational progression (Fig. 3a). This is a typical emission feature of corroles, which is also similar to the emission of porphyrin compounds. 1PF2-I4 has a similar emission profile in the same solvents (Fig. 3b), but the intensity is much weaker (Fig. 3c). Instead, a new emission band at 796 nm was observed in deaerated solutions of 1PF2-I4 at room temperature, which was completely quenched in aerated solution (Fig. 3d), consistent with phosphorescence.41 No phosphorescence was observed for the non-iodinated corroles under the same conditions (Fig. S28†), consistent with previous demonstration that phosphorescence of free base corroles is only recognizable at low temperature (77 K).50 This observation can be attributed to the enhanced ISC in the iodinated corroles, i.e. the heavy atom effect.51–53 The photophysical parameters for the phosphorus corroles in toluene are compiled in Table 1. 1PF2 and 2PF2 are fluorescent with quantum yields of 10% and 13% and fluorescence lifetimes of 3.3 ns and 3.7 ns, respectively. Iodination of these complexes at the corrole macrocycle reduces the fluorescence quantum yields and the fluorescence lifetimes dramatically, and the complexes become phosphorescent at longer wavelengths (about 800 nm) with lifetimes of several hundred microseconds (Table 1). An in-depth investigation of the fluorescence lifetimes revealed a mono-exponential decay with a lifetime of 3.3 ns for 1PF2 (Fig. 4a), corresponding to the decay of the bright S1 state of the corrole. Interestingly, for 1PF2-I4, a bi-exponential decay was observed for the emission at 598 nm (fluorescence, Fig. 4b). A fast component (64%) is with a lifetime of 0.065 ns, which is close to the instrument response function (IRF). A long-lived (36%) component was observed with a lifetime of 3.0 ns. We attribute the fast-decaying component to the ISC, and the relatively long-lived component is due to the presence of the activation barrier for ISC, i.e., a minor component of the excited molecules does not undergo ISC.55 This is consistent with the low-intensity fluorescence emission seen in Fig. 3c. In other words, there is a non-unity transition probability for ISC. Similar phenomena are also very often observed for molecular systems used for photo-induced electron transfer.56 The decay at 795 nm was also monitored and its lifetime was determined as 412 μs (Fig. 4c). Considering the large Stokes shift, the long lifetime, and the sensitivity of this emission band to O2, the emission at 795 nm is assigned to the phosphorescence of the iodinated corrole. Room temperature phosphorescence is rarely obtained for organic chromophores with a large π-conjugation framework, but is displayed for iodinated chromophores such as iodinated Bodipy derivatives.19 Multiple iodo-substitutions on the corrole framework are clearly beneficial for obtaining phosphorescence.57 The luminescence lifetimes of the compounds in different solvents are summarized in Table S1,† which reveals that 2PF2 without iodo substituents has a fluorescence lifetime that is similar to that of 1PF2. Similar results were observed for 2PF2-I3 and 2PF2-I4, compared with 1PF2-I4: all of them have fluorescence lifetimes that are 10–20 times smaller relative to their non-substituted analogs, and phosphorescence lifetimes in the range of several hundred microseconds.
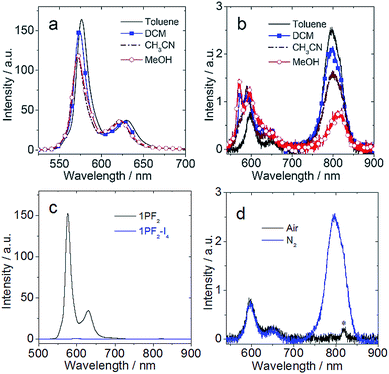 |
| | Fig. 3 Emission spectra (λex = 410 nm, optically matched solutions were used (A = 0.454), 20 °C) of (a) 1PF2 in four N2 saturated solvents; (b) 1PF2-I4 in four N2 saturated solvents; (c) 1PF2 and 1PF2-I4 in N2 saturated toluene; (d) 1PF2-I4 in air- and N2- saturated toluene (“*” denotes the secondary transmission of the monochromator, due to the excitation wavelength). | |
Table 1 Photophysical properties of the compounds
|
|
λ
abs
/nm |
ε
|
λ
em
/nm |
τ
L
|
Φ
L
(%) |
τ
T
(μs) |
Φ
Δ
(%) |
|
In toluene, c = 1.0 × 10−5 M.
Molar absorption coefficient × 105, M−1 cm−1.
Emission wavelength, in toluene.
Luminescence lifetimes.
Luminescence (fluorescence and phosphorescence) quantum yields, with Ru(bpy)3[PF6]2 (in deaerated CH3CN, ΦP = 9.5%) as in ref. 54.
Triplet life time, in deaerated toluene.
Quantum yield of singlet oxygen (1O2). MB (methylene blue) was used as the standard (ΦΔ = 0.57 in DCM).
Fluorescence emission wavelength.
Fluorescence lifetimes (ns).
Fluorescence quantum yields.
Phosphorescence emission wavelength.
Phosphorescence lifetimes (μs).
Phosphorescence quantum yields.
|
|
1PF2
|
404, 556, 573 |
3.31, 0.30, 0.33 |
577h, 630h |
3.3i |
10j |
312 |
49 |
|
1PF2-I4
|
423, 544, 589 |
2.80, 0.15, 0.62 |
598h, 647h, 796k |
1.2i, 412l |
0.05j, 0.27m |
179 |
67 |
|
2PF2
|
405, 557, 576 |
3.12, 0.30, 0.42 |
578h, 632h |
3.7i |
13j |
192 |
46 |
|
2PF2-I3
|
415, 570, 587 |
2.88, 0.42, 0.61 |
591h, 645h, 786k |
0.48i, 338l |
0.12j, 0.15m |
127 |
60 |
|
2PF2-I4
|
417, 570, 587 |
3.06, 0.42, 0.61 |
592h, 638h, 795k |
0.14i, 530l |
0.11j, 0.36m |
181 |
71 |
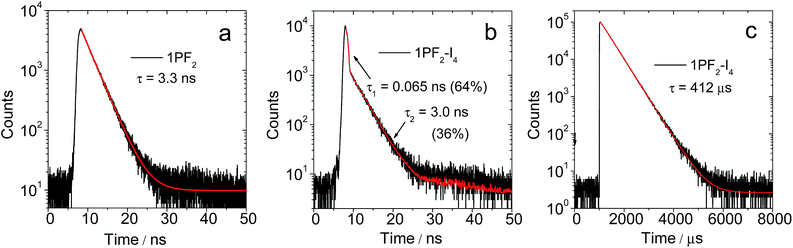 |
| | Fig. 4 Decay traces of the luminescence of (a) 1PF2 at 577 nm (fluorescence band) in toluene; (b) 1PF2-I4 at 598 nm (fluorescence band) in deaerated toluene; (c) 1PF2-I4 at 795 nm (phosphorescence band) in deaerated toluene. (b) Excited with a picosecond pulsed laser for the fluorescence band (λex = 405 nm), and excited with a microsecond flash lamp for the phosphorescence peak at λex = 420 nm, c = 1.0 × 10−5 M, 20 °C. | |
As a preliminary evaluation of the ISC efficiency of the corroles, the singlet oxygen quantum yields (ΦΔ) were measured. For the non-iodinated corroles 1PF2 and 2PF2, the ΦΔ values are 49% and 46%, respectively. These results indicate that while ISC is quite efficient even for 1PF2 and 2PF2, upon iodination, the ΦΔ increases to 60–71% (Table 1), which is clearly attributed to the heavy atom effect of the iodo-substituents.
Nanosecond transient absorption spectroscopy: triplet excited states
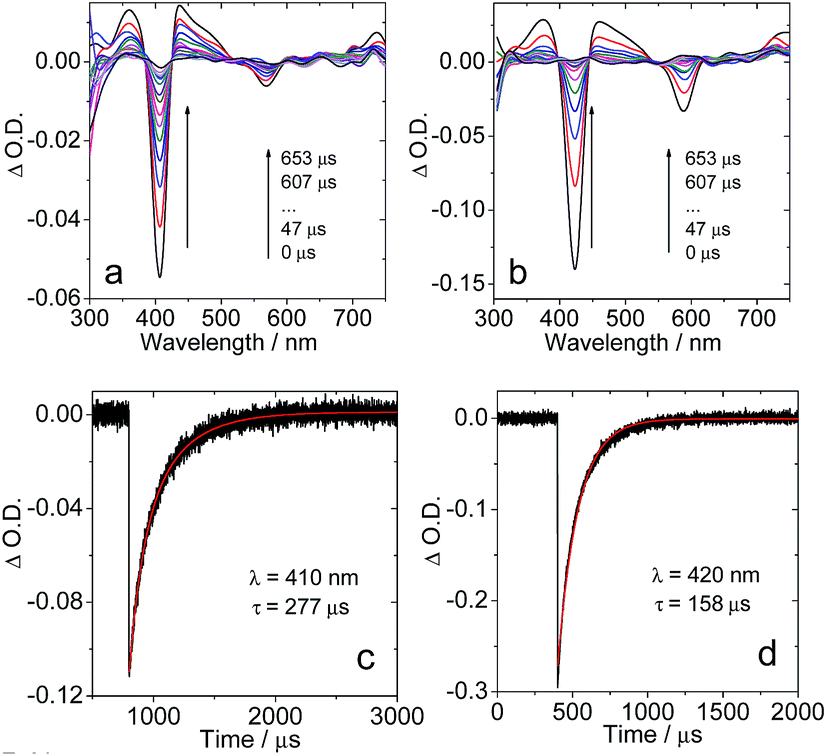
In order to confirm the triplet state production of the corroles upon photoexcitation, the nanosecond transient absorption spectra of the compounds were studied (Fig. 5). For 1PF2, a significant ground state bleaching band at 405 nm was observed upon photo-excitation. However, the ground state band (GSB) at 567 nm is much weaker, apparently due to the much smaller molar extinction coefficients of the ground state absorption of the compound in this region (Fig. 2), as well as the possible superimposition of the GSB with the excited state absorption (ESA) band in this region. This effect is much more significant in the range of 350–500 nm, where the ESA band also overlaps with the GSB. The transient was determined to have a lifetime of 277 μs in deaerated solution (Fig. 5c). This was reduced by 3 orders of magnitude (266 ns) in aerated solution (Fig. S35†), thus confirming that the transient absorption is of the triplet-excited state of the corrole. It should be pointed out that for a long-lived triplet state that is formed with high quantum yield (Table 1), self-quenching of the triplet state might be significant, i.e. that the observed triplet state lifetime might be concentration dependent. The intrinsic triplet state was determined by extrapolation to indefinite diluted concentration, uncovering it to be 312 μs (Table 1 and Fig. S58, S59†).
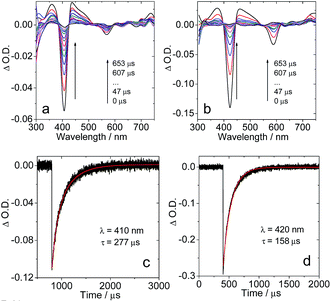 |
| | Fig. 5 Nanosecond transient absorption spectra of (a) 1PF2 (λex = 556 nm) and (b) 1PF2-I4 (λex = 589 nm); decay trace of the transients of (c) 1PF2 at 410 nm and (d) 1PF2-I4 at 420 nm. Excited with a nanosecond pulsed laser. c = 2.5 × 10−6 M in deaerated toluene, 20 °C. | |
Similar transient absorption spectra were observed for 1PF2-I4 (Fig. 5b), and by extrapolation to indefinite diluted concentration, its triplet state lifetime was determined as 179 μs (Table 1). The shorter triplet state lifetime of 1PF2-I4 as compared to 1PF2 may be attributed to the heavy atom effect in 1PF2-I4. The triplet states of other compounds were also studied with nanosecond transient absorption spectra: long-lived triplet states were observed in all cases (Fig. S33†). The ESA bands of the compounds in the region of 300–550 nm can be assigned to the transition of T1 to higher triplet states, i.e. T1 → Tn based on TD-DFT computations (Table S2†). Interestingly, some previously reported corroles show much shorter triplet state lifetimes. For instance, corroles with non-fluorinated phenyl rings show triplet state lifetimes in the range of 0.82–3.50 μs.58 Comparison with other heavy atom containing corroles (Br, Au, Ir, Os, and Re) discloses typical excited state life times in the range of 19–183 μs, which identifies the iodinated P corroles as among the complexes with the largest excited-state lifetimes.32,35,46,59,60 It should be pointed out that the strong absorption of visible light and the long-lived triplet states of these corroles are beneficial for these compounds to be used as triplet photosensitizers, because these are the desired photophysical properties for intermolecular energy- or electron-transfer (vide infra).
The spatial confinement of the triplet state wavefunction of the corroles was investigated by DFT optimization studies (Fig. 6). For both 1PF2 and 1PF2-I4, the triplet state wavefunction is largely confined on the corrole π-conjugation framework, although it also spreads slightly to the peripheral perfluorinated phenyl moieties (Fig. 6).
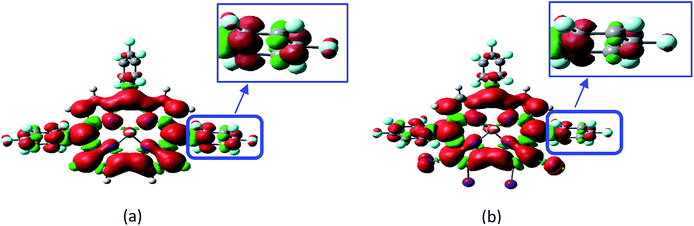 |
| | Fig. 6 Spin density surfaces of (a) 1PF2 (calculated at the B3LYP/6-31G(d) level with Gaussian 09W) and (b) 1PF2-I4 (calculated at the B3LYP/GENECP level with Gaussian 09W) at the optimized triplet state geometry. | |
Although different torsion angles between the peripheral C6F5 ring and the corrole core of the two compounds were observed, i.e. the dihedral angles are 65° and 78° for 1PF2 and 1PF2-I4, respectively, the confinement of the triplet states is almost the same. These different dihedral angles may safely be attributed to the iodine substitution in 1PF2-I4, which exerts some steric hindrance on the rotation of the aryl rings. The confinement of the triplet state wavefunction of the triplet state of corroles was previously studied by time-resolved electron paramagnetic resonance (TREPR), but the confinement of the triplet state was not discussed in detail.61
Production of the corrole radical anions by reversible photoreduction
The motivation for investigating photo-driven intermolecular electron transfer between the corroles and a sacrificial electron donor (triethylamine, Et3N) was to gain fundamental information that is relevant for potential applications of the corroles as photocatalysts. UV-vis examination upon continuous white light irradiation of an anaerobic solution of 1PF2/Et3N disclosed that the absorption bands at 400 nm and 567 nm decreased, whereas new absorption bands in the region of 350–450 nm and 575–650 nm appeared (Fig. 7a, the ground state difference absorption spectra). These new absorption bands may safely be attributed to the radical anion of the corroles.62 Interestingly, the photo-reduction process is reversible, clearly evident by the recovery of the pink color of the corrole solution after exposure of the irradiated solution to air (Fig. 7b) i.e., 1PF2−˙ is oxidized to the neutral form of the corroles by dioxygen in air. The process of light-induced reduction of 1PF2 to 1PF2−˙ and reoxidation of the latter by oxygen may in fact be repeated for several cycles without degradation of the corrole, as illustrated in Fig. 7c.
 |
| | Fig. 7 (a) UV-vis difference absorption spectra of 1PF2 in the presence of Et3N in the deaerated MeCN with white light continuous photoirradiation using a xenon lamp, light power density: 5.2 mW cm−2. (b) Photos showing the consecutive color change of the 1PF2/Et3N mixed solution upon alternating photoirradiation and exposure to air: (1). Without photoirradiation under an air atmosphere; (2) Upon 8 second photoirradiation under a N2 atmosphere; (3) Exposure to air after photoirradiation; (4) Upon another 8 second photoirradiation under a N2 atmosphere. (c) Reversibility of the formation of 1PF2−˙ monitored at the 594 nm absorption band (after generation of 1PF2−˙, re-exposure to air will oxidize the radical anion to the neutral form 1PF2). Photoirradiation power density is 23 mW cm−2. c [1PF2] = 1 × 10−5 M and c [Et3N] = 0.25 M, 20 °C. | |
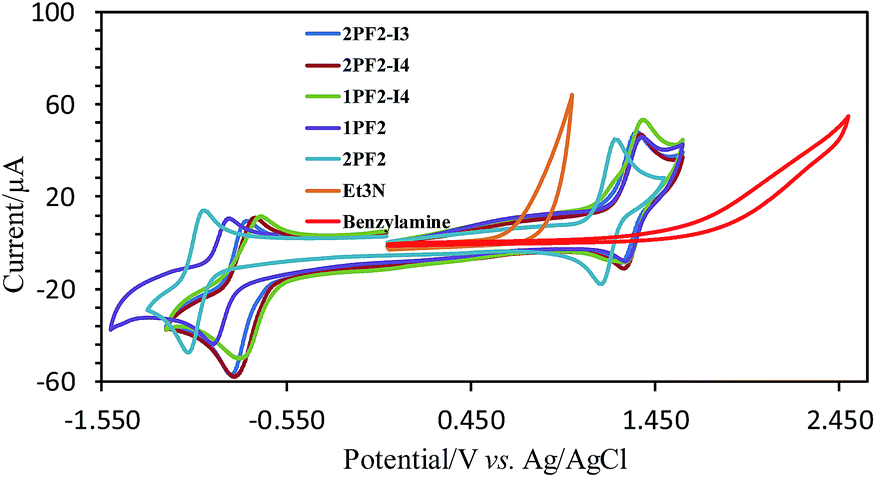
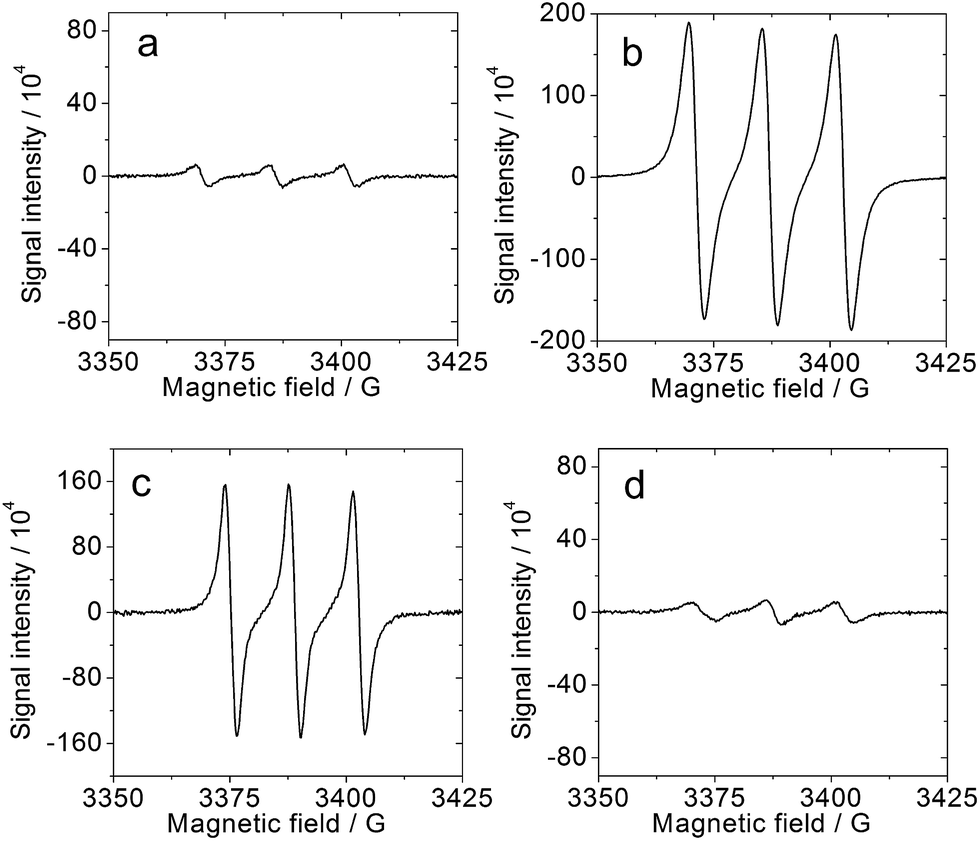
We also studied the interaction of molecular oxygen with the triplet excited state and the photo-generated radical anion of the corroles (Fig. 8): singlet oxygen (1O2) and superoxide radical anions (O2−˙) are expected to be formed by the two species, respectively. Spin traps selective for 1O2 (2,2,6,6-tetramethyl-4-piperidone, TEMP) and O2−˙ (5,5-dimethyl-1-pyrroline-N-oxide, DMPO) were used in combination with 1PF2 and photoirradiation, in the presence/absence of Et3N. The results depicted in Fig. 8 reveal the formation of the 1O2-scavenged product of TEMP only when Et3N was absent (Fig. 8a and b) and of the O2−˙-DMPO product only when Et3N was present (Fig. 8c and d). These studies clarify that both 1O2 and O2−˙ can be produced under appropriate conditions with the phosphorus corroles.63 Similar results were obtained for 2PF2-I3 and 2PF2-I4 (Fig. S49 and S50†). The ability of these phosphorus corroles to photooxidize Et3N is attributed to their high triplet reduction potential (Table 2) relative to the onset oxidation potential of Et3N (0.65 V, Fig. 9). The triplet excited state reduction potential of 2PF2 (0.52 V, Table 2) is however lower than the onset oxidation potential of Et3N. This explains the inability of 2PF2 to be photo-reduced by TEA in the absence of O2 (Fig. S38†).
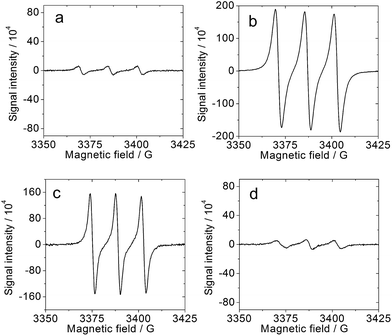 |
| | Fig. 8 ESR spectrum of the mixtures of 1PF2 and TEMP (spin trapping reagent for 1O2) or DMPO (spin trapping reagent for O2−˙) upon photo-irradiation. (a) 1PF2 (1.0 × 10−5 M), Et3N (0.25 M) and TEMP (0.1 M); (b) 1PF2 (1.0 × 10−5 M) and TEMP (0.1 M); (c) 1PF2 (1.0 × 10−5 M), DMPO (0.01 M), Et3N (0.25 M); (d) 1PF2 (1.0 × 10−5 M), DMPO (0.01 M). In air-saturated CH3CN. The samples were photo-irradiated with a Xe lamp for 60 s (23 mW cm−2), 20 °C. | |
Table 2 Redox potentials of phosphorus corroles
|
|
Ground state redox potentials, V vs. Ag/AgCl |
E
S
(eV) |
E
T
(eV) |
Excited state redox potentials, V vs. Ag/AgCl |
|
E
ox1/2(corrole+/corrole) |
E
red1/2(corrole/corrole−) |
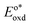
|
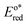
|
E
S = [1240/(λmax(onset) in nm)], λ is the fluorescence wavelength.
E
T = [1240/(λmax(onset) in nm)], λ is the phosphorescence wavelength.
The triplet energy levels were calculated using B3LYP/6-31G(d).
In the singlet state.
In the triplet state. 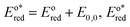 is the reduction potential of the excited state species, Eored is the reduction potential of the ground state using electrochemical measurements. is the reduction potential of the excited state species, Eored is the reduction potential of the ground state using electrochemical measurements. 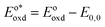 , where , where 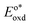 is the oxidation potential of the excited state species, Eooxd is the oxidation potential of the ground state using electrochemical measurements. E0,0 is the zero–zero excitation energy estimated from the fluorescence (E0,0 = ES)/phosphorescence (E0,0 = ET) spectra. is the oxidation potential of the excited state species, Eooxd is the oxidation potential of the ground state using electrochemical measurements. E0,0 is the zero–zero excitation energy estimated from the fluorescence (E0,0 = ES)/phosphorescence (E0,0 = ET) spectra.
|
|
1PF2
|
1.34 |
−0.90 |
2.15 |
1.57c |
−0.81d (−0.23)e |
1.25d (0.67) e |
|
1PF2-I4
|
1.34 |
−0.74 |
2.07 |
1.56 (1.49)c |
−0.73d (−0.22)e |
1.33d (0.82)e |
|
2PF2
|
1.20 |
−1.03 |
2.13 |
1.55c |
−0.83d (−0.35)e |
1.11d (0.52)e |
|
2PF2-I3
|
1.31 |
−0.81 |
2.10 |
1.58 (1.55)c |
−0.79d (−0.27)e |
1.29d (0.77)e |
|
2PF2-I4
|
1.33 |
−0.77 |
2.10 |
1.56 (1.51)c |
−0.77d (−0.23)e |
1.33d (0.79)e |
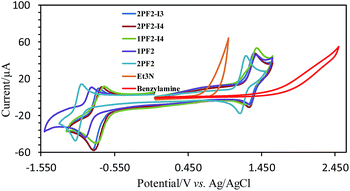 |
| | Fig. 9 Cyclic voltammograms of phosphorus corroles, triethylamine (Et3N), and benzylamine in acetonitrile (1 mM substrate, 0.1 M TBAP as the electrolyte, glassy carbon as the working electrode, Ag/AgCl as the reference electrode, and Pt wire as the supporting electrode; scan rate: 100 mV s−1). | |
Phosphorus corroles as photocatalysts for redox-involving organic reactions
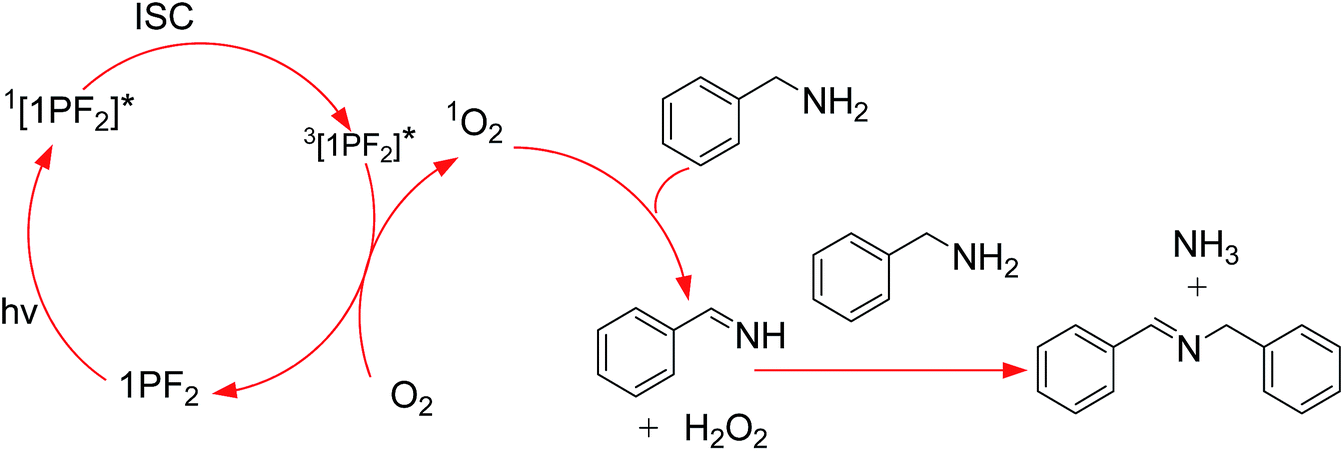
Encouraged by the photo- and electro-chemical properties uncovered for the phosphorus corroles, it is suggested that they might be good photocatalysts for redox-involving organic reactions. The Aza Henry reaction was selected as it is an oxidative coupling of benzylamine with itself. The literature points towards two possible mechanisms for the role of the photocatalyst: (a) production of singlet oxygen (1O2) that reacts with the benzylamine;64 (b) direct electron transfer from the amine to the photoexcited dye.65 Porphyrins, porphycences, and mainly the Ru(bpy)3[PF6]2 (bpy = 2,2′-bipyridine) or Ir(ppy)3 complex (ppy = 2-phenylpyridine) were reported as suitable photocatalysts.65–75 However, Ru(bpy)3[PF6]2 has only very weak absorption in the visible spectral region, and its triplet state lifetime is quite short (1.10 μs in CH3CN).76 These photophysical features are not optimal and precious metal complex photosensitizers are also expensive. Our results show that phosphorus corroles, free of any precious or rare elements, are efficient photocatalysts for the Aza Henry reaction (Table 3). With only 1 mol% phosphorus corrole, most of the benzylamine was converted with high selectivity to N-benzylidenebenzylamine (67–100%), at RT and after short time visible light irradiation (1 h). No reaction took place in the absence of oxygen and 2PF2 appeared to be the best photocatalyst among the phosphorus corroles that were tested in this study, despite displaying the lowest reduction potential of its photo-excited triplet state (0.52 V). This result indicates that the photocatalytic reaction mechanism in our system may be attributed to the involvement of 1O2 and not the electron transfer mechanism (Scheme 2).64 In fact, a comparison of the oxidation potential of benzylamine and the redox potentials of the phosphorus corroles suggests that all the phosphorus corroles tested in this study cannot photo-oxidize the amine via the electron transfer mechanism. The onset potential for oxidation of benzylamine is 1.62 V, which is much higher than the photoreduction potential of the triplet excited state and even higher than the ground state oxidation potentials of all the phosphorus corroles (Table 2). We hence propose that the oxidation of benzylamine by the phosphorus corroles as photosensitizers occurs via the singlet oxygen mechanism, which is also consistent with the superiority of 2PF2 as its photoexcited state is highest in energy.
Table 3 Transformation of benzylamine catalyzed by different triplet photosensitizersa
| Entry |
Photosensitizer |
T (°C) |
T (h) |
Conv.b,c (%) |
Φ
UC
/% |
Reaction conditions: benzylamine (0.5 mmol), photosensitizers (catalyst, 0.005 mmol, 1 mol%), solvent (5 mL), under an air atmosphere, λ > 380 nm (20 mW cm−2), 1 h, 20 °C, in MeCN/CH2Cl2 (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v).
Yield was determined by 1H NMR. The conversion of the reaction was calculated by integrating the singlet peak of the featured proton in the products (at about 4.82 ppm for –CH :1, v/v).
Yield was determined by 1H NMR. The conversion of the reaction was calculated by integrating the singlet peak of the featured proton in the products (at about 4.82 ppm for –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–CH2–) and that of the corresponding proton in the starting materials (at about 3.86 ppm as the singlet for H2N–CH2–).
Reaction was monitored by TLC. Only two spots (one point was attributed to the starting material and the other was attributed to the product) were found (except the photosensitizer), and therefore the selectivity of the photoreaction is good.
TTA upconversion quantum yield with diiodo-distyryl-bodipy as the standard (ΦF = 0.101 in DCM). The laser power density is 70.6 mW cm−2.
Not measured. N–CH2–) and that of the corresponding proton in the starting materials (at about 3.86 ppm as the singlet for H2N–CH2–).
Reaction was monitored by TLC. Only two spots (one point was attributed to the starting material and the other was attributed to the product) were found (except the photosensitizer), and therefore the selectivity of the photoreaction is good.
TTA upconversion quantum yield with diiodo-distyryl-bodipy as the standard (ΦF = 0.101 in DCM). The laser power density is 70.6 mW cm−2.
Not measured.
|
| 1 |
1PF2
|
20 |
1.0 |
87 |
33.6 |
| 2 |
1PF2-I4
|
20 |
1.0 |
67 |
24.8 |
| 3 |
2PF2
|
20 |
1.0 |
100 |
38.9 |
| 4 |
2PF2-I3
|
20 |
1.0 |
75 |
26.4 |
| 5 |
2PF2-I4
|
20 |
1.0 |
87 |
34.1 |
| 6 |
Ru(bpy)3[PF6]2
|
20 |
1.0 |
100 |
—e |
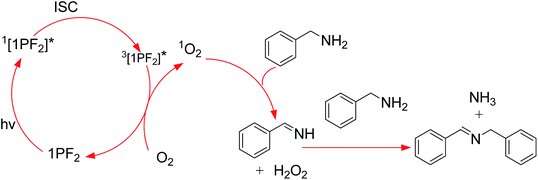 |
| | Scheme 2 Proposed reaction mechanism for the photocatalytic aerobatic oxidation of benzylamine with the triplet photosensitizers 1PF2, 1PF2-I4, 2PF2, 2PF2-I3 and 2PF2-I4. | |
Triplet–triplet annihilation upconversion with the phosphorus corroles as triplet photosensitizers
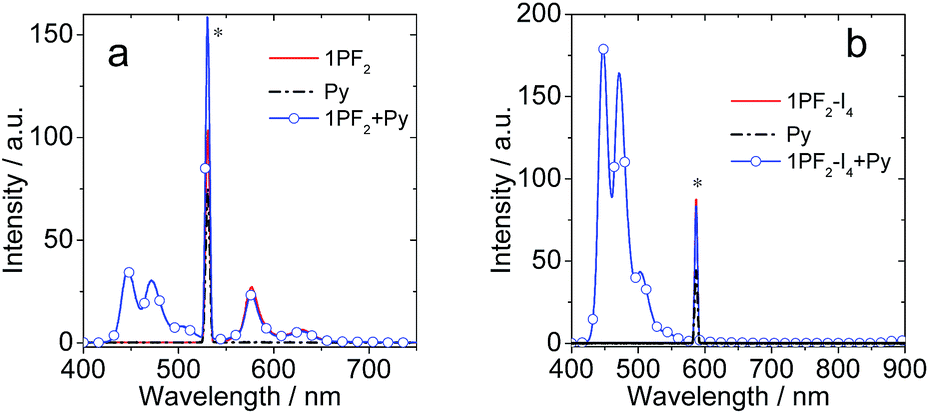
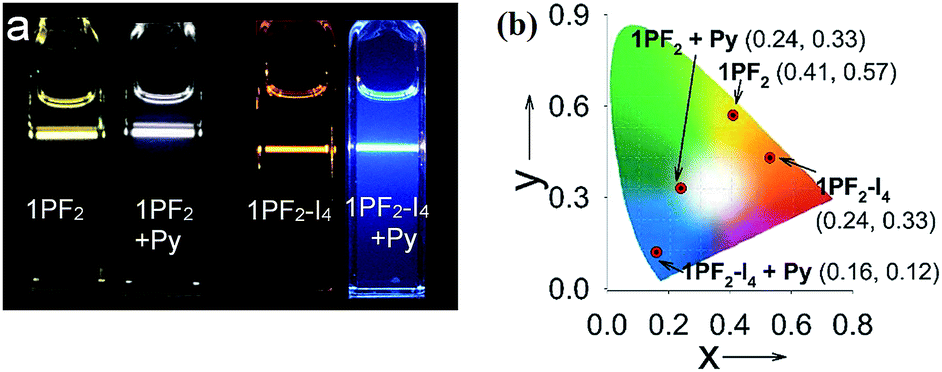
Considering that the corroles are characterized by strong absorption of visible light and the long-lived triplet excited state, the compounds were tested as triplet photosensitizers for TTA upconversion (Fig. 10). Nitridoosmium(VI) and oxorhenium(V) corroles were previously reported for TTA upconversion, with SG 5 (diisobutyl 3,9-perylenedicarboxylate) as the triplet acceptor, but the upconversion quantum yields were very low: 0.16%–2.0%.32,46 Based on the triplet state energy level of the corroles (approximated as 1.56 eV, by the phosphorescence emission of 1PF2-I4), perylene was selected as the triplet acceptor and the emitter (with a T1 state energy level of 1.54 eV).77 For 1PF2-I4, no emission was observed upon 589 nm cw laser excitation (Fig. 10b). This is due to the weak emission of 1PF2-I4 (luminescence quantum yield: 0.27%, Table 1). In the presence of perylene, however, strong emission was observed in the range of 420–550 nm, which is the characteristic emission profile of perylene. Taking into account that the excitation wavelength is 589 nm, which is insufficient for the direct excitation of perylene, the emission is safely attributed to TTA. The upconversion quantum yield was determined as 24.8% (Table 3, note that the factor of 2 was used in the equation). Similarly, upconversion was also observed with 1PF2 as the triplet photosensitizer (Fig. 10a), but its anti-Stoke's shift was much smaller. The visual perception of the upconversion systems is shown in Fig. 11. It is clear that the blue upconverted emission with 1PF2-I4 is more significant than that of 1PF2. The highest upconversion quantum yield was observed with 2PF2 (38.9%)
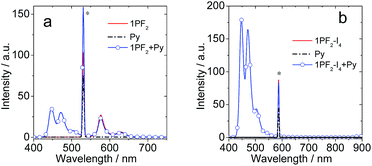 |
| | Fig. 10 The upconversion fluorescence spectra of (a) 1PF2 and (b) 1PF2-I4 as photosensitizers in toluene. Py (perylene) was the acceptor. Excitation was done with a continuous laser of 532 nm for 1PF2 and 589 nm for 1PF2-I4 and the power density was 70.6 mW cm−2 under a deaerated atmosphere. c (sensitizer) = 2.0 × 10−6 M, and c (Py) was 2.4 × 10−5 M and 2.8 × 10−5 M, respectively, 20 °C. | |
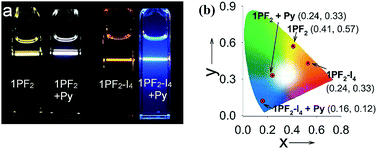 |
| | Fig. 11 (a) Photographs of the emission of triplet photosensitizers alone and the upconversion. The CIE coordinate of (b) the luminescence of the triplet sensitizers (1PF2 and 1PF2-I4) before and after adding Py. Excited with a continuous laser of 532 nm for 1PF2 and a 589 nm laser for 1PF2-I4. The power density was 4.8 mW. c (sensitizers) = 2.0 × 10−6 M in deaerated toluene. Py was at 2.4 × 10−5 M and 2.8 × 10−5 M for upconversion, respectively, 20 °C. | |
In order to study the efficient TTA upconversion capability, the upconversion intensity–excitation power relationship was investigated (Fig. S43–S47†). A linear relationship, instead of the typical quadratic relationship, was observed. This is indicative of a highly efficient TTA upconversion system, with efficient triplet–triplet-energy-transfer (TTET).78
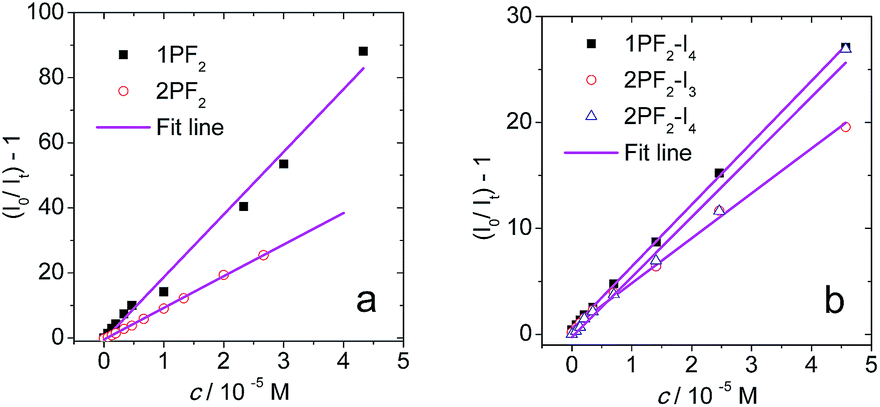
The intermolecular triplet state energy transfer between the phosphorus corrole photosensitizers and the triplet energy acceptor perylene was also studied by nanosecond transient absorption spectroscopy, focusing on the quenched triplet state lifetime in the presence of increasing amounts of perylene (Fig. 12). Analysis of the Stern–Volmer plots provides the Stern–Volmer quenching constants (KSV values), in the range of 4.2–19.3 × 105 M−1 (Table 4). The bimolecular quenching constants (kq) calculated based on the variation of the triplet state lifetimes are in the range of 3.1–6.2 × 109 M−1 s−1. As these values are close to the diffusion-controlled bimolecular rate constants (k0), we conclude that the intermolecular triplet–triplet energy transfer is very efficient. We further note that, to the best of our knowledge, this is the first report of TTA upconversion with corroles not chelated by transition metals.
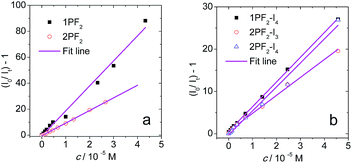 |
| | Fig. 12 Stern–Volmer plots based on fluorescence intensity-quenching of (a) 1PF2 and 2PF2 (λex = 532 nm); and phosphorescence intensity-quenching of (b) 1PF2-I4, 2PF2-I3 and 2PF2-I4 (λex = 589 nm), with perylene as the quencher. c [sensitizers] = 2.0 × 10−6 M in toluene, at 20 °C. | |
Table 4 Upconversion-related parameters for 1PF2, 2PF2, 1PF2-I4, 2PF2-I3 and 2PF2-I4, for their utility as photosensitizersa
|
|
1PF2
|
1PF2-I4
|
2PF2
|
2PF2-I3
|
2PF2-I4
|
|
In deaerated toluene, c (sensitizers) = 2.0 × 10−6 M, λex = 532 nm for 1PF2 and 2PF2, λex = 589 nm for 1PF2-I4, 2PF2-I3 and 2PF2-I4.
Stern–Volmer quenching constants.
Bimolecular quenching constants. KSV = kqτp.
The radius of the quencher molecules.
Diffusion coefficients. In toluene, η = 5.9 × 10−4 Pa s; T = 293.15 K; Rf (1PF2) = Rf (1PF2) = Rf (1PF2-I4) = Rf (2PF2-I3) = Rf (2PF2-I4) = 5.01 × 10−10 m; Df is for the energy donor, Df = 5.54 × 10−6 cm2 s−1 for all three compounds, Dq is for the quencher molecules.
Diffusion-controlled bimolecular quenching rate constants.
The quenching efficiency.
The bimolecular TTET efficiency (ΦTTET) occurred between perylene and the triplet dyad, ΦTTET = 1 − τ/τ0, where τ0 and τ are the lifetimes of the triplet dyad in the absence and presence of perylene, respectively.
|
|
K
SV
/105 M−1 |
19.3 |
5.8 |
9.7 |
4.2 |
5.7 |
|
k
q
/109 M−1 s−1 |
6.2 |
3.3 |
5.1 |
3.3 |
3.1 |
|
R
q
/10−10 m |
3.7 |
3.7 |
3.7 |
3.7 |
3.7 |
|
D
q
/10−6 cm2 s−1 |
7.4 |
7.4 |
7.4 |
7.4 |
7.4 |
|
k
0
/109 M−1 s−1 |
8.1 |
8.1 |
8.1 |
8.1 |
8.1 |
|
f
Q
/% |
75.8 |
40.0 |
62.1 |
40.6 |
38.7 |
|
Φ
TTET
/% |
97.9 |
94.4 |
95.1 |
92.6 |
94.4 |
Conclusions
A series of phosphorus corroles with peripheral fluorinated phenyl moieties were prepared, with focus on those that are also with iodo-substituents on the π-conjugation core. The photophysical properties were studied by both steady state and time-resolved spectroscopies. The corroles display intense absorption in the visible spectral region (centered at 404 nm for 1PF2), and iodo-substitution on the core induces up to 19 nm red-shifts of the absorption band. Near IR phosphorescence (centered as 796 nm) was observed in fluid solution at room temperature for the iodo-substituted derivatives, whereas the regular phosphorus corroles emit only fluorescence. Transient absorption spectroscopy was used for determining photoexcited triplet state formation and lifetimes (τT), which ranged from = 412 to 179 μs. Singlet oxygen quantum yields (ΦΔ) of the compounds were elucidated to be in the range of 46–71%, with higher quantum yields for the iodo-containing corroles. Properly tuned photo-excited corroles react with both reductive and oxidative quenchers, as exemplified by their reactions with a frequently used sacrificial electron donor (triethylamine) and the formation of superoxide radical anions (O2−˙), respectively. They may hence be very useful as photoredox catalysts for many organic transformations.79–81 This was exemplified by utilizing the phosphorus corroles as triplet photosensitizers for efficient photocatalytic oxidative coupling of benzylamine (the Aza Henry reaction). Efficient triplet–triplet annihilation upconversion was observed with the phosphorus corroles as triplet photosensitizers and perylene as the triplet acceptor. The upconversion quantum yield is record high, with up to ΦUC = 38.9% (a factor of 2 was used in the equation) for 2PF2-I4 and an anti-Stokes effect of 0.5 eV, an outcome of appropriate adjustment of excited-state lifetimes and redox potentials. This work demonstrates that phosphorus corroles can be developed as a new type of efficient triplet photosensitizers for photocatalytic organic reactions and photon upconversions.
Experimental
General methods
All the chemicals used in synthesis were analytically pure and were used as received. 1P(OH)2, 1PF2, 2PF2, 2P(OH)2-I3, and 2P(OH)2-I4 were synthesized as previously reported.41 Fluorescence quantum yields were measured in toluene, with Ru(bpy)3[PF6]2 in deaerated CH3CN (Φp = 9.5%) as in ref. 54. The molecular structures of the corroles are shown in Scheme 1. The fluorescence lifetimes of the compounds were measured with an OB920 luminescence lifetime spectrometer (in TCSPC detection mode. Edinburgh Instrument Ltd., UK). The fluorescence lifetimes were measured with 405 nm EPL picosecond pulsed laser excitation (pulse width: 51.9 ps, maximum average power: 5 mW). The phosphorescence lifetimes were measured with a μs flash lamp (maximum average power: 100 μW). Fluorescence and triplet–triplet annihilation (TTA) upconversion spectra were recorded with a RF 5301PC spectrofluorometer (Shimadzu, Japan). 1H and 19F NMR spectra were recorded using a Bruker Advance III 400 MHz spectrometer (operating at 400.4 MHz for 1H and at 376.7 MHz for 19F). Chemical shifts are reported in ppm relative to the residual hydrogen atoms in the CDCl3 solvent (δ = 7.26), and relative to CFCl3 (δ = 0.00) in the 19F NMR spectra. Mass spectra for the compounds were recorded on a Bruker MaXis Impact mass spectrometer, using an APCI (atmospheric pressure chemical ionization) direct probe in either positive or negative mode.
Synthesis and characterization
Synthesis of 2PF2-I3.
A sample of 2P(OH)2-I3 (20 mg, 18.2 μmol) was dissolved in CH2Cl2 (5 mL) in a small plastic flask. 2 mL of 40% hydrofluoric acid (caution! HF should be handled with extreme care. It is a highly corrosive inorganic acid and can cause deep tissue damage) was added and the mixture was stirred overnight. The CH2Cl2 solution was separated and washed twice with distilled water, and the organic layer was dried over anhydrous sodium sulfate and evaporated. The purple solid material was crystalized in n-hexane/CH2Cl2 (1:1, v/v) to afford pure 2PF2-I3 in 92% yield (18 mg, 16.7 μmol). 1H NMR (400.4 MHz, CDCl3): δ = 9.99 (broad s, 1H), 8.89 (broad s, 2H), 8.69 (broad s, 2H), 7.75 (unresolved m, 3H), 7.30 (t, J = 6.80 Hz, 6H). 19F NMR (376.75 MHz, CDCl3): −36.90 (d, J = 829.23 Hz, 2F), −108.01 (unresolved t, 2F), −108.08 (broad s, 4F). 31P NMR (162 MHz, CDCl3) δ = −180.3 (t, J = 828 Hz). 13C NMR (CDCl3) δ = 164.11, 161.69, 140.61, 137.86, 132.18, 131.69, 124.50, 115.55, 114.47, 111.98, 111.79, 111.74, 101.11. UV-vis (toluene) λmax (ε) [nm (×105 cm−1 M−1)]: 415 (2.88), 570 (0.42), 587 (0.61). HR-MS (APCI+) for C37H14N4F8I3P: m/z = 1077.7957 (calculated), 1077.8015 (observed).
Synthesis of 2PF2-I4.
The procedure for axial ligand substitution was performed on complex 2P(OH)2-I4, by the same method and for the same amounts as for complex 2P(OH)2-I3. 17 mg (85% yield, 16.7 μmol) was obtained after crystallization from n-hexane/CH2Cl2 = 1:1. 1H NMR (400.4 MHz, CDCl3): δ = 8.93 (t, J = 4.8 Hz, 2H), 8.71 (t, J = 4.8 Hz, 2H), 7.75 (unresolved m, 3H), 7.30 (unresolved m, 6H). 19F NMR (376.75 MHz, CDCl3): −37.52 (d, J = 832.61 Hz, 2F), −108.01 (unresolved m, 4F), −108.14 (unresolved m, 2F). 31P NMR (162 MHz, CDCl3) δ = −181.9 (t, J = 832 Hz). 13C NMR (CDCl3) δ = 161.50, 132.18, 131.68, 128.00, 127.92, 124.92, 124.155, 124.07, 112.08, 111.83. UV-vis (toluene) λmax (ε) [nm (×105 cm−1 M−1)]: 417 (3.06), 570 (0.42), 576 (0.61). HR-MS (APCI+) for C37H13N4F8I4P: m/z = 1203.6924 (calculated), 1203.6954 (observed).
X-ray quality crystals of 2PF2-I4 were obtained from recrystallization from a mixture of CH2Cl2: n-hexane (1:1, v/v) solution. Crystal data for C38H15Cl2F8I4N4P (M = 1289.01 g mol−1): monoclinic, space group P21/c (no. 14), a = 21.925(2) Å, b = 13.1020(11) Å, c = 13.146(3) Å, β = 97.96(5)°, V = 3740.0(11) Å3, Z = 4, T = 200.15 K, μ(MoKα) = 3.596 mm−1, Dcalc = 2.289 g cm−3, 6114 reflections measured (3.752° ≤ 2Θ ≤ 49.058°), 6114 unique (Rint = 0.0650, Rsigma = 0.0384) which were used in all calculations. The final R1 was 0.0569 (I > 2σ(I)) and wR2 was 0.1438 (all data). CCDC 1888513.†
Synthesis of 1P(OH)2–I4.
A sample of 1P(OH)2 (40 mg, 46.6 μmol), 50 eq. of N-iodosuccinimide (NIS, 524 mg), and TFA (200 μL) were dissolved in acetonitrile (10 mL). The solution was stirred for 4 h at room temperature in the dark. The reaction mixture was diluted with CH2Cl2 (40 mL), washed with distilled water (50 mL), dried over anhydrous sodium sulfate, and evaporated. The reaction products were chromatographed on silica gel by using hexanes/ethyl acetate (100:5, v/v) as the eluent. The product was further purified by silica gel 60 preparative thin-layer chromatography (PTLC) with n-hexane/ethyl acetate (100:3, v/v) as the eluent. The product was eluted as the third fraction with purple color (20 mg, 32% yield). 1H NMR (400.4 MHz, CDCl3): δ = 8.80 (dd, J = 4.8, 3.2 Hz, 2H), 8.62 (dd, J = 4.8, 3.2 Hz, 2H), −4.11 (d, 7.6 Hz, 2H). 19F NMR (376.75 MHz, CDCl3): −136.10 (dd, J = 23.0 Hz, 7.5 Hz, 4F; ortho-F), −136.33 (dd, J = 23.0, 7.5 Hz, 2F; ortho-F), −150.68 (t, J = 21.1 Hz, 1F; para-F), −150.94 (t, J = 21.5, 2F; para-F), −160.48 (m, 2F; meta-F), −161.46 (m, 4F; meta-F). 31P NMR (162 MHz, CDCl3) δ = −188.0 (s). UV-vis (toluene) λmax (ε) [nm (×105 cm−1 M−1)]: 428 (2.6), 551 (0.17), 596 (0.60). HR-MS (APCI+) for C37H6F15N4I4PO2: m/z = 1361.6162 (calculated), 1361.6148 (observed).
Synthesis of 1PF2-I4.
The procedure for axial ligand substitution was performed on complex 1P(OH)2-I4, by the same method as for complex 2P(OH)2-I3. 18 mg (90% yield, 13.2 μmol) was obtained after crystallization from n-hexane/CH2Cl2 (1:1, v/v).1 H NMR (400.4 MHz, CDCl3): δ = 8.98 (broad s, 2H), 8.78 (broad s, 2H). 19F NMR (376.75 MHz, CDCl3): −27.20 (d, J = 818.19 Hz, 2F), −135.72 (unresolved m, 2F; ortho-F), −136.47 (unresolved m, 4F; ortho-F), −150.41 (t, J = 21.80 Hz, 1F; para-F), −150.92 (t, J = 21.80 Hz, 2F; para-F), −160.75 (m, 2F; meta-F), −160.92 (m, 4F; meta-F). 31P NMR (162 MHz, CDCl3) δ = −183.5 (t, J = 819 Hz). 13C NMR (CDCl3) δ = 139.00, 136.53, 129.05, 126.70, 126.62, 126.55, 126.44, 123.15, 123.07, 100.23, 100.17, 95.92. UV-vis (toluene) λmax (ε) [nm (×105 cm−1 M−1)]: 423 (2.8), 544 (0.15), 589 (0.62). HR-MS (APCI+) for C37H4F17N4I4P–2H+: m/z = 1363.5919 (calculated), 1363.6068 (observed).
X-ray quality crystals of 1PF2-I4 were obtained from recrystallization from a mixture of CH2Cl2: n-hexane (1:1, v/v) solution. Crystal data for C39H4Cl4F17I4N4P (M = 1531.83 g mol−1): monoclinic, space group P21/m (no. 11), a = 17.2430(13) Å, b = 7.806(5) Å, c = 17.503(3) Å, β = 110.42(3)°, V = 2207.9(15) Å3, Z = 2, T = 200.15 K, μ(MoKα) = 3.211 mm−1, Dcalc = 2.304 g cm−3, 3932 reflections measured (4.11° ≤ 2Θ ≤ 49.42°), 3932 unique (Rint = 0.0770, Rsigma = 0.0485) which were used in all calculations. The final R1 was 0.1059 (I > 2σ(I)) and wR2 was 0.2462 (all data). CCDC 1888512.†
Crystal structure determination.
The single-crystal material was immersed in Paratone-N oil and was quickly fished with a glass rod and mounted on a Nonius Kappa CCD diffractometer under a cold stream of nitrogen at 200 K. Using Olex2,82 the structure was solved with the SIR2004 (ref. 83) structure solution program using Direct Methods and refined with the ShelXL84 refinement package using least squares minimization. The software Mercury 3.10.1 was used for molecular graphics.
Electrochemistry.
The cyclic voltammograms were obtained utilizing a PlamStat3+ connected to a personal computer using PSTrace 5.5 software. A three-electrode system was used consisting of a mini glassy carbon electrode (diameter of the active zone: 2.8 mm; Metrohm) as a working electrode, a platinum wire as a counter electrode, and an Ag/AgCl electrode as a reference electrode. The CV measurements were performed in acetonitrile solutions (HPLC grade), 0.1 M tetrabutylammonium perchlorate (TBAP, Fluka, recrystallized twice in methanol), and 1 mM substrate at ambient temperature with 100 mV s−1 scan rates. The E1/2 value for the ferrocene/ferrocenium (Fc/Fc+) couple under these conditions was 0.42 V.
Singlet oxygen quantum yield (ΦΔ).
1,3-Diphenylisobenzofuran (DPBF) was used as the 1O2 scavenger (Fig. S37†). The 1O2 production was monitored by following the absorbance of DPBF at 414 nm upon photo-irradiation of the photosensitizer/DPBF mixed solution. To determine the singlet oxygen quantum yield (ΦΔ), a relative method was used according to eqn (1):| | 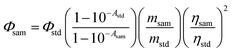 | (1) |
where ‘sam’ and ‘std’ represent the sample and the standard. Φ, A, m and η represent the singlet oxygen quantum yield, the absorbance at the excitation wavelength, the slope of the absorbance of DPBF changing over time, and the refractive index of the solvent used for measurement, respectively. Optically matched solutions were used (the solutions of the sample and the standard give the same absorbance at the excitation wavelength). Singlet oxygen quantum yields (ΦΔ) were measured in toluene. The excitation wavelength was 573 nm (A = 0.222) for 1PF2, 576 nm (A = 0.218) for 2PF2, and 589 nm (A = 0.203) for the other compounds. The monochromatic light source includes a xenon lamp and a monochromator. Methylene blue (MB) was used as the standard (ΦΔ = 0.57 in DCM).
Nanosecond transient absorption spectroscopy.
The nanosecond transient absorption spectra were obtained with a LP980-K Laser Flash Photolysis Spectrometer (kinetic mode, Edinburgh Instruments, UK). The analogue signal was digitized with a Tektronix TDS 3012C oscilloscope. The samples were purged with N2 for 15 min and excited with a nanosecond pulsed laser (Opolette™, the wavelength is tunable in the range of 210–2400 nm. OPOTEK, USA). The raw data were analyzed by using LP900 software.
The general procedure for transformation of benzylamine catalyzed by different triplet photosensitizers.
The mixture of photosensitizer (1 mol%) and benzylamine (0.5 mmol) in CH3CN (5 mL) was stirred at room temperature (RT) under an air atmosphere. The solution was irradiated using a 35 W xenon lamp through a cut off filter (0.72 M NaNO2 aqueous solution, which is transparent for light > 380 nm). Thin layer chromatography (TLC) was used to monitor the progress of the reaction. After completion of the reaction, the solvent was evaporated under reduced pressure. The conversion of the photoreactions was determined by 1H NMR: calculated by integrating the singlet peak of the unique proton in the product (at about 4.82 ppm for –CHN–CH2–) relative to that of the corresponding proton in the starting materials (singlet at 3.86 ppm for H2N–CH2–).
Photoreduction of the corroles to produce radical anions.
UV-vis difference absorption spectra of the corroles were recorded in the presence of Et3N in deaerated MeCN with white light photoirradiation using a xenon lamp. Et3N was used as the sacrificial electron donor and corrole was used as the electron acceptor. Photos were taken to show the color change using a digital camera.
Triplet–triplet annihilation upconversion.
A 532 nm and 589 nm continuous wave (cw) diode-pumped solid state laser was used as the excitation source for the upconversion. The mixed solution of the triplet photosensitizers and the triplet acceptors was degassed with N2 for at least 15 min before the upconversion experiments. The upconverted fluorescence was recorded with a RF 5301PC spectrofluorometer. In order to depress the laser scattering in the sample chamber of the spectrometer, a small black box was put behind the cuvette as the beam dump to trap the transmitted laser. The upconversion quantum yields (ΦUC) of the corroles in toluene were determined, relative to diiodo-distyryl-bodipy as the standard: ΦF = 0.101 in DCM. Plots of UC intensity vs. power density, in the range of 30–900 mW cm−2, were linear with unity slopes (Fig. S43–S47†). The upconversion quantum yields were calculated with the following equation (eqn (2)), where Φ, A, I and η represent the quantum yield, absorbance, integrated photoluminescence intensity and refractive index, respectively. Symbols with ‘std’ and ‘sam’ present the corresponding parameter for the standard and sample (eqn (2)). Note a factor of 2 was used in the equation.2| | 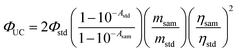 | (2) |
Theoretical computations.
Density Functional Theory (DFT) and Time-dependent Density Functional Theory (TD-DFT) were used for geometry optimization and energy calculation of all the compounds in a vacuum. All the calculations were performed with the Gaussian 16 program. The geometry optimization of the ground state and the excited state was conducted at the B3LYP/6-31G(d) level for non-iodo-substituted compounds and the B3LYP/GENECP level for iodo-substituted compounds and frequency calculations guarantee that the optimal structures are valid. Vertical excitation energies of singlet and triplet states were calculated at the B3LYP/6-31G(d) level for non-iodo-substituted compounds and the B3LYP/GENECP level for iodo-substituted compounds. The Gaussian 09W program package was used for the calculations.85
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
ZG and JZ acknowledge the ISF/NSF Israel-China co-funding project (21761142005) for financial support. JZ also thanks the NSFC (21673031, 2181101275 and 21421005), the Fundamental Research Funds for the Central Universities (DUT16TD25, DUT15ZD224, and DUT2016TB12) and the State Key Laboratory of Fine Chemicals (ZYTS201801) for financial support.
Notes and references
- P. Ceroni, Chem.–Eur. J., 2011, 17, 9560–9564 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- Y. Y. Cheng, B. Fückel, R. W. MacQueen, T. Khoury, R. G. C. R. Clady, T. F. Schulze, N. J. Ekins-Daukes, M. J. Crossley, B. Stannowski, K. Lips and T. W. Schmidt, Energy Environ. Sci., 2012, 5, 6953–6959 RSC.
- J. Liu, Y. Liu, Q. Liu, C. Li, L. Sun and F. Li, J. Am. Chem. Soc., 2011, 133, 15276–15279 CrossRef CAS PubMed.
- L. Xiong, Z. Chen, Q. Tian, T. Cao, C. Xu and F. Li, Anal. Chem., 2009, 81, 8687–8694 CrossRef CAS PubMed.
- H. M. Kim and B. R. Cho, Acc. Chem. Res., 2009, 42, 863–872 CrossRef CAS PubMed.
-
C. J. Fahrni, in Reviews in Fluorescence 2007, Springer New York, New York, NY, 2009, pp. 249–269 Search PubMed.
- Q. Liu, T. Yang, W. Feng and F. Li, J. Am. Chem. Soc., 2012, 134, 5390–5397 CrossRef CAS PubMed.
- S. H. C. Askes and S. Bonnet, Nat. Rev. Chem., 2018, 2, 437–452 CrossRef.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed.
- J. Zhao, S. Ji and H. Guo, RSC Adv., 2011, 1, 937–950 RSC.
- S. Ji, W. Wu, W. Wu, H. Guo and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 1626–1629 CrossRef CAS PubMed.
- Q. Yan, R. Zhou, C. Fu, H. Zhang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2011, 50, 4923–4927 CrossRef CAS PubMed.
- S. Guo, W. Wu, H. Guo and J. Zhao, J. Org. Chem., 2012, 77, 3933–3943 CrossRef CAS PubMed.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2018, 9, 6670–6678 RSC.
- F. Auzel, Chem. Rev., 2004, 104, 139–174 CrossRef CAS PubMed.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- G. Bergamini, P. Ceroni, P. Fabbrizi and S. Cicchi, Chem. Commun., 2011, 47, 12780–12782 RSC.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, J. Org. Chem., 2011, 76, 7056–7064 CrossRef CAS PubMed.
- Y. Shang, S. Hao, C. Yang and G. Chen, Nanomaterials, 2015, 5, 1782–1809 CrossRef CAS PubMed.
- M. González-Béjar, L. Francés-Soriano and J. Pérez-Prieto, Frontiers in Bioengineering and Biotechnology, 2016, 4, 47 CrossRef PubMed.
- W. Wu, J. Zhao, J. Sun and S. Guo, J. Org. Chem., 2012, 77, 5305–5312 CrossRef CAS PubMed.
- P. Yang, W. Wu, J. Zhao, D. Huang and X. Yi, J. Mater. Chem., 2012, 22, 20273–20283 RSC.
- C. A. Parker and J. Bowen Edmund, Proc. R. Soc. London, Ser. A, 1963, 276, 125–135 CrossRef CAS.
- C. A. Parker and C. G. Hatchard, Proc. Chem. Soc., 1962, 386–387 CAS.
- C. Ye, L. Zhou, X. Wang and Z. Liang, Phys. Chem. Chem. Phys., 2016, 18, 10818–10835 RSC.
- J. Peng, X. Guo, X. Jiang, D. Zhao and Y. Ma, Chem. Sci., 2016, 7, 1233–1237 RSC.
- S. Baluschev, V. Yakutkin, T. Miteva, Y. Avlasevich, S. Chernov, S. Aleshchenkov, G. Nelles, A. Cheprakov, A. Yasuda, K. Müllen and G. Wegner, Angew. Chem., Int. Ed., 2007, 46, 7693–7696 CrossRef CAS PubMed.
- R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 3776–3778 RSC.
- H.-C. Chen, C.-Y. Hung, K.-H. Wang, H.-L. Chen, W. S. Fann, F.-C. Chien, P. Chen, T. J. Chow, C.-P. Hsu and S.-S. Sun, Chem. Commun., 2009, 4064–4066 RSC.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. A, 2009, 113, 5912–5917 CrossRef CAS PubMed.
- S. M. Borisov, A. Alemayehu and A. Ghosh, J. Mater. Chem. C, 2016, 4, 5822–5828 RSC.
- J. H. Palmer, A. C. Durrell, Z. Gross, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 9230–9231 CrossRef CAS PubMed.
- A. B. Alemayehu, N. U. Day, T. Mani, A. B. Rudine, K. E. Thomas, O. A. Gederaas, S. A. Vinogradov, C. C. Wamser and A. Ghosh, ACS Appl. Mater. Interfaces, 2016, 8, 18935–18942 CrossRef CAS PubMed.
- C. M. Lemon, D. C. Powers, P. J. Brothers and D. G. Nocera, Inorg. Chem., 2017, 56, 10991–10997 CrossRef CAS PubMed.
- M. Soll, K. Sudhakar, N. Fridman, A. Müller, B. Röder and Z. Gross, Org. Lett., 2016, 18, 5840–5843 CrossRef CAS PubMed.
- R. Orłowski, D. Gryko and D. T. Gryko, Chem. Rev., 2017, 117, 3102–3137 CrossRef PubMed.
- X. Miao, W. Hu, T. He, H. Tao, Q. Wang, R. Chen, L. Jin, H. Zhao, X. Lu, Q. Fan and W. Huang, Chem. Sci., 2019, 10, 3096–3102 RSC.
- E. l. G. Azenha, A. C. Serra, M. Pineiro, M. M. Pereira, J. Seixas de Melo, L. G. Arnaut, S. J. Formosinho and A. M. d. A. Rocha Gonsalves, Chem. Phys., 2002, 280, 177–190 CrossRef CAS.
- J. Vestfrid, I. Goldberg and Z. Gross, Inorg. Chem., 2014, 53, 10536–10542 CrossRef CAS PubMed.
- J. Vestfrid, R. Kothari, A. Kostenko, I. Goldberg, B. Tumanskii and Z. Gross, Inorg. Chem., 2016, 55, 6061–6067 CrossRef CAS PubMed.
- X. Liang, J. Mack, L.-M. Zheng, Z. Shen and N. Kobayashi, Inorg. Chem., 2014, 53, 2797–2802 CrossRef CAS PubMed.
- Z. Zhang, H.-H. Wang, H.-J. Yu, Y.-Z. Xiong, H.-T. Zhang, L.-N. Ji and H.-Y. Liu, Dalton Trans., 2017, 46, 9481–9490 RSC.
- F. Wu, J. Liu, P. Mishra, T. Komeda, J. Mack, Y. Chang, N. Kobayashi and Z. Shen, Nat. Commun., 2015, 6, 7547 CrossRef CAS PubMed.
- B. Brizet, N. Desbois, A. Bonnot, A. Langlois, A. Dubois, J.-M. Barbe, C. P. Gros, C. Goze, F. Denat and P. D. Harvey, Inorg. Chem., 2014, 53, 3392–3403 CrossRef CAS PubMed.
- S. M. Borisov, R. F. Einrem, A. B. Alemayehu and A. Ghosh, Photochem. Photobiol. Sci., 2019, 18, 1166–1170 RSC.
- A. Ghosh and M. Ravikanth, Chem.–Eur. J., 2012, 18, 6386–6396 CrossRef CAS PubMed.
- M. L. Naitana, S. Nardis, G. Pomarico, M. Raggio, F. Caroleo, D. O. Cicero, S. Lentini, L. Prodi, D. Genovese, S. Mitta, A. Sgarlata, M. Fanfoni, L. Persichetti and R. Paolesse, Chem.–Eur. J., 2017, 23, 905–916 CrossRef CAS PubMed.
- X. Zhan, P. Teplitzky, Y. Diskin-Posner, M. Sundararajan, Z. Ullah, Q.-C. Chen, L. J. W. Shimon, I. Saltsman, A. Mahammed, M. Kosa, M.-H. Baik, D. G. Churchill and Z. Gross, Inorg. Chem., 2019, 58, 6184–6198 CrossRef CAS PubMed.
- V. N. Knyukshto, T. H. Ngo, W. Dehaen, W. Maes and M. M. Kruk, RSC Adv., 2016, 6, 43911–43915 RSC.
- E. Pomarico, P. Pospíšil, M. E. F. Bouduban, J. Vestfrid, Z. Gross, S. Záliš, M. Chergui and A. Vlček, J. Phys. Chem. A, 2018, 122, 7256–7266 CrossRef CAS PubMed.
- T. Stensitzki, Y. Yang, A. Berg, A. Mahammed, Z. Gross and K. Heyne, Struct. Dyn., 2016, 3, 043210 CrossRef CAS PubMed.
- C. Zahn, T. Stensitzki, M. Gerecke, A. Berg, A. Mahammed, Z. Gross and K. Heyne, Molecules, 2017, 22, 1174 CrossRef PubMed.
- H. Ishida, S. Tobita, Y. Hasegawa, R. Katoh and K. Nozaki, Coord. Chem. Rev., 2010, 254, 2449–2458 CrossRef CAS.
-
J. R. Lakowicz, Principles of fluorescence spectroscopy, Kluwer Academic/Plenum, New York, 2nd edn, 1999 Search PubMed.
- K. Pettersson, K. Kilså, J. Mårtensson and B. Albinsson, J. Am. Chem. Soc., 2004, 126, 6710–6719 CrossRef CAS PubMed.
- G. Baryshnikov, B. Minaev and H. Ågren, Chem. Rev., 2017, 117, 6500–6537 CrossRef CAS PubMed.
- S. S. K. Raavi, J. Yin, G. Grancini, C. Soci, V. R. Soma, G. Lanzani and L. Giribabu, J. Phys. Chem. C, 2015, 119, 28691–28700 CrossRef CAS PubMed.
- C. M. Lemon, R. L. Halbach, M. Huynh and D. G. Nocera, Inorg. Chem., 2015, 54, 2713–2725 CrossRef CAS PubMed.
- J. Vestfrid, M. Botoshansky, J. H. Palmer, A. C. Durrell, H. B. Gray and Z. Gross, J. Am. Chem. Soc., 2011, 133, 12899–12901 CrossRef CAS PubMed.
- M. Tanabe, H. Matsuoka, Y. Ohba, S. Yamauchi, K. Sugisaki, K. Toyota, K. Sato, T. Takui, I. Goldberg, I. Saltsman and Z. Gross, J. Phys. Chem. A, 2012, 116, 9662–9673 CrossRef CAS PubMed.
- B. S. Achary, A. R. Ramya, J. B. Nanubolu, S. Seetharaman, G. N. Lim, Y. Jang, F. D'Souza and L. Giribabu, New J. Chem., 2018, 42, 8230–8240 RSC.
- Y.-G. Wang, Z. Zhang, H. Wang and H.-Y. Liu, Bioorg. Chem., 2016, 67, 57–63 CrossRef CAS PubMed.
- L. Huang, J. Zhao, S. Guo, C. Zhang and J. Ma, J. Org. Chem., 2013, 78, 5627–5637 CrossRef CAS PubMed.
- A. Berlicka and B. König, Photochem. Photobiol. Sci., 2010, 9, 1359–1366 RSC.
- M. Ertl, E. Wöβ and G. Knör, Photochem. Photobiol. Sci., 2015, 14, 1826–1830 RSC.
- K. Rybicka-Jasińska, B. König and D. Gryko, Eur. J. Org. Chem., 2017, 2017, 2104–2107 CrossRef.
- Y. Cheng, J. Yang, Y. Qu and P. Li, Org. Lett., 2012, 14, 98–101 CrossRef CAS PubMed.
- D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094–8097 CrossRef CAS PubMed.
- K. Hanson, D. L. Ashford, J. J. Concepcion, R. A. Binstead, S. Habibi, H. Luo, C. R. K. Glasson, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2012, 134, 16975–16978 CrossRef CAS PubMed.
- K. Mori, M. Tottori, K. Watanabe, M. Che and H. Yamashita, J. Phys. Chem. C, 2011, 115, 21358–21362 CrossRef CAS.
- Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034–9037 CrossRef CAS PubMed.
- Y.-Q. Zou, L.-Q. Lu, L. Fu, N.-J. Chang, J. Rong, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2011, 50, 7171–7175 CrossRef CAS PubMed.
- K. Rybicka-Jasińska, W. Shan, K. Zawada, K. M. Kadish and D. Gryko, J. Am. Chem. Soc., 2016, 138, 15451–15458 CrossRef PubMed.
- C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010–1014 CrossRef CAS PubMed.
- D. P. Rillema, J. K. Nagle, L. F. Barringer and T. J. Meyer, J. Am. Chem. Soc., 1981, 103, 56–62 CrossRef CAS.
- R. H. Clarke and R. M. Hochstrasser, J. Mol. Spectrosc., 1969, 32, 309–319 CrossRef CAS.
- A. Haefele, J. Blumhoff, R. S. Khnayzer and F. N. Castellano, J. Phys. Chem. Lett., 2012, 3, 299–303 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937–1945 CrossRef CAS PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M.
C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori, D. Siliqi and R. Spagna, J. Appl. Crystallogr., 2007, 40, 609–613 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
-
M. J. Frisch, G. W. Truncks and H. B. e. a. Schlegel, Gaussian 03, revision E.01, Gaussian Inc., Wallingford, CT, 2007 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, computational details, molecular structure characterization, crystal structures, crystallographic data, electrochemical data and additional spectra. CCDC 1888512–1888513. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01463b |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Jenya
Vestfrid
c,
Jianzhang
Zhao
b,
Jenya
Vestfrid
c,
Jianzhang
Zhao



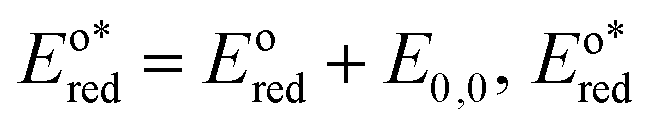 is the reduction potential of the excited state species, Eored is the reduction potential of the ground state using electrochemical measurements.
is the reduction potential of the excited state species, Eored is the reduction potential of the ground state using electrochemical measurements. 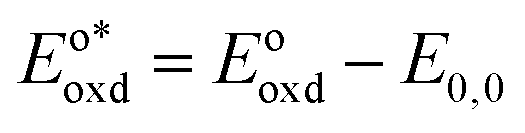 , where
, where 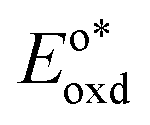 is the oxidation potential of the excited state species, Eooxd is the oxidation potential of the ground state using electrochemical measurements. E0,0 is the zero–zero excitation energy estimated from the fluorescence (E0,0 = ES)/phosphorescence (E0,0 = ET) spectra.
is the oxidation potential of the excited state species, Eooxd is the oxidation potential of the ground state using electrochemical measurements. E0,0 is the zero–zero excitation energy estimated from the fluorescence (E0,0 = ES)/phosphorescence (E0,0 = ET) spectra.