
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly regioselective complexation of tungsten with Eu@C82/Eu@C84: interplay between endohedral and exohedral metallic units induced by electron transfer†
Lipiao
Bao
a,
Pengyuan
Yu
a,
Ying
Li
b,
Changwang
Pan
a,
Wangqiang
Shen
 a,
Peng
Jin
*b,
Shuquan
Liang
*c and
Xing
Lu
*a
a,
Peng
Jin
*b,
Shuquan
Liang
*c and
Xing
Lu
*a
aState Key Laboratory of Materials Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, 1037 Luoyu Road, Wuhan, 430074, China. E-mail: lux@hust.edu.cn
bSchool of Materials Science and Engineering, Hebei University of Technology, Tianjin, 300130, China. E-mail: china.peng.jin@gmail.com
cDepartment of Materials Science and Engineering, Central South University, Changsha, 410083, China. E-mail: lsq@csu.edu.cn
First published on 26th April 2019
Abstract
Interactions between the inner and outer units through a fullerene cage are of fundamental importance for the creation of molecular spintronics and machines, but the mechanism of such through-cage interplay is still unclear. In this work, we have designed and synthesized two prototypical compounds which contain only a single europium atom inside the cage and merely a tungsten atom coordinating outside to clarify the interactions between the endohedral and exohedral metallic units. They are obtained by reacting a tungsten complex W(CO)4(Ph2PC2H4PPh2) (1) with the corresponding metallofullerenes in a highly regioselective manner (2a for Eu@C2(5)-C82 and 2b for Eu@C2(13)-C84). On the one hand, the endohedral Eu-doping has changed the LUMO distribution on C2(5)-C82/C2(13)-C84 dramatically, via electron transfer, which governs the addition pattern of the exohedral tungsten resulting in surprisingly high regioselectivity. On the other hand, the exohedral tungsten coordination with Eu@C2(5)-C82/Eu@C2(13)-C84 has restricted the motion of the internal europium ion to some extent by changing the electrostatic potentials, as confirmed by the X-ray results of 2a, 2b and the corresponding pristine metallofullerenes cocrystallized with Ni(OEP) (OEP is the dianion of octaethylporphyrin). We now make it clear that the interplay between the endohedral and exohedral metallic units can be realized in a single system by means of intramolecular charge transfer, which may arouse interest in the design and utilization of novel metallofullerene-based molecular devices.
Introduction
Confining metal atom(s) or metallic units inside fullerene cages affords a collection of novel hybrid molecules named endohedral metallofullerenes (EMFs).1–6 Results show that the internal metallic units are able to influence the chemical properties of the cage carbon atoms through, for example, charge transfer and/or the geometric effect. For instance, Sc2C2-doping of D2d(23)-C84 altered its chemical reactivity dramatically by raising the HOMO and LUMO levels so that D2d(23)-C84 and Sc2C2@D2d(23)-C84 follow different routes when reacting with 2-adamantane-2,3′-[3H]-diazirine.7 Besides, the size effect of the internal cluster has been observed in the 1,3-dipolar reactions of M3N@Ih-C80 (M = Sc, Y, Er and Gd) where EMFs with a small M3N cluster (M = Sc and Y) give [5,6]-adducts but that containing a larger Gd3N or Er3N cluster favors the formation of [6,6]-adducts.8–11On the other hand, the impact of the external addition on the position and even the configuration of the internal metallic units has also been demonstrated. Systematic studies of Akasaka and coworkers evidenced that the dynamic behavior of the encaged metal atoms in M2@Ih-C80 (M = Ce and La) is drastically restrained upon chemical functionalization of the cage mainly by geometric distortion.12 Recently, a collaborative study demonstrated that the attachment of a benzyl radical on Sc3C2@C80 alters the configuration of the internal Sc3C2 cluster from a bat-ray-like shape to a trifoliate one13 whereas another brilliant study reported that the magnetic properties of Sc3C2@C80 can also be altered by attaching a remote nitroxide radical.14
Even though the impact from the outer on the inner moieties or the reverse process has been clearly demonstrated, it is still blurry whether the endohedral–exohedral interactions can be realized simultaneously in a single system. In their pioneering work, Popov and coworkers reported that the addition of 14 or 16 CF3 groups onto Sc3N@C80 confines the geometry of the internal Sc3N cluster and the multi-addition pattern seems to be a result of cluster–cage interactions.15 Similar results were also obtained in the YCN@C84(CF3)n (n = 16 and 18) system recently.16 However, because of the existence of multiple endohedral atoms and many exohedral addends in these examples, it is still difficult to clarify the fundamental mechanism of the endohedral–exohedral interactions.
Herein, we have synthesized two coordination complexes (2a and 2b) by reacting a tungsten complex W(CO)4(Ph2PC2H4PPh2) (1) with either Eu@C2(5)-C82 or Eu@C2(13)-C84. 2a and 2b are ideal prototypes for understanding through-cage interactions because only one europium atom is encapsulated inside the cage with merely a tungsten atom coordinating outside. Based on the concrete crystallographic and theoretical results, the interactions between the inner and outer metallic units have been well documented for the first time by considering intramolecular charge transfer as the main driving force.
Results and discussion
Chemical functionalization of Eu@C82/Eu@C84
Photo-irradiation of a toluene solution containing Eu@C2(5)-C82 and W(CO)4(Ph2PC2H4PPh2) (1) with a mercury-arc lamp (cutoff < 350 nm) at room temperature produced the corresponding complex 2a (Scheme 1). The reaction process was traced by high performance liquid chromatography (HPLC) and the corresponding profiles are shown in Fig. 1a. After irradiation for three minutes, a new peak at 5.9 min was observed in addition to the peaks of the precursors (1 and Eu@C2(5)-C82), indicative of high regioselectivity. The same procedures conducted on Eu@C2(13)-C84 also showed high regioselectivity and presented a single new peak at 6.1 min (Fig. 1b). Subsequent HPLC separations afforded only one product for each EMF (2a for Eu@C2(5)-C82 and 2b for Eu@C2(13)-C84). The conversion yields of 2a and 2b are estimated to be 51% and 49% according to the HPLC peak areas, respectively.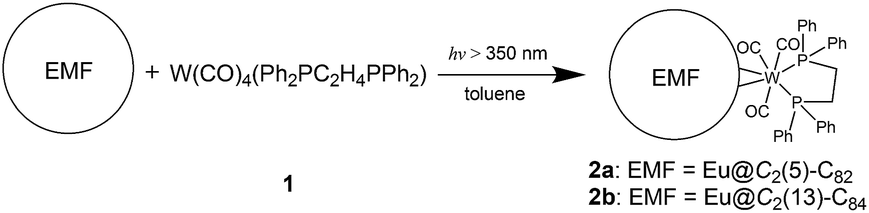 | ||
| Scheme 1 Reaction of 1 with Eu@C2(5)-C82 or Eu@C2(13)-C84. | ||
 | ||
| Fig. 1 HPLC profiles of the reaction mixture containing 1 and (a) Eu@C2(5)-C82 or (b) Eu@C2(13)-C84 probed at different reaction times. HPLC conditions: Buckyprep column (φ = 4.6 × 250 mm); 20 μL injection volume; 1.0 mL min−1 toluene flow; 40 °C; 330 nm detection wavelength. | ||
The electronic structures of the two EMFs have been altered dramatically by the attachment of the tungsten addend, as evidenced by their absorption spectra (Fig. 2a). Since the absorptions of fullerenes and their derivatives are dominated by the π → π* excitation of the cage π-systems,2 the changes in absorptions can be ascribed to the charge donation from the exohedral tungsten units onto the fullerene cages. Consistently, our theoretical natural population analysis (NPA) suggests that the fullerene moieties in 2a and 2b both receive a negative charge of −0.35e from tungsten. Surprisingly, the two pristine EMFs together with 2a and 2b in CS2 solution show strong photoluminescence (PL) emission. Upon excitation at 406 nm at room temperature, two obvious emission bands at ca. 450 and 475 nm are generally observed for all four compounds (Fig. 2b). This phenomenon is quite interesting and the origin is currently under investigation in our lab.
 | ||
| Fig. 2 (a) Vis-NIR absorption spectra and (b) photoluminescence emission and excitation spectra of Eu@C2(5)-C82, 2a, Eu@C2(13)-C84 and 2b dissolved in CS2 at room temperature. The curves are vertically shifted for ease of comparison. | ||
Motional control of the internal metal ion by external complexation
The molecular structures of 2a and 2b together with those of pristine Eu@C2(5)-C82 and Eu@C2(13)-C84 co-crystalized with Ni(OEP) (OEP is the dianion of octaethylporphyrin) are unambiguously established by single crystal X-ray crystallography. The results show an obvious impact of the exohedral complexation on the motional behavior of the internal Eu atom.The X-ray structures of Eu@C2(5)-C82/Ni(OEP) are depicted in Fig. 3a. The fullerene cage is unambiguously assigned as C2(5)-C82. Inside the cage, five disordered Eu sites are distinguished with occupancy values ranging from 0.17 to 0.23, indicative of a moving metal atom. In particular, three Eu sites with a total occupancy of 0.64, including the major one (Eu1, occupancy: 0.23), stay under a hexagonal ring away from the C2 axis (highlighted with a red circle in Fig. 3a).
 | ||
| Fig. 3 Molecular structures of (a) Eu@C2(5)-C82·Ni(OEP) and (b) 2a. The endohedral Eu atom is depicted with thermal ellipsoids set at the 20% probability level. Only one cage orientation and the major metal site are shown. Solvent molecules and the minor metal sites are omitted for clarity. The prohibited area inside the cage upon exohedral functionalization is highlighted with a red dashed circle. The C2 axis of the cage is indicated with a black dashed line. The electrostatic potential maps of (c) [C2(5)-C82]2− and (d) [C2(5)-C82W(CO)3(Ph2PC2H4PPh2)]2−. | ||
In 2a, the tungsten moiety coordinates to a [6,6]-bond in an η2-fashion (Fig. 3b and Fig. S1†). The carbon atoms at the sites of addition (C1 and C2) are pulled out from the cage. Their pyramidalization angles17 increase from 10.65° and 10.66° in the pristine EMF to 15.10° and 15.99° in 2a, respectively (Table S1†). The C1–C2 bond is coplanar with the two W–P bonds and is elongated from 1.36(2) Å in Eu@C2(5)-C82 to 1.478(6) Å in 2a, demonstrating substantial back-donation from tungsten to the π*-orbital of the C1–C2 bond.18 Besides, π–π interaction is found between one of the exohedral phenyl rings and the cage with a plane-to-cage distance of 3.301 Å (Fig. S1†). It is noteworthy that 2a represents the first example of a functionalized C2(5)-C82 cage, though functionalizations on other C82 cages including Cs(6)-C82, C3v(7)-C82, C3v(8)-C82 and C2v(9)-C82 have been reported.2,19
Inside the cage of 2a, four disordered Eu positions with occupancy values ranging from 0.14 to 0.40 are identified and the major Eu site (Eu1) is located under the hexagonal ring perpendicular to the C2 cage-axis. These disordered sites all locate away from the addition sites, suggesting a repulsive effect of tungsten on europium through the cage. In contrast, the internal metal atom generally stays closer to the addition sites in the carbene derivatives of M@C82 (M = Sc, Y, La, and Gd) as a consequence of bond cleavage of the corresponding cages.20–23 In addition, our theoretical calculations reveal that the electrostatic potential map of [C82·W(CO)3(Ph2PC2H4PPh2)]2− (nearly two electrons are transferred from Eu to the cage, see Table S2†) shows a minimum in the position where the major Eu in 2a stays (Fig. 3d). Moreover, appreciable differences in the electrostatic potentials inside the cages are observed between [C82·W(CO)3(Ph2PC2H4PPh2)]2− and [C82]2− (Fig. 3), theoretically corroborating the charge-transfer-induced motional control of the endohedral europium atom by the exohedral tungsten coordination as observed by crystallographic analysis.
The co-crystals of Eu@C2(13)-C84 and Ni(OEP) belong to the monoclinic space group C2/m, and as a result two disordered cage orientations with an equal occupancy of 0.50 are presented. The fullerene cage is unambiguously assigned as C2(13)-C84. Fig. 4a shows the molecular structure of Eu@C2(13)-C84 together with Ni(OEP). Within the cage, 13 disordered Eu sites (occupancy values: 0.02–0.18) are distinguished, with the major Eu position (Eu1, 0.18 occupancy) locating under a hexagonal ring away from the C2 axis. The disordered phenomenon suggests moving nature of the endohedral europium atom.
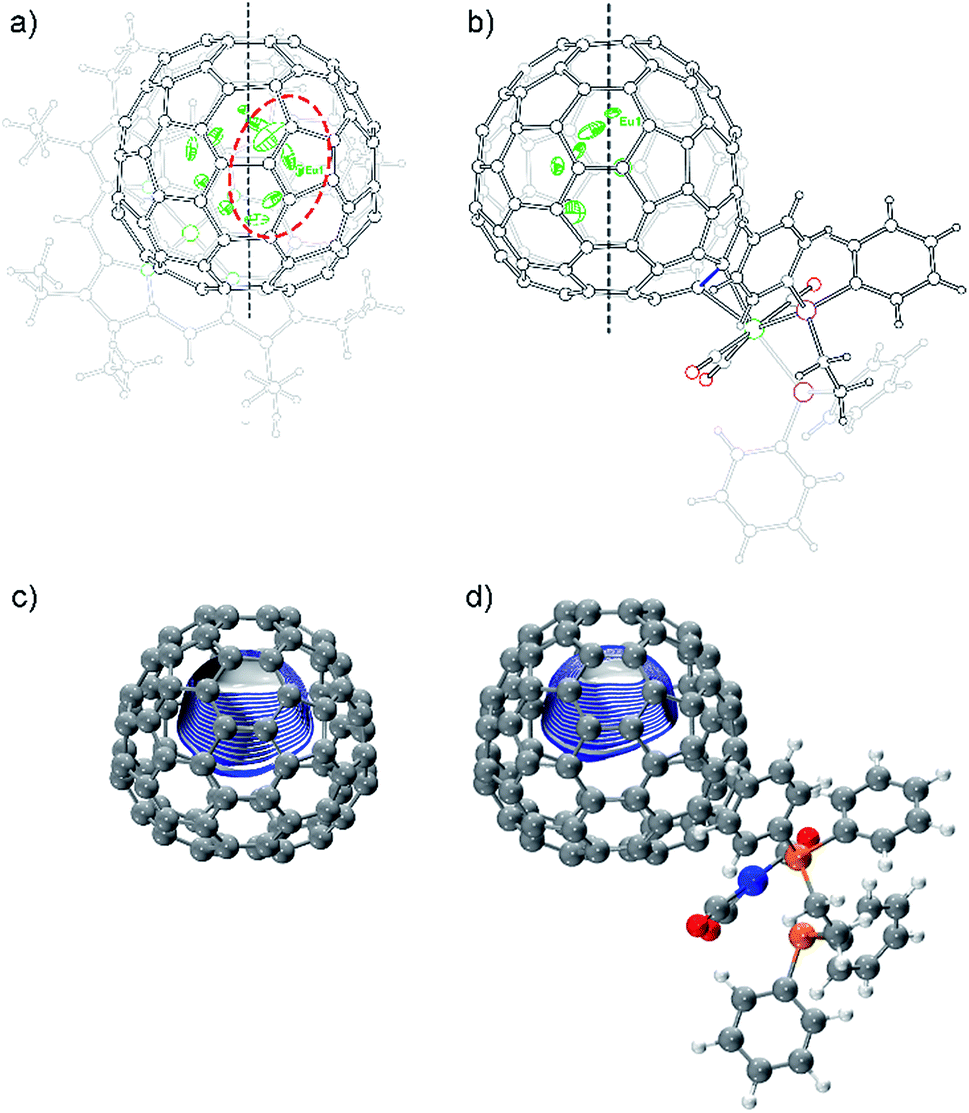 | ||
| Fig. 4 Molecular structures of (a) Eu@C2(13)-C84·Ni(OEP) and (b) 2b. The endohedral Eu ions are depicted with thermal ellipsoids set at the 20% probability level. Only one cage orientation and the major metal sites are shown with solvent molecules and the minor metal sites omitted for clarity. The prohibited area inside the cage upon exohedral functionalization is highlighted with a red dashed circle. The C2 axis of the C2(13)-C84 cage is marked with a black dashed line. The electrostatic potential maps of (c) [C2(13)-C84]2− and (d) [C2(13)-C84 W(CO)3(Ph2PC2H4PPh2)]2−. | ||
In 2b, the tungsten moiety adds onto a [6,6]-bond in an η2-fashion to afford a closed structure (Fig. 4b and Fig. S2†), which is different from the [5,6]-fulleroid structure of the reported carbene adduct of Yb@C2(13)-C84 (ref. 24) bearing the same C2(13)-C84 cage, suggesting different reaction mechanisms. The exohedral functionalization has induced a geometric change in the cage. In detail, the pyramidalization angles of the cage carbons at the sites of addition (C1 and C2) are amplified (from 11.44° and 9.31° to 15.40° and 16.83° for C1 and C2, respectively), and the C1–C2 bond length is elongated from 1.38(2) Å in parent Eu@C2(13)-C84 to 1.49(1) Å in 2b (Table S1†), indicative of remarkable back-donation from tungsten onto the π*-orbital of the C1–C2 bond. Besides, π–π interaction is found between one of the exohedral phenyl rings and the C2(13)-C84 cage with a plane-to-cage distance of 3.306 Å (Fig. S2†).
Within the cage of 2b, the number of disordered europium sites is reduced to only six (occupancy values: 0.36–0.05). They locate at an area far away from the addition sites with the major position Eu1 staying under a hexagonal ring perpendicular to the C2 axis of the cage (Fig. 4b). The crystallographic results confirm that the motional behavior of the internal Eu ion has been constrained by the external tungsten addition. Consistently, our calculations suggest that such a motional control is a consequence of the alteration of the electrostatic potential inside the cage upon exohedral coordination (Fig. 4).
Regiochemical control of exohedral addition by internal metal doping
DFT calculations are carried out to explore the origin of the high regioselectivity observed here. Due to the two electron-transfer from Eu onto the cage (Table S2†) and the moving state of the Eu ion in Eu@C2(5)-C82/Eu@C2(13)-C84, it is reasonable to consider [C2(5)-C82]2− and [C2(13)-C84]2− as the prototypes. The coordination reactions of alkenes18 and C6025,26 with metallic complexes can be well understood by considering the frontier orbitals where the LUMO of alkenes/C60 accepts electrons from metallic complexes to form coordination bonds. Accordingly, the LUMO distributions of [C2(5)-C82]2− and [C2(13)-C84]2− along with the HOMO distribution of the tungsten addend are calculated (Fig. 5). Clearly, the LUMOs at the C1–C2 bonds on both [C2(5)-C82]2− and [C2(13)-C84]2− possess pronounced lobes and overlap with the HOMOs of the tungsten addend (mainly the 5d orbital of the tungsten center), implying that the C1–C2 sites are suitable for tungsten addition. | ||
| Fig. 5 LUMO distributions of (a) [C2(5)-C82]2− and (b) [C2(13)-C84]2− and (c) HOMO distributions of the tungsten addend. | ||
Although LUMO distributions can describe the high regioselectivity in a qualitative way, it is hard to accurately explain the formation of merely one adduct for each EMF (2a and 2b) since as many as 62/63 different types of nonequivalent C–C bonds exist in the C2(5)-C82/C2(13)-C84 cages, respectively (Fig. S3†). Accordingly, another factor, POAV (p-orbital axis vector) which represents the local strain on the cage carbon atoms17 is introduced to rationalize the reaction mechanism herein because it has shown great success in elucidating the chemical reactivities of EMFs.2,17,19 For the ease of comparison, the average LUMO and POAV values of the two bonded cage carbons for each type of C–C bond are calculated and presented in Fig. 6. The C1–C2 bond in [C2(5)-C82]2− features both pronounced LUMO coefficients and high pyramidal angles, and therefore it is made favorable to accept the electron from the tungsten center to form the coordination bonds with negligible steric hindrance. Similarly, the C1–C2 bond on [C2(13)-C84]2− is also preferred owing to its high POAV and LUMO values. The above discussions suggest a synergistic effect of both the electron distribution and geometric structure of the cages on the addition patterns.
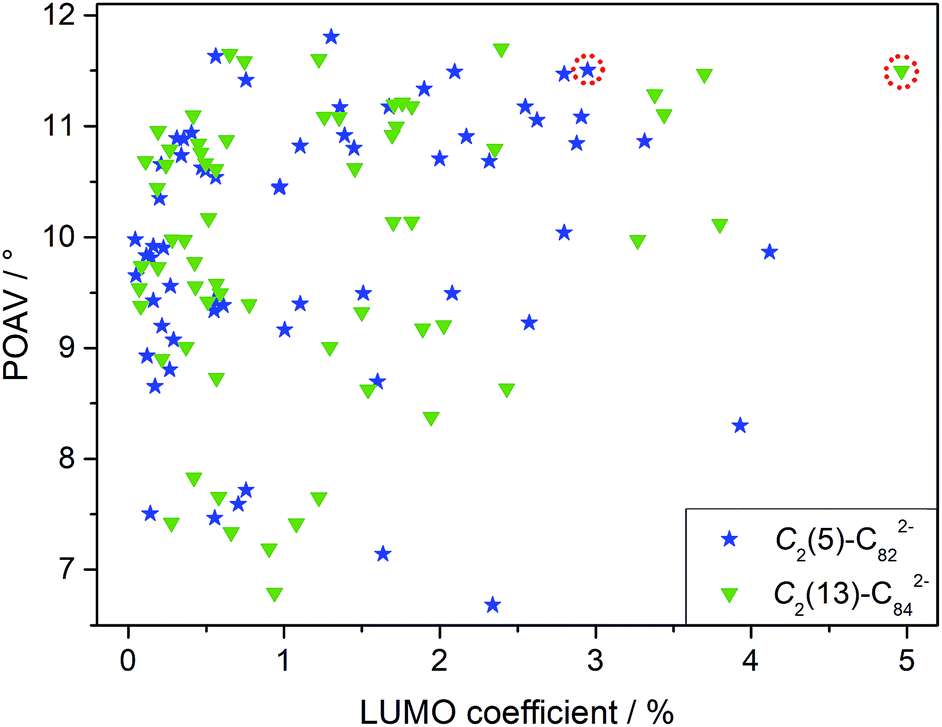 | ||
| Fig. 6 The average LUMO coefficients and the π-orbital axis vector (POAV) angles of two bonded cage carbons on [C2(5)-C82]2− and [C2(13)-C84]2−. The bonds at the sites of coordination are marked with a red dotted circle. Equivalent C–C bonds on the cages are omitted for clarity. | ||
In fact, our calculations reveal that the two-electron transfer does not alter the POAV values of the cage atoms but changes the LUMO distributions dramatically for both C2(5)-C82 and C2(13)-C84 (Fig. S4†). In particular, the LUMO distributions at the addition sites (C1–C2 in Fig. S4 and S5†) are remarkably enhanced by Eu-encapsulation with the values increasing from 0.58% to 2.95% for C2(5)-C82 and from 0.15% to 4.97% for C2(13)-C84, which eventually governs the high regioselectivity observed in the reactions. Accordingly, the addition pattern in 2a and 2b and the high regioselectivity in the reactions are controlled by the endohedral metal doping by changing the electronic structures of the corresponding hollow cages.
Another remarkable feature of the reactions is the formation of merely mono-adducts, whilst the reactions of EMFs usually produced mixtures containing mono-, bis- and multi-adducts.2,19,27,28 According to the reaction mechanism stated above, the LUMO and POAV distributions of the 2a and 2b were calculated (Fig. S6†). It is clear that the sites featuring both large POAV values and pronounced LUMO distributions are all close to the existing tungsten addend. Accordingly, further additions are prevented by the steric hindrance of the bulky tungsten addend. Thus, this work presents a practical strategy for the controlled synthesis of EMF-derivatives in a highly regioselective manner.
Conclusions
Reacting a tungsten complex W(CO)4(Ph2PC2H4PPh2) (1) with either Eu@C2(5)-C82 or Eu@C2(13)-C84 affords a sole mono-adduct for each metallofullerene (2a for Eu@C2(5)-C82 and 2b for Eu@C2(13)-C84). The X-ray results of 2a, 2b and the two pristine EMFs cocrystallized with Ni(OEP) clearly show that the motional behavior of the Eu atom inside the cage is significantly restricted upon exohedral tungsten addition, as a result of electrostatic potential redistribution. Meanwhile, theoretical results demonstrate that the endohedral Eu-doping has changed the LUMO distributions on the cages via charge transfer, which eventually leads to the regioselective addition of the exohedral tungsten addend. It is now evident that it is possible for the endohedral and exohedral metallic units in the complexes of EMFs to interact with each other via intramolecular charge transfer, highlighting the potential for the synthesis of EMF-based molecular devices with novel magnetic or electronic properties.Experimental
W(CO)4(Ph2PC2H4PPh2) (1) was synthesized according to the procedures described in the literature.29 Eu@C2(5)-C82 and Eu@C2(13)-C84 were produced with an arc-discharge method and were isolated with multi-stage HPLC (Fig. S7†).30 The reaction between 1 and Eu@C2(5)-C82/Eu@C2(13)-C84 was monitored with an HPLC system (LC-16, SHIMADZU LIMITED) equipped with an analytical Buckyprep column (∅ 4.6 × 250 mm). The separation was conducted on an HPLC machine (LC-908; Japan Analytical Industry Co. Ltd.) equipped with a preparative Buckyprep column (∅ 20 × 250 mm) with toluene as the eluent. The Vis-NIR experiments were carried out on a PE Lambda 750S UV-vis-NIR spectrophotometer and photoluminescence spectra were recorded on a Jasco FP-6500 machine.Crystallographic characterization
Single-crystal X-ray data were collected at 100 K using a radiation wavelength of 0.65250 Å with a MarCCD detector at beamline BL17B of the Shanghai Synchrotron Radiation Facility (China). A multi-scan method (SADABS) was used for absorption corrections. The structures were solved with direct methods and were refined with SHELXL-2016.31 Cocrystals of Eu@C2(5)-C82 or Eu@C2(13)-C84 were obtained by slow diffusion of a benzene solution of NiII(OEP) (OEP = octaethylporphyrin) into a CS2 solution of the respective EMF in a glass tube at room temperature for four weeks. Crystals of 2a or 2b were obtained by slow diffusion of n-hexane into a CS2 solution of 2a or 2b in glass tubes at room temperature for two weeks, respectively.Theoretical calculations
Density functional theory calculations were carried out by using the M06-2X functional in conjunction with the SDD basis set and the corresponding effective core potential for W and Eu and the standard 6-31G* basis set for all other atoms (denoted as 6-31G*∼SDD) as implemented in the Gaussian 09 software package.32–35 A spin multiplicity of M = 8 was employed according to the half-filled 4f shell of the Eu2+ ion.Chemical functionalization of Eu@C2(5)-C82/Eu@C2(13)-C84
In a typical reaction, a toluene solution containing ca. 2 mg (1.8 μmol) of Eu@C2(5)-C82 and 24 mg (34.6 μmol) of 1 was degassed with the freeze–pump–thaw method for three cycles. The mixture was then photo-irradiated with a mercury-arc lamp (cutoff < 350 nm) at room temperature for six minutes. The reaction mixture was concentrated and subjected to HPLC separations to afford 2a. The same procedures performed on Eu@C2(13)-C84 produced 2b.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the NSFC (Grants 51672093, 51602112 and 51602097) is gratefully acknowledged. We thank the staff from the BL17B beamline of the National Center for Protein Sciences Shanghai at the Shanghai Synchrotron Radiation Facility, for assistance during data collection, and the Analytical and Testing Center in the Huazhong University of Science and Technology for all related measurements.Notes and references
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CrossRef CAS PubMed.
- A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS PubMed.
- X. Lu, L. Echegoyen, A. L. Balch, S. Nagase and T. Akasaka, Endohedral Metallofullerenes: Basics and Applications, CRC Press, 2014 Search PubMed.
- R. D. Bolskar, A. F. Benedetto, L. O. Husebo, R. E. Price, E. F. Jackson, S. Wallace, L. J. Wilson and J. M. Alford, J. Am. Chem. Soc., 2003, 125, 5471–5478 CrossRef CAS PubMed.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed.
- S. Yang, T. Wei and F. Jin, Chem. Soc. Rev., 2017, 46, 5005–5058 RSC.
- M. Yamada, Y. Tanabe, J.-S. Dang, S. Sato, N. Mizorogi, M. Hachiya, M. Suzuki, T. Abe, H. Kurihara, Y. Maeda, X. Zhao, Y. Lian, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2016, 138, 16523–16532 CrossRef CAS PubMed.
- T. Cai, Z. X. Ge, E. B. Iezzi, T. E. Glass, K. Harich, H. W. Gibson and H. C. Dorn, Chem. Commun., 2005, 3594–3596 RSC.
- C. M. Cardona, A. Kitaygorodskiy and L. Echegoyen, J. Am. Chem. Soc., 2005, 127, 10448–10453 CrossRef CAS PubMed.
- C. M. Cardona, B. Elliott and L. Echegoyen, J. Am. Chem. Soc., 2006, 128, 6480–6485 CrossRef CAS PubMed.
- N. Chen, E. Y. Zhang, K. Tan, C. R. Wang and X. Lu, Org. Lett., 2007, 9, 2011–2013 CrossRef CAS PubMed.
- M. Yamada, T. Akasaka and S. Nagase, Acc. Chem. Res., 2010, 43, 92–102 CrossRef CAS PubMed.
- H. Fang, H. Cong, M. Suzuki, L. Bao, B. Yu, Y. Xie, N. Mizorogi, M. M. Olmstead, A. L. Balch, S. Nagase, T. Akasaka and X. Lu, J. Am. Chem. Soc., 2014, 136, 10534–10540 CrossRef CAS PubMed.
- B. Wu, T. Wang, Y. Feng, Z. Zhang, L. Jiang and C. Wang, Nat. Commun., 2015, 6, 6468 CrossRef CAS PubMed.
- N. B. Shustova, Y. S. Chen, M. A. Mackey, C. E. Coumbe, J. P. Phillips, S. Stevenson, A. A. Popov, O. V. Boltalina and S. H. Strauss, J. Am. Chem. Soc., 2009, 131, 17630–17637 CrossRef CAS PubMed.
- F. Jin, S. Wang, N. B. Tamm, S. Yang and S. I. Troyanov, Angew. Chem., Int. Ed., 2017, 56, 11990–11994 CrossRef CAS PubMed.
- R. C. Haddon, Science, 1993, 261, 1545–1550 CrossRef CAS PubMed.
- A. Ariafard, J. Organomet. Chem., 2004, 689, 2275–2283 CrossRef CAS.
- X. Lu, L. Feng, T. Akasaka and S. Nagase, Chem. Soc. Rev., 2012, 41, 7723–7760 RSC.
- Y. Maeda, Y. Matsunaga, T. Wakahara, S. Takahashi, T. Tsuchiya, M. O. Ishitsuka, T. Hasegawa, T. Akasaka, M. T. H. Liu, K. Kokura, E. Horn, K. Yoza, T. Kato, S. Okubo, K. Kobayashi, S. Nagase and K. Yamamoto, J. Am. Chem. Soc., 2004, 126, 6858–6859 CrossRef CAS PubMed.
- T. Akasaka, T. Kono, Y. Takematsu, H. Nikawa, T. Nakahodo, T. Wakahara, M. O. Ishitsuka, T. Tsuchiya, Y. Maeda, M. T. H. Liu, K. Yoza, T. Kato, K. Yamamoto, N. Mizorogi, Z. Slanina and S. Nagase, J. Am. Chem. Soc., 2008, 130, 12840–12841 CrossRef CAS PubMed.
- X. Lu, H. Nikawa, L. Feng, T. Tsuchiya, Y. Maeda, T. Akasaka, N. Mizorogi, Z. Slanina and S. Nagase, J. Am. Chem. Soc., 2009, 131, 12066–12067 CrossRef CAS PubMed.
- M. Hachiya, H. Nikawa, N. Mizorogi, T. Tsuchiya, X. Lu and T. Akasaka, J. Am. Chem. Soc., 2012, 134, 15550–15555 CrossRef CAS PubMed.
- W. Zhang, M. Suzuki, Y. Xie, L. Bao, W. Cai, Z. Slanina, S. Nagase, M. Xu, T. Akasaka and X. Lu, J. Am. Chem. Soc., 2013, 135, 12730–12735 CrossRef CAS PubMed.
- J. A. López and C. Mealli, J. Organomet. Chem., 1994, 478, 161–171 CrossRef.
- H. Zheng, X. Zhao and S. Sakaki, Inorg. Chem., 2017, 56, 6746–6754 CrossRef CAS PubMed.
- W. Zhang, M. Suzuki, Y. Xie, L. Bao, W. Cai, Z. Slanina, S. Nagase, M. Xu, T. Akasaka and X. Lu, J. Am. Chem. Soc., 2013, 135, 12730–12735 CrossRef CAS PubMed.
- Y. P. Xie, M. Suzuki, W. Cai, N. Mizorogi, S. Nagase, T. Akasaka and X. Lu, Angew. Chem., Int. Ed., 2013, 52, 5142–5145 CrossRef CAS PubMed.
- S. O. Grim, W. L. Briggs, R. C. Barth, C. A. Tolman and J. P. Jesson, Inorg. Chem., 1974, 13, 1095–1100 CrossRef CAS.
- L. Bao, P. Yu, C. Pan, W. Shen and X. Lu, Chem. Sci., 2019, 10, 2153–2158 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC profiles and theoretical calculation results. CCDC 1579794, 1579799, 1579800 and 1851767. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01479a |
| This journal is © The Royal Society of Chemistry 2019 |