
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUO22+-mediated ring contraction of pyrihexaphyrin: synthesis of a contracted expanded porphyrin-uranyl complex†
James T.
Brewster‡
II
 ,
Harrison D.
Root‡
,
Daniel
Mangel‡
,
Adam
Samia
,
Hadiqa
Zafar
,
Adam C.
Sedgwick
,
Vincent M.
Lynch
and
Jonathan L.
Sessler
*
,
Harrison D.
Root‡
,
Daniel
Mangel‡
,
Adam
Samia
,
Hadiqa
Zafar
,
Adam C.
Sedgwick
,
Vincent M.
Lynch
and
Jonathan L.
Sessler
*
Department of Chemistry, The University of Texas at Austin, 105 East 24th St., Stop A5300, Austin, Texas 78712, USA. E-mail: sessler@cm.utexas.edu
First published on 30th April 2019
Abstract
A new mixed hexaphyrin, pyrihexaphyrin (0.1.0.0.1.0) (1), was prepared via an acid catalyzed cyclization between 5,5′-(pyridine-2,6-diyl)bis(pyrrole-2-carbaldehyde) (2) and terpyrrole (3). This expanded porphyrin undergoes a ring contraction upon metallation with uranyl silylamide [UO2[N(SiMe3)2]2] under anaerobic conditions followed by purification over basic aluminum oxide exposed to air. The uranyl-contracted pyrihexaphyrin (0.0.0.0.1.0) complex (4) produced as a result contains a unique structural architecture and possesses a formally 22 π-electron globally aromatic periphery, as inferred from NMR spectroscopy, single crystal X-ray diffraction, and computational analyses. Support for the proposed contraction mechanism came from experimental data and DFT calculations. Proton NMR and mass spectroscopic analysis provided the first insight into expanded porphyrin-mediated activation of the uranyl dication (UO22+).
1. Introduction
Metalloporphyrins and ostensibly related corrin species are central to a variety of biochemical processes essential for life.1–3 Not surprisingly, the biosynthetic pathways and function of these and related conjugated tetrapyrrolic species remain a topic of fundamental interest.4–6 Insights obtained from model systems have helped elucidate the molecular origins of naturally occurring tetrapyrroles while synthetic analogues, such as corrole and amethyrin (Fig. 1), have revealed unique chemical and spectroscopic features not seen in the case of naturally occurring tetrapyrroles.7–15 A recent research focus has involved probing the influence of modified heterocyclic constituents within expanded and contracted porphyrins.16 Obtaining such species generally requires dedicated synthetic efforts wherein key precursors are constructed with structural components of the target in mind (Fig. 1).17,18 | ||
| Fig. 1 The core scaffold of porphyrin, corrole, and amethyrin with key structural components highlighted [dipyrromethane (blue), bipyrrole (red), terpyrrole (purple)]. | ||
Nature uses uroporphyrinogen III, a common porphyrin precursor, to prepare the structurally unique corrin core found in cobalamin (vitamin B12) (Scheme 1).20 Within this biosynthetic sequence, an incipient cobalt–corrin complex facilitates ring contraction.21 Biomimetic ring contraction–oxidation approaches involving porphyrin starting materials have allowed for the construction of substituted corroles.18,19 An early route by Johnson and co-workers utilized sulfur extrusion from thiaphlorins to yield corrole, often in good yields.22 However, the requisite sulfur-based starting materials are unstable, which limited the effectiveness of this approach. More recent examples have further mimicked nature by using a metal-mediated strategy, wherein a metalloporphyrin undergoes ring contraction.23–25
 | ||
| Scheme 1 Ring contraction sequence in the anaerobic biosynthesis of cobalamin. | ||
Expanded porphyrins, species containing a larger internal cavity, have received considerable attention in recent years.26–29 Modification of coordinating moieties within the internal lacuna has provided a number of systems displaying rich coordination chemistry and extended conjugation pathways. However, expanded porphyrins displaying meso-reactivity are rare. Early on Sessler and co-workers reported meso-activation of sapphyrin, wherein U(VI)O22+ coordination enabled addition of methanol and dearomatization.30 Continued efforts by the groups of Yoneda and Neya have shown that metallation of sapphyrin with Ag(I) results in a structural rearrangement with attendant incorporation of two methanol molecules into a neo-confused sapphyrin core.31 A recent example by Osuka and co-workers demonstrated that water can insert into the meso-position of hexaphyrin (1.1.1.1.1.1) upon Cu(II)- or Ni(II)-metallation.32 Setsune and co-workers have also reported that dipyriamethyrin can undergo meso-selective nucleophilic addition with cyanide, amines, and alkoxides.33 Elegant demonstrations of ring contraction have been observed by the Furuta group with N-confused dihydrosapphyrin to give neo-confused corrole,34 as well as by Maruyama, Fujita, Osuka, and co-workers with bis-Cu(II)octaphyrin to give Cu(II)-porphyrin.35 Similar chemistry has been applied by both groups in other selected systems.36 Metal-mediated contractions have also been reported by Latos-Grażyński et al. in the case of azuli- and benziporphyrinoids.37 Interestingly, metal-mediated meso-extrusion has been primarily limited to porphycene.38
Not surprisingly, meso-activation generally results in the formation of high-energy intermediates inducing structural modification (i.e., ring contraction) as a means to reach an energetic minimum. Owing to the complex architectures and difficulty in observing often transient intermediates, mechanistic rationales for such transformations are rarely detailed.18,23,25,36 Furthermore, the proposed mechanisms usually fail to include computational analyses as additional support. Here, we report the synthesis of a new hexaphyrin, pyrihexaphyrin (0.1.0.0.1.0) (1), that upon uranyl dication (UO22+) complexation under anaerobic conditions and purification on the bench-top over basic aluminum oxide undergoes ring contraction. The resulting contracted pyrihexaphyrin (0.0.0.0.1.0) displayed an asymmetric architecture with connectivity markedly different than that of previously reported expanded porphyrinoids.18,23,25–29,36–39 Structural reorganization was also accompanied by electronic reconfiguration with the uranyl-contracted pyrihexaphyrin (0.0.0.0.1.0) complex displaying Hückel-type aromatic features. Experimental data and computational analyses, in tandem, provide support for the proposed activation of a uranyl(VI) dication (UO22+) accessing a U(V) intermediate that facilitates the observed ring contraction.
2. Results and discussion
Pyrihexaphyrin (1) was designed to combine within one molecular framework key features of amethyrin40 and dipyriamethyrin.41 It was prepared in 86% yield via a trifluoroacetic acid (TFA) catalyzed cyclization of 5,5′-(pyridine-2,6-diyl)bis(pyrrole-2-carbaldehyde) (2)42 and terpyrrole (3)43 (Scheme 2).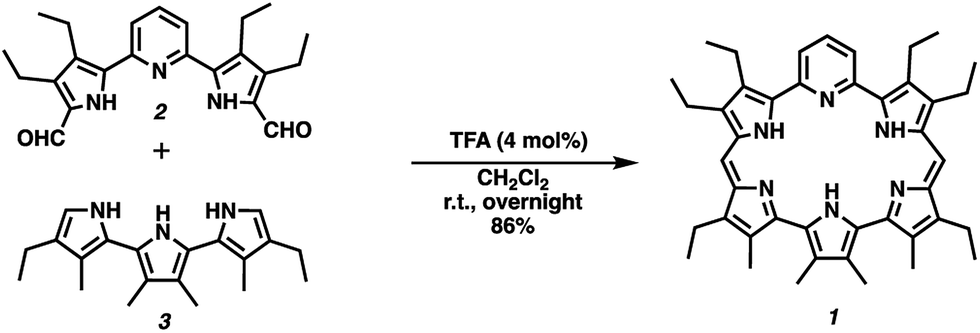 | ||
| Scheme 2 Synthesis of pyrihexaphyrin (1). | ||
Proton NMR spectroscopic analyses of pyrihexaphyrin 1 in CDCl3 revealed features consistent with a non-aromatic porphyrinoid core. In particular, resonances attributed to the pyridine CH protons and pyrrole NH protons were not shifted (ESI S3†). The UV-vis absorption spectrum of 1 recorded in CHCl3 is characterized by a Soret-like band at λmax = 452 nm (ε = 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) and two smaller peaks at λ = 372 nm (ε = 17000 M−1 cm−1) and λ = 278 nm (ε = 14000 M−1 cm−1) (Fig. 2). A broad shoulder extending from λ = 600 to 850 nm is also seen. A cyclic voltammogram (CV) of 1 recorded in acetonitrile ([1] = 1 mM) revealed a quasi-reversible oxidation feature at 27 mV relative to Fc/Fc+ and two quasi-reversible reduction waves at −1267 mV and −1992 mV. An irreversible reduction was also observed at −1600 mV. An irreversible oxidation was observed on the return sweep at −518 mV and two irreversible oxidation peaks were observed above 250 mV. These peaks are attributed to reversible two electron oxidation and reduction processes involving the pyrihexaphyrin ring with irreversible events being observed at higher redox potentials.44,45
000 M−1 cm−1) and two smaller peaks at λ = 372 nm (ε = 17000 M−1 cm−1) and λ = 278 nm (ε = 14000 M−1 cm−1) (Fig. 2). A broad shoulder extending from λ = 600 to 850 nm is also seen. A cyclic voltammogram (CV) of 1 recorded in acetonitrile ([1] = 1 mM) revealed a quasi-reversible oxidation feature at 27 mV relative to Fc/Fc+ and two quasi-reversible reduction waves at −1267 mV and −1992 mV. An irreversible reduction was also observed at −1600 mV. An irreversible oxidation was observed on the return sweep at −518 mV and two irreversible oxidation peaks were observed above 250 mV. These peaks are attributed to reversible two electron oxidation and reduction processes involving the pyrihexaphyrin ring with irreversible events being observed at higher redox potentials.44,45
 | ||
| Fig. 2 Stacked (a) UV-vis spectra in CHCl3 and (b) CV of 1 and 4 ([c] = 1 mM) in dry acetonitrile containing 0.1 M [(n-Bu)4N][ClO4] as the electrolyte. All potentials are reported relative to the Fc/Fc+ couple. | ||
Initial efforts at probing the coordination chemistry with transition metal and lanthanide cation salts returned mixtures of starting material and putative incorporation of methoxide within the meso-position with and without metal insertion, as inferred from mass spectrometric analyses (ESI S6–S10†). Cation sources tested in this regard included AgOAc, Cu(OAc)2, Co(OAc)2, Fe(OAC)2, In(OAc)3, ZrCl4, Ni(OAc)2, Gd(OAc)3, and VO(acac)2.
Both amethyrin and dipyriamethyrin are known to stabilize complexes with certain actinyl ions.40,41 This led us to explore whether a uranyl(VI) (UO22+) complex could be stabilized using 1. However, in contrast with what was previously seen, a ring-contracted product was obtained. Specifically, reaction of compound 1 with three equivalents of uranyl silylamide [UO2[N(SiMe3)2]2]46 in tetrahydrofuran followed by purification over basic aluminum oxide§ gave the 22 π-electron aromatic contracted pyrihexaphyrin (0.0.0.0.1.0)-uranyl complex (4) in 63–65% yield (Scheme 3).¶ Attempts to remove the coordinated U(VI)O22+ ion by acidic, 1 N HCl in MeOH at room temperature overnight, and basic, 10 equiv. 1,8-diazobicyclo[5.4.0]undec-7-ene in methanol at room temperature overnight, conditions returned complex 4 and a mixture of decomposition products. Such results are not surprising as the constricted internal cavity prevents significant decomplexation. The coordinated UO22+ ion is also expected to provide stabilization for the high-lying HOMO of a 22 π-electron Hückel-type aromatic core.
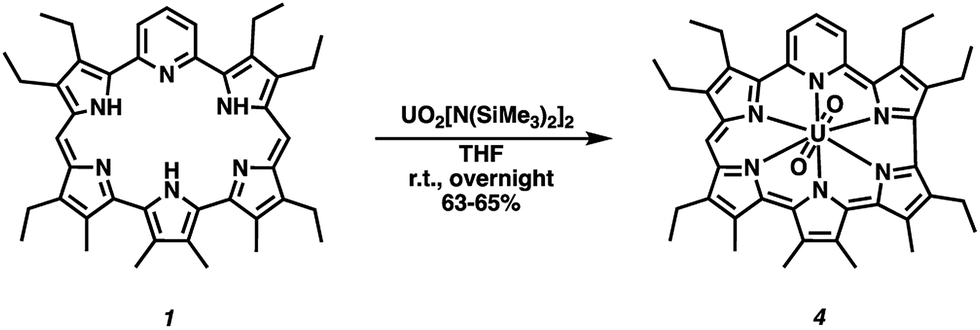 | ||
| Scheme 3 Synthesis of expanded corrole–uranyl complex (4). | ||
Proton NMR spectroscopic analyses revealed that 4 possessed relatively low symmetry, as inferred from the large number of observable signals. Downfield shifts in the proton resonances corresponding to the pyridine CH peaks and meso-CH peak were also seen. These shifts mirror what had been observed in the case of napthoisoamethyrin,47 isoamethyrin,48 and amethyrin40 upon uranyl coordination, findings that were attributed to formation of a globally aromatic core.49 UV-vis spectroscopic analyses revealed marked differences in comparison to previous hexaphyrin systems, as well as to the parent ligand 1. In particular, a relatively weak Soret-like band was observed at λmax = 549 nm (ε = 18400 M−1 cm−1), along with features at λ = 365 nm (ε = 13300 M−1 cm−1), λ = 734 nm (ε = 4800 M−1 cm−1), and λ = 1003 nm (ε = 9300 M−1 cm−1) (Fig. 2). CV studies of complex 4 (1 mM in acetonitrile) revealed two quasi-reversible peaks at −916 mV and −1270 mV vs Fc/Fc+, respectively. A slight shoulder preceding the reduction event at −1270 mV was observed at −960 mV. Two quasi-reversible oxidation events were observed at 236 mV and 445 mV (Fig. 2). These complex redox features suggest instability of the radical intermediates with concomitant decomposition via chemical reaction. A separate mechanism wherein electronic communication between the uranyl(VI) and the porphyrinoid ligand stabilizes intermediate radical species with distinct redox events might also be operative.
Single crystals suitable for X-ray diffraction analysis of 1 and 4 were grown via slow evaporation of a CH2Cl2: hexanes mixture (1:1, v/v). As shown in Fig. 3, the metal-free pyrihexaphyrin (1) adopts a twisted conformation wherein the central pyridine (N1) and pyrrole (N4) point in toward the concave face. The distortion largely involves torsion between the central heterocycles and dipyrromethene subunits. The cavity size, as measured from the in-plane dipyrromethene subunits, was found to be 5.846 Å (N3 to N6). On this basis, the cavity size of 1 is similar to that of dipyriamethyrin, but smaller than amethyrin, for which opposing N–N distances of 5.858 Å and 6.055 Å were found, respectively.
 | ||
| Fig. 3 ORTEP plot (thermal ellipsoids set at 50% probability) of the structures obtained from single crystal X-ray diffraction analyses of (a) 1 and (b) 4 viewed from top and side (upper and lower views, respectively). Hydrogen atoms are removed for clarity. | ||
As expected for an aromatic system, upon uranyl cation complexation and contraction, a structure (4) with greater overall planarity was obtained (Fig. 3). Complex 4 displays an atypical asymmetric coordination environment characterized by a distorted hexagonal bipyramidal geometry. Relevant bond angles were: 59.32° (N1 to N2), 58.26° (N2 to N3), 58.74° (N3 to N4), 59.28° (N4 to N5), 67.18° (N5 to N6), and 59.60° (N6 to N1). Uranyl-to-nitrogen bond distances were: 2.532 (6) Å (N4), 2.557 (6) Å (N3), 2.561 (6) Å (N2), 2.622 (6) Å (N6), 2.658 (6) Å (N5), and 2.716 (6) Å (N1). The U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds also showed slight asymmetry with U–O distances of 1.780 (5) Å and 1.796 (5) Å.
O bonds also showed slight asymmetry with U–O distances of 1.780 (5) Å and 1.796 (5) Å.
In an effort to understand the unique reactivity of 1, density functional theory (DFT) calculations were carried out using the Gaussian 16 package.50 Geometric relaxations were performed using the X-ray crystal structural parameters so as to obtain energy minimized structures (Fig. S1†) and simulated spectra (Fig. S2†). Concordance with the experimental data was seen in both cases.
The aromatic nature of 1 was probed using nucleus independent chemical shifts (NICS)51 and anisotropy induced current density (ACID)52 plots. As expected, 1 gave an NICS value of 1.119, indicative of a non-aromatic species, while the ACID plot displayed a node between the meso-bridge and adjacent pyrroles, indicating weak conjugation (Fig. S3†). An electrostatic potential map showed neutral to high levels of electron density throughout the macrocycle (Fig. S4†). In contrast, the ACID plot for the uranyl complex 4 revealed a clockwise current characteristic of a diatropic π-system (Fig. S5†). This was taken as further evidence that complex 4 is best considered as being an aromatic species rather than one with a more localized conjugation (e.g., 4′ in Scheme 4).||
A proposed mechanism for the metallation-contraction involved in the conversion of 1 to 4 is shown in Scheme 4. Briefly, pyrihexaphyrin is expected to undergo deprotonation-mediated metal insertion with concomitant oxidation to give the cationic uranyl(VI) complex A. Formation of A is postulated to proceed via an intramolecular uranyl(VI) [(L*U(IV)O2)+] facilitated ring oxidation to yield water and a uranium (IV) species 5 that is re-oxidized to the uranyl(VI) complex A upon exposure to air.30,53 However, the alternative formation of a ligand-based radical-U(V) intermediate cannot be excluded. Bart, Schelter, and co-workers have recently reported the ability of redox-active iminoquinone ligands to facilitate oxo-activation and reduction of U(VI) to U(IV).54 Furthermore, in the presence of a reductant and proton source, the reduction of U(VI)O22+ under anaerobic conditions to yield U(VI) and water can occur.54,55
 | ||
| Scheme 4 Proposed base-mediated mechanism for the observed ring contraction that yields pyrihexaphyrin (0.0.0.0.1.0)-uranyl complex 4. | ||
In loose analogy to the ring-contraction central to the biosynthesis of vitamin B12, the activated metallo-expanded porphyrin A is expected to facilitate nucleophilic addition of a hydroxide anion into the meso-position to give dearomatized intermediate B.56 Subsequent pyrrole α-addition into the uranyl-activated CN bond, followed by rearrangement of the 1,2-donor-acceptor cyclopropanolate (C), would yield the exo-carbaldehyde D.57 The porphyrinoid ring in conjunction with in-plane uranyl(VI) center are expected to act as an electron sink and provide resonance stabilization to this intermediate, as illustrated in D′. A final sequence of base-mediated fragmentation,56b,58 proceeding via intermediate E and resonance stabilized intermediates F or F′ with the uranyl(V) center acting as an electron sink with attendant delocalization across the porphyrinoid core, followed by aerobic oxidation would give the aromatic product 4.
An alternative mechanism may also be operative wherein hydroperoxide, present from the reduction of molecular oxygen and deprotonation by basic aluminum oxide, adds into the meso-position to give a hydroperoxide dearomatized intermediate.56 Subsequent rearrangement of the 1,2-donor–acceptor peroxycyclopropane with attendant cleavage of the O–O peroxide bond would again yield an exo-carbaldehyde on an α-carbon of the bipyrrole subunit.57 A final sequence of base-mediated fragmentation, followed by oxidation would give the aromatic ring-contracted product 4 (Scheme S1†).
Support for the initial step of this proposed mechanism came from 1H NMR analysis of intermediate 5 (Fig. S6†), as well as low and high-resolution mass spectrometric analyses and the observation of peaks consistent with intermediate A (Fig. S7 & S8†).** In particular, the crude reaction mixture, filtered through Celite and dried under reduced pressure, yielded a proton NMR spectrum devoid of proton resonances with a signals attributed to water, THF, hexanes, NaHMDS, HN(SiMe3)2, and CHCl3. While not definitive proof, these observations are consistent with the presence of a U(IV)-pyrihexaphyrin complex produced via reduction of U(VI)O22+ with the lack of resonances attributed to the through-contact or -space transfer of spin density from the actinide ion to the porphyrinoid ligand.59 An alternative formulation wherein the electrons are delocalized across a porphyrinoid-U(V) intermediate is also possible.†† The same solid was also utilized immediately for mass spectrometric analyses as a methanolic solution. In our hands, the use of non-aqueous uranyl silylamide [UO2[N(SiMe3)2]2] under anaerobic conditions was required to induce this transformation. Efforts to effect U(VI)O22+ insertion using UO2(OAc)2 dihydrate in CH2Cl2:MeOH (1:1, v/v) failed to yield isolable quantities of the metalated product.
Computational analysis via DFT calculations revealed an electrostatic potential map for A characterized by low levels of electron density throughout the macrocycle (Fig. S9†). An ACID plot of A proved consistent what would be expected for a non-aromatic species with a non-planar geometry (Fig. S9†). Also of note is that a stable porphycene-based ruthenium complex and porphycene-free base analogous to intermediate D have been isolated after purification over basic aluminum oxide or in the presence of base, respectively. The resultant exo-carbaldehyde porphycenes displayed aromatic features and thus had no thermodynamic driving force to excise the pendant aldehyde.38a,c Furthermore, uranyl (UO22+)-mediated oxidation to furnish globally aromatic expanded porphyrins has been observed with amethyrin,40 naphthoisoamethyrin,46 isoamethyrin,47 cyclo[6]pyrrole,60 and cyclo[1]furan[1]pyridine[4]pyrrole.61,‡‡ However, no intermediate species or postulated mechanisms have been reported for any of these systems.
3. Conclusions
In conclusion, a new expanded porphyrin, pyrihexaphyrin (0.1.0.0.1.0) (1) has been prepared. Treatment of 1 with non-aqueous uranyl silylamide under anaerobic conditions, followed by purification on basic aluminum oxide exposed to air, yielded the ring contracted pyrihexaphyrin (0.0.0.0.1.0)-uranyl complex (4) with a 22 π-electron Hückel-type aromatic core. A postulated mechanism proceeding via a putative U(IV) intermediate followed by basic aluminum oxide mediated dearomatization-ring contraction is supported by proton NMR and MS spectroscopic analyses as well as DFT calculations. The present constructs thus provide insight into expanded porphyrinoid activation of the uranyl(VI) dication (UO22+) while highlighting the unique reactivity of mixed porphyrinoid structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Office of Basic Energy Sciences, U.S. Department of Energy (DOE) (Grant DOE-FG02-01ER15186 to J. L. S.) and the Robert A. Welch Foundation (F-0018 to J. L. S.). The authors acknowledge the Texas Advanced Computing Center (TACC) for providing HPC resources that have contributed to the research results reported within this paper. The authors would like to acknowledge NIH funding (NIH Grant Number 1-S10-OD021508-01) for the Bruker AVANCE III 500. J. T. B. and H. D. R. would like to thank The University of Texas at Austin for a Scientist in Residence Fellowship. J. T. B. would like to thank Los Alamos National Laboratory for a G. T. Seaborg Fellowship.References
- A. R. Battersby, Nat. Prod. Rep., 2000, 17, 507 RSC.
- A. Vannotti, Porphyrins. Their Biological and Chemical Importance, Hilger and Watts, London, 1955 Search PubMed.
- S. Lesage, H. Hu and L. Durham, Hydrolog. Sci. J., 1993, 38, 343 CrossRef CAS.
- D. Shemin and C. S. Russell, J. Am. Chem. Soc., 1953, 75, 4873 CrossRef CAS; J. M. Tomio and M. Grinstein, Eur. J. Biochem., 1968, 6, 80 CrossRef; H. L. Bonkovsky, T. J. Guo, W. Hou, T. Li, T. Narang and M. Thapar, Compr. Physiol., 2013, 3, 365 Search PubMed.
- K. Iida, K. Ohtaka and M. Kajiwara, FEBS J., 2007, 274, 3475 CrossRef CAS PubMed; D. Heldt, A. D. Lawrence, M. Lindenmeyer, E. Deery, P. Heathcote, S. E. Rigby and M. J. Warren, Biochem. Soc. Trans., 2005, 33, 815 CrossRef PubMed.
- M. K. Geno and J. Halpern, J. Am. Chem. Soc., 1987, 109, 1238 CrossRef CAS.
- M. R. Wasielewski, Chem. Rev. Soc., 1992, 92, 435 CrossRef CAS.
- S. Schulz, R. J. Wong, H. J. Vreman and D. K. Stevenson, Front. Pharmacol., 2012, 3, 68 Search PubMed.
- M. Imran, M. Ramzan, A. K. Qureshi, M. A. Khan and M. Tariq, Biosensors, 2018, 8, 95 CrossRef PubMed.
- I. Aviv and Z. Gross, Chem. Commun., 2007, 1987 RSC.
- A. Mahmood, J.-Y. Hu, B. Xiao, A. Tang, X. Wang and E. Zhou, J. Mater. Chem. A., 2018, 6, 16769 RSC.
- S. Nardi, D. Monti and R. Paolesse, Mini Rev. Org. Chem., 2005, 2, 355 CrossRef.
- C. I. M. Santos, E. Oliveira, J. F. B. Barata, M. A. F. Faustino, J. A. S. Cavaleiro, M. G. P. M. S. Neves and C. Lodeiro, J. Mater. Chem., 2012, 22, 13811 RSC.
- W. Auwarter, D. Ecija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105 CrossRef PubMed.
- R. D. Teo, J. Y. Hwang, J. Termini, Z. Gross and H. B. Gray, Chem. Rev., 2017, 117, 2711 CrossRef CAS PubMed.
- J. L. Sessler, S. J. Weghorn, Expanded, Contracted & Isomeric Porphyrins, Pergamon, New York, 1997 CrossRef CAS PubMed; T. Sarma and P. K. Panda, Chem. Rev., 2017, 117, 2785 CrossRef CAS PubMed; T. Chatterjee, V. S. Shetti, R. Sharma and M. Ravikanth, Chem. Rev., 2017, 117, 3254 CrossRef PubMed; M. d. G. H. Vincente and K. M. Smith, Curr. Org. Synth., 2014, 11, 3–28 CrossRef.
- S. Hiroto, Y. Miyake and H. Shinokubo, Chem. Rev., 2017, 117, 2910 CrossRef CAS PubMed.
- R. Orlowski, D. Gryko and D. T. Gryko, Chem. Rev., 2017, 117, 3102 CrossRef CAS PubMed; J. F. B. Barata, M. G. P. M. S. Neves, A. C. Faustino, J. Tome and A. S. Cavaleiro, Chem. Rev., 2017, 117, 3192 CrossRef PubMed.
- P. G. Bulger, S. K. Bagal and R. Marquez, Nat. Prod. Rep., 2008, 25, 254 RSC; M. C. de la Torre and M. A. Sierra, Angew. Chem., Int. Ed., 2004, 43, 160–181 CrossRef CAS PubMed.
- A. I. Scott and C. A. Roessner, Biochem. Soc. Trans., 2002, 30, 613 CrossRef CAS.
- C. A. Roessner, P. J. Santander and A. I. Scott, Vitam. Horm., 2001, 61, 267 CrossRef CAS.
- M. J. Broadhurst, R. Grigg and A. W. Johnson, J. Chem. Soc., Perkin Trans. 1, 1972, 1124 RSC; M. J. Broadhurst, R. Grigg and A. W. Johnson, J. Chem. Soc. D, 1970, 807 RSC.
- H. J. Callot, Dalton Trans., 2008, 6346 RSC.
- B. Szyszko and L. Latos-Grazynski, Chem. Soc. Rev., 2015, 44, 3588–3616 RSC.
- B. Patra, S. Sobottka, S. Mondal, B. Sarkar and S. Kar, Chem. Commun., 2018, 54, 9945 RSC.
- J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134 CrossRef CAS PubMed.
- V. V. Roznyatovskiy, C.-H. Lee and J. L. Sessler, Chem. Soc. Rev., 2013, 42, 1921 RSC.
- J. Mack, Chem. Rev., 2017, 117, 3444 CrossRef CAS.
- S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 4342 CrossRef CAS PubMed.
- A. K. Burrell, M. J. Cyr, V. Lynch and J. L. Sessler, J. Chem. Soc., Chem. Commun., 1991, 1710 RSC.
- D. Mori, T. Yoneda, T. Hoshino and S. Neya, Chem. Asian. J., 2018, 13, 934 CrossRef CAS PubMed.
- S. Shimizu, V. G. Anard, R. Taniguchi, K. Furukawa, T. Kato, T. Yokoyama and A. Osuka, J. Am. Chem. Soc., 2004, 126, 12280 CrossRef CAS PubMed; M. Inoue, C. Ikeda, Y. Kawata, S. Venkatraman, K. Furukawa and A. Osuka, Angew. Chem., Int. Ed., 2007, 46, 2306 CrossRef PubMed.
- J.-i. Setsune and K. Yamato, Chem. Commun., 2012, 48, 4447 RSC.
- Y. Xie, P. Wei, X. Li, T. Hong, K. Zhang and H. Furuta, J. Am. Chem. Soc., 2013, 135, 19119 CrossRef CAS PubMed.
- Y. Tanaka, W. Hoshino, S. Shimizu, K. Youfu, N. Aratani, N. Maruyama, S. Fujita and A. Osuka, J. Am. Chem. Soc., 2004, 126, 3046 CrossRef CAS PubMed.
- M. Toganoh and H. Furuta, Chem. Commun., 2012, 48, 937 RSC; R. Sakamoto, S. Saito, S. Shimizu, Y. Inokuma, N. Aratani and A. Osuka, Chem. Lett., 2010, 39, 439 CrossRef CAS; K. Moriya, S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 4297 CrossRef PubMed; Q. Li, M. Ishida, H. Kai, T. Gu, C. Li, X. Li, B. Baryshnikov, X. Liang, W. Zhu, H. Agren, H. Furuta and Y. Xie, Angew. Chem., Int. Ed., 2019, 58, 5925 CrossRef PubMed.
- (a) M. J. Bialek and L. Latos-Grażyński, Chem. Commun., 2018, 54, 1837 RSC; (b) M. J. Bialek and L. Latos-Grażyński, Inorg. Chem., 2016, 55, 1758 CrossRef CAS PubMed; (c) K. Hurej, M. Pawlicki, L. Szterenberg and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2016, 55, 1427 CrossRef CAS PubMed; (d) K. Hurej, M. Pawlicki and L. Latos-Grażyński, Chem.–Eur. J., 2018, 24, 115 CrossRef CAS PubMed; (e) K. Hurej, M. Pawlicki and L. Latos-Grażyński, Chem.–Eur. J., 2017, 23, 2059 CrossRef CAS PubMed.
- (a) A. Anguera, W.-Y. Cha, M. D. Moore, J. T. Brewster II, M. Y. Zhao, V. M. Lynch, D. H. Kim and J. L. Sessler, Angew. Chem., Int. Ed., 2018, 57, 2575 CrossRef PubMed; (b) J. T. Brewster, G. Anguera and J. L. Sessler, Synlett, 2019, 30, 765 CrossRef CAS; (c) S. Will, A. Rahbar, H. Schmickler, J. Lex and E. Vogel, Angew. Chem., Int. Ed., 1990, 12, 1390 CrossRef.
- U. Englich and K. Ruhlandt-Senge, Org. Lett., 1999, 1, 587 CrossRef CAS; R. Kumar, R. Misra, V. Prabhuraja and T. K. Chandrashekar, Chem.–Eur. J., 2005, 11, 5695 CrossRef PubMed; R. Paolesse, R. G. Khoury, F. D. Sala, C. D. Natale, F. Sagone and K. M. Smith, Angew. Chem., Int. Ed., 1999, 38, 2577 CrossRef PubMed.
- J. T. Brewster II, A. Aguilar, G. Anguera, H. Zafar, M. D. Moore and J. L. Sessler, J. Coord. Chem., 2018, 71, 1808 CrossRef CAS; J. L. Sessler, A. E. V. Gorden, D. Seidel, S. Hannah, V. Lynch, P. L. Gordon, R. J. Donohoe, C. D. D. Tait and D. W. W. Keogh, Inorg. Chim. Acta, 2002, 341, 54 CrossRef.
- J. T. Brewster, Q. He, G. Anguera, M. Moore, X.-S. Ke, V. M. Lynch and J. L. Sessler, Chem. Commun., 2017, 53, 4981 RSC.
- J.-i. Setsune and K. Watanabe, J. Am. Chem. Soc., 2008, 130, 2404 CrossRef CAS PubMed.
- J. L. Sessler, S. J. Weghorn, Y. Hiseada and V. Lynch, Chem.–Eur. J., 1995, 1, 56 CrossRef CAS.
- A. G. Nepomnyashchi, M. Broring, J. Ahrens and A. J. Bard, J. Am. Chem. Soc., 2011, 133, 19498 CrossRef PubMed.
- Y. Fang, Z. Ou and K. M. Kadish, Chem. Rev., 2017, 117, 3377 CrossRef CAS PubMed.
- C. J. Burns, D. L. Clark, R. J. Donohoe, P. B. Duval and B. L. Scott, Inorg. Chem., 2000, 39, 5464 CrossRef CAS PubMed.
- G. Anguera, J. T. Brewster II, M. D. Moore, J. Lee, G. I. Vargas-Zuniga, H. Zafar, V. M. Lynch and J. L. Sessler, Inorg. Chem., 2017, 56, 9409 CrossRef CAS PubMed.
- J. L. Sessler, D. Seidel, A. E. Vivian, V. Lynch, B. L. Scott and D. W. Keogh, Angew. Chem., Int. Ed., 2001, 40, 591 CrossRef CAS PubMed.
- A. L. Sargent, I. C. Hawkins, W. E. Allen, H. Liu, J. L. Sessler and C. J. Fowler, Chem.–Eur. J., 2003, 9, 3065 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. von Rague Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- D. Geuenich, K. Hess, F. Kohler and R. Herges, Chem. Rev., 2005, 105, 3758 CrossRef CAS PubMed.
- H. S. Moon, J. Komlos and P. R. Jaffe, J. Contam. Hydrol., 2009, 105, 18 CrossRef CAS PubMed.
- E. J. Coughlin, Y. Qiao, E. Lapsheva, M. Zeller, E. J. Schelter and S. C. Bart, J. Am. Chem. Soc., 2019, 141, 1016 CrossRef CAS PubMed.
- E. L. Muhr-Ebert, F. Wagner and C. Walther, Appl. Geochem., 2019, 100, 213 CrossRef.
- (a) Basic aluminium oxide mediated elimination, dehydration, condensation, and epoxide ring opening are well-documented processes, see: V. H. Rawal, S. Iwasa, A. S. Florjancic, A. Faber, Alumina Encyclopedia of Reagents for Organic Synthesis, Wiley, New York, 1995, vol. 1, p. 141 Search PubMed; (b) G. H. Posner, Angew. Chem., Int. Ed., 1978, 17, 487 CrossRef; (c) S. Siddhan and K. Narayanan, J. Catal., 1979, 59, 405 CrossRef CAS; (d) H. A. Dabbagh and J. M. Salehi, J. Org. Chem., 1998, 63, 7619 CrossRef CAS; (e) H. Knozinger, Angew. Chem., Int. Ed., 1968, 7, 791 CrossRef and references cited therein.
- O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597 CrossRef CAS PubMed.
- Base-mediated decarbonylation has also been observed in the reaction between aluminium oxide and steroid substrates, see: M. Kaufman, P. Morand and S. A. Samad, J. Org. Chem., 1972, 37, 1067 CrossRef CAS; J. F. Bagli, P. F. Morand and R. Gaudry, J. Org. Chem., 1963, 28, 1207 CrossRef; R. M. Moriarty and K. Kapadia, Tet. Lett., 1964, 5, 1165–1170 CrossRef An aqueous base-mediated method using 4% NaOH has also been reported, see: H. Hagiwara, S. Noguchi and M. Nishikawa, Chem. Pharm. Bull., 1960, 8, 84 CrossRef.
- J. Lisowski, J. L. Sessler, V. Lynch and T. D. Mody, J. Am. Chem. Soc., 1995, 117, 2273 CrossRef CAS.
- P. J. Melfi, S. K. Kim, J. T. Lee, F. Bolze, D. Seidel, V. M. Lynch, J. M. Veauthier, A. J. Gaunt, M. P. Neu, Z. Ou, K. M. Kadish, S. Fukuzumi, K. Ohkubo and J. L. Sessler, Inorg. Chem., 2007, 46, 5143 CrossRef CAS PubMed.
- I.-T. Ho, Z. Zhang, M. Ishida, V. M. Lynch, W.-Y. Cha, Y. M. Sung, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2014, 136, 4281 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray crystal structure for 1893382 (1) and 1893883 (4) (CIF); detailed experimental procedures and characterization data for all new compounds (PDF). CCDC 1893382 and 1893883. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01593k |
| ‡ These authors contributed equally. |
| § Commercial basic alumina is treated with base until the pH of a 10% slurry (w/v in water) is ∼10. Base-mediated ring contraction yielding an exo-carbaldehyde has also been observed in porphycene (ref. 37c). |
| ¶ Repeat attempts using UO2(OAc)2 (5 equiv.) in CH2Cl2:MeOH (1:1, v/v) open to air returned only starting material. |
| || The calculated molecular orbital energies also revealed a reduced HOMO–LUMO gap between 1 (2.3 eV) and 4 (1.6 eV; Δ 16.1 kcal/mol). Furthermore, the LUMO+1, LUMO+2, and LUMO+3 of uranyl complex 4 were found to be metal centered orbitals with similar energies, ranging −2.82 eV to −2.61 eV (Fig. S10†). |
| ** Attempts at isolating intermediate A under anaerobic conditions failed to yield X-ray quality crystals. |
| †† The formation of U(V) intermediates are generally viewed as unstable due to the 5f1 electron configuration. However, in the presence of an appropriate ligand environment stabilization of uranyl(V) species is possible; see: R. Faizova, R. Scopelliti, A.-S. Chauvin, M. Mazzanti. J. Am. Chem. Soc., 2018, 140, 13554. |
| ‡‡ No ring contraction or other structural modifications were observed in these instances. |
| This journal is © The Royal Society of Chemistry 2019 |