
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two non-identical twins in one unit cell: characterization of 34π aromatic core-modified octaphyrins, their structural isomers and anion bound complexes†
Arindam
Ghosh‡
 a,
Syamasrit
Dash‡
a,
A.
Srinivasan
a,
Cherumuttathu H.
Suresh
b and
Tavarekere K.
Chandrashekar
*a
a,
Syamasrit
Dash‡
a,
A.
Srinivasan
a,
Cherumuttathu H.
Suresh
b and
Tavarekere K.
Chandrashekar
*a
aSchool of Chemical Sciences, National Institute of Science Education and Research (NISER), HBNI, Bhubaneswar-752050, Odisha, India. E-mail: tkc@niser.ac.in
bInorganic and Theoretical Chemistry Section, Chemical Sciences and Technology Division, CSIR-National Institute of Interdisciplinary Science and Technology, Trivandrum-695019, Kerala, India
First published on 15th May 2019
Abstract
Four different core-modified planar 34π octaphyrins (10, 11, 13, and 15) which exhibit rotational isomerism have been synthesized and characterized both in solution and solid states. Octaphyrins 10, 11 and 13 show two inseparable isomers A and B which crystallize in the same unit cell. However, 15 forms two identical isomers of A. Structurally, the two isomers in 10, 11 and 13 (A and B) are different only in the ring inversion of one of the thiophene or selenophene rings present in the terthiophene subunit of the macrocycle. In isomer A, the middle thiophene or selenophene rings are inverted, while in isomer B, the terminal thiophene rings are inverted. The 1H NMR spectrum of these macrocycles shows peaks assignable to protons of both the isomers in toluene D8. The single crystal structure analysis of 10 reveals the presence of both isomers 10A and 10B in a single unit cell with the P21/n space group. Both the isomers exhibit aromatic behaviour in the freebase form. Protonation of pyrrole nitrogens leads to exclusive formation of isomer B for 10 and 11. However, both the isomers are present upon protonation of 13 where the central heterocyclic ring of terthiophene subunits has thiophene and selenophene rings. Octaphyrin 15 crystallizes in the P21/c space group and exclusively isomer A was formed in the reaction. Protonation of pyrrole nitrogens leads to significant increases in aromaticity as revealed by 1H NMR chemical shift data. The NICS values calculated for the individual heterocyclic rings before and after protonation support such a conclusion. The AICD plots exhibit clockwise orientation of current density vectors suggesting the presence of diatropic ring current in the octaphyrins. Energy calculations at the M06L/CC-pVTZ//M06L/6-31G** level qualitatively account for exclusive stabilization of a particular isomer relative to the other upon protonation. To the best of our knowledge 10 represents the first example in expanded porphyrin chemistry where two different structural isomers crystallize in a single unit cell.
1. Introduction
An octaphyrin is an eight-pyrrole containing macrocycle in which pyrrole rings are connected in a cyclic fashion via meso carbon bridges and/or with some direct pyrrole–pyrrole links.1 Octaphyrins are known to be conformationally flexible and are shown to adopt twisted figure-eight,2 dumbbell,3 aromatic planar,4 and antiaromatic planar5 conformations. The attainment of a figure-eight conformation results in loss of aromaticity.6 Hence, to synthesize planar aromatic octaphyrins, various synthetic approaches have been adopted. We used an approach to substitute sterically bulky mesityl substituents at the meso positions to avoid twisting of the macrocycle to synthesize 1 (ref. 4) which is a 34π planar aromatic core-modified octaphyrin. Later Osuka and co-workers followed a bridging approach to tie a bridge across the meso carbons to avoid twisting and were successful in synthesizing 2a which turned out to be non-aromatic.7 Later, we used a similar bridging approach to synthesize 34π aromatic bridged octaphyrin 2b.8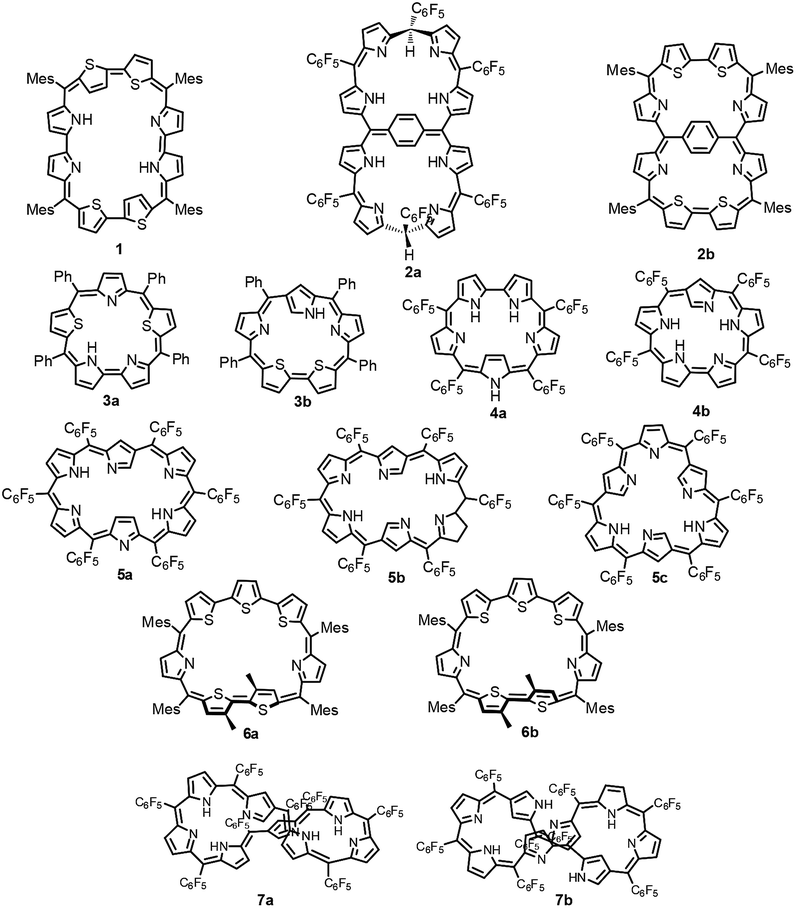
Structural isomers of porphyrins such as porphycenes,9 corrphycenes,10 hemiporphycenes11 and N-confused porphyrins12 and their diverse chemistry have been well documented in the literature.13–15 However, there are only limited reports on the structural isomers of expanded porphyrins in general and octaphyrins in particular. Latos-Grażyński and co-workers reported the first synthesis of core-modified sapphyrin 3a.16 Later, Chandrashekar and coworkers reported synthesis of 3a and along with Furuta demonstrated the synthesis of its N-confused isomer 3b.17 In 2008, Furuta reported all aza N-confused sapphyrin 4b (ref. 19), which is a structural isomer of 4a (ref. 18) in which one of the pyrrole rings has a β-connectivity. The structural isomers of hexaphyrins, singly, doubly and triply N-confused hexaphyrins 5a,205b (ref. 21) and 5c (ref. 22) respectively, were reported by Furuta and Xie in 2009. We have characterized two conformers of heptaphyrin 6a and 6b.23 Furthermore, the existence of two tautomers of 1 where the pyrrole NH proton is exchanging between imine and amine pyrrolic nitrogens was structurally characterized by our group.4 Recently, Furuta and Xie reported the synthesis of a neo-confused octaphyrin which is an isomer of all aza figure-eight octaphyrin.24 More recently, Ishida and Furuta reported the synthesis of a doubly N-confused 36π octaphyrin which exhibits isomerization between figure-eight 7a and dumbbell 7b structures.3 However, upon bis-metallation only the figure-eight structure is stabilized relative to the dumbbell structure. In all the above examples the structural isomers have been independently synthesized by various synthetic routes and their chemistry has been reported.
The brief literature survey described above clearly reveals that the characterization of structural isomers of expanded porphyrins in general and octaphyrins in particular is in its infancy and more studies are needed to understand the structural diversity of expanded porphyrins to exploit their rich and diverse chemistry.25 Recently, we and others have shown that aromatic octaphyrins are good NLO materials26 and they exhibit bicyclic Baird type aromaticity.27 Thus, in this article we wish to report the characterization of new structural isomers of 34π modified octaphyrins which exhibit rotational isomerization through C–C bond rotation. Three 34π aromatic modified octaphyrins have been synthesized and characterized by varying the heteroatom present in the expanded porphyrin core.
The sulphur analogue 10 exhibits two rotational isomers 10A and 10B in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. However, replacing one of the central thiophene rings by a selenophene ring in the terthiophene moiety gave two new isomers 13A and 13B in a 4:1 ratio. Replacement of both central thiophene rings by selenophene rings results in the formation of two identical isomers (15A and 15A′) in which both the central selenophene rings are inverted. Characterization of all these isomers has been done using UV-VIS, 1H and 2D-NMR and single crystal X-ray structure analyses. It has been shown that isomers 10A and 10B crystallized in a single unit cell with the P21/n space group which is unprecedented to the best of our knowledge.
:1 ratio. However, replacing one of the central thiophene rings by a selenophene ring in the terthiophene moiety gave two new isomers 13A and 13B in a 4:1 ratio. Replacement of both central thiophene rings by selenophene rings results in the formation of two identical isomers (15A and 15A′) in which both the central selenophene rings are inverted. Characterization of all these isomers has been done using UV-VIS, 1H and 2D-NMR and single crystal X-ray structure analyses. It has been shown that isomers 10A and 10B crystallized in a single unit cell with the P21/n space group which is unprecedented to the best of our knowledge.
2. Results and discussion
2.1. Syntheses
The synthesis of a terthiophene based [34]π octaphyrin is outlined in Scheme 1. We have adopted a [5 + 3] acid-catalyzed MacDonald type condensation reaction. The required precursors 5,5′′-bis(mesityl(1H-pyrrol-2-yl)methyl)-2,2′:5′,2′′-terthiophene 8 and [2,2′:5′,2′′-terthiophene]-5,5′′-diylbis(arylmethanol) 9 were synthesized using our earlier reported procedures.28 Thus the condensation of 8 and 9 in the presence of 1 equiv. of trifluoroacetic acid (TFA) in dry CH2Cl2 followed by oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) gave octaphyrins 10 and 11 in 10–12% yield. | ||
| Scheme 1 Synthesis of octaphyrins 10 and 11. | ||
The macrocycles 10 and 11 are stable and their composition was confirmed through ESI mass spectrometry (m/z, 1145.3190 for 10 and m/z, 1089.2703 for 11) (Fig. S1 and S2, ESI†).
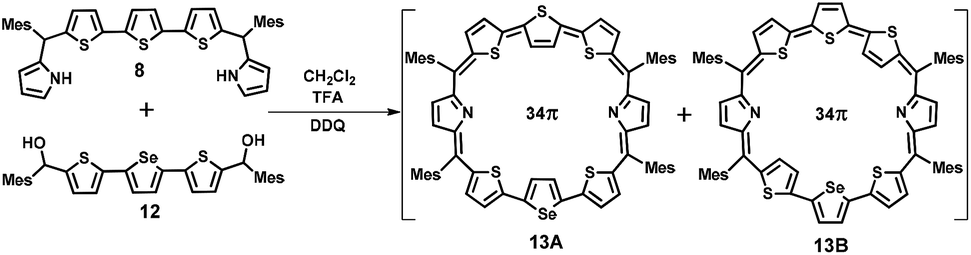
For the synthesis of 13 in which one of the central thiophene rings of the terthiophene moiety is replaced by a selenophene ring, the required precursor 12 was synthesized by using a reported procedure.29 Thus, the condensation of 8 and 12 under acid-catalyzed conditions in CH2Cl2 followed by DDQ oxidation (Scheme 2) gave 13, a dark blue band on a silica gel column (100–200 mesh) eluted with CH2Cl2/n-hexane (58:42 v/v) in 9% yield. Here also isomers 13A and 13B were inseparable and 13A was a major isomer while 13B is minor. The composition was confirmed through ESI mass spectrometry (m/z, 1193.2602 for 13) (Fig. S3, ESI†).
 | ||
| Scheme 2 Synthesis of octaphyrin 13. | ||
The third octaphyrin 15 was synthesized by the [5 + 3] approach (Scheme 3). The required precursor 14 was synthesized using our earlier reported procedure.28 Acid-catalyzed condensation of 12 and 14 in CH2Cl2 followed by DDQ oxidation gave 15. The compound was eluted with CH2Cl2/n-hexane (52:48 v/v) in 10% yield. The reaction afforded isomer A exclusively whereas isomer B was not formed. The composition was confirmed through ESI mass spectrometry (m/z, 1241.2308 for 15) (Fig. S4, ESI†).
 | ||
| Scheme 3 Synthesis of octaphyrin 15. | ||
2.2. Spectral characterization
 | ||
| Fig. 1 Electronic absorption spectra of 10 and 10·2H+ in CH2Cl2. | ||
:1 ratio (Fig. S27–S30, ESI†).
 | ||
| Fig. 2 1H NMR spectrum: (a) 10 (213 K) and (b) 10·2H+(298 K) in toluene D8 (* residual solvent peaks). | ||
Protonation of pyrrolic nitrogens with dilute solution of TFA leads to stabilization of isomer B and the 1H NMR spectrum of 10B·2H+ shown in Fig. 2b confirms this observation (Fig. S22–S26, ESI†). Specifically, the pyrrolic NH protons which are experiencing the diatropic ring current appear as a broad signal at −2.76 ppm and a D2O experiment confirms the assignment. The terminal inverted thiophene ring β-CH protons (1 and 2) are further shielded relative to the freebase form and appear at −4.29 ppm and −4.70 ppm. The β-CH protons of normal thiophene rings (3, 4, 5, and 6) are further deshielded and appear between 10.47 ppm and 11.28 ppm. The β-CH protons of the pyrrole ring (7 and 8) appear as a doublet at 9.29 ppm and 9.34 ppm. The CH protons of mesityl rings appear between 7.45 and 7.61 ppm and methyl signals between 2.01 and 2.75 ppm. Thus, comparison of proton NMR of freebase and protonated forms of 10 clearly suggests stabilization of 10B upon protonation (Scheme S4, ESI†). 11 shows similar behaviour upon protonation and because of lowering of symmetry separate signals are observed for the two inverted thiophene protons (Fig. S31–S36, ESI†).
The proton NMR spectra of freebase and protonated forms of 13 are shown in Fig. 3 and 4. The presence of two different heteroatoms (S and Se) lowers the symmetry of the macrocycle, where one would expect individual signals for the ring protons (Fig. S37–S40, ESI†). For isomer 13A, the β-CH protons of inverted thiophene (a) and selenophene rings (h) appear at 0.14 ppm and 0.49 ppm respectively, while for 13B, the terminally inverted thiophene ring protons (1, 2, 9 and 10) appear at 0.49 (10), 0.14 (9), −1.01 (2) and −1.22 (1), suggesting that these protons are experiencing the diatropic ring current of the macrocycle. The β-CH protons of normal thiophene rings of 13A (c, b, f, and g) appear in the region of 8.73–9.22 ppm, while those of isomer 13B (3, 4, 5, 6, 11, 12, 13 and 14) appear between 9.4 ppm and 10.03 ppm. The pyrrole β-CH protons of 13A (d and e) appear in the region of 8.02–8.08 ppm, while those of 13B (7, 8, 15 and 16) are slightly deshielded and appear in the region from 8.42 ppm to 8.56 ppm. Based on the spectral analysis, both the isomers (13A and 13B) were found in a 4:1 ratio. The protonation of pyrrole protons leads to significant shielding and deshielding effects expected for an aromatic macrocycle (Fig. S41 and S42, ESI†). However, both the isomers 13A and 13B retain their identity unlike in 10. The inverted thiophene (a) and selenophene (h) β-CH protons experience significant shielding upon protonation and appear at −3.35 ppm (h) and −4.02 ppm (a). The terminally inverted β-CH protons of thiophene rings (1, 2, 9 and 10) of 13B·2H+ appear between −4.13 ppm and −4.92 ppm. The normal thiophene protons of 13B·2H+ also experience the deshielding effect and appear between 10.47 and 11.6 ppm as eight doublets. The pyrrole β-CH protons of 13A·2H+ (d and e) appear at 8.99–9.02 ppm as two doublets while those of 13B·2H+ (7, 8, 15 and 16) appear in the region of 9.28–9.4 ppm. The pyrrole NH protons of 13·2H+ appear as a broad signal at −6.5 ppm (13A·2H+) and −7.5 ppm (13B·2H+). The shielding and deshielding of different protons suggest the aromatic nature of the macrocycles.
 | ||
| Fig. 3 1H NMR spectrum of 13 (298 K) in toluene D8. | ||
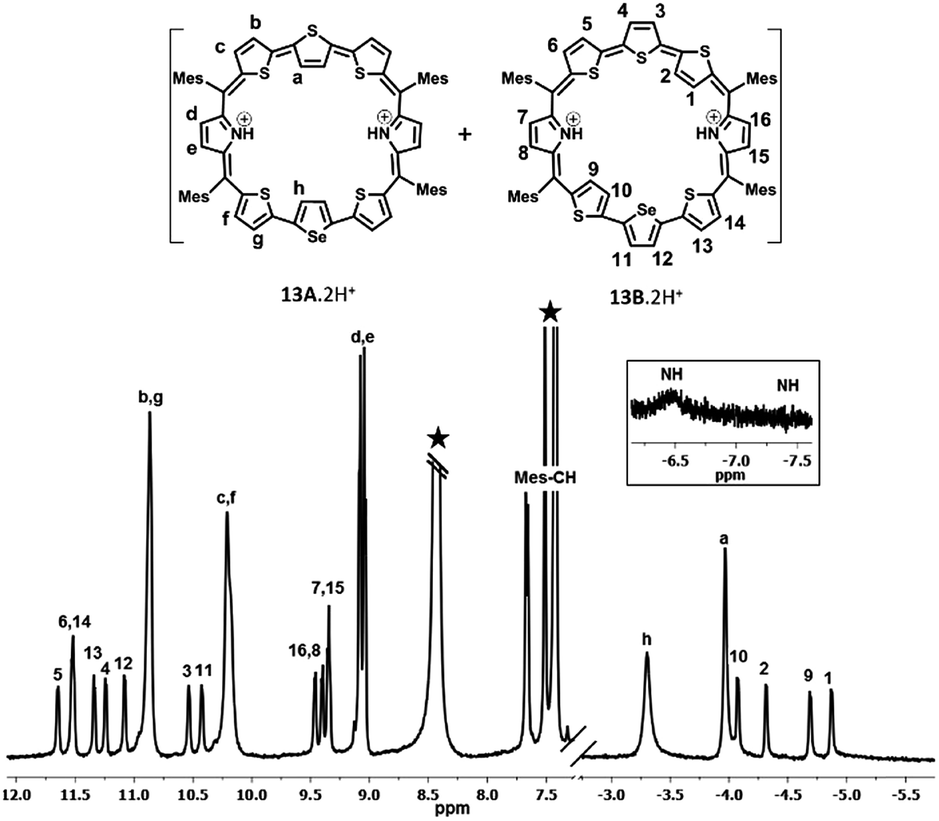 | ||
| Fig. 4 1H NMR spectrum of 13·2H+ (298 K) in toluene D8. | ||
In the case of 15, since only one isomer, i.e. the centrally inverted symmetric isomer A was formed, and the 1H NMR spectrum was simple to interpret (Fig. S43 and S44, ESI†). Fig. 5 depicts the 1H NMR spectra of 15A and its protonated derivative 15A·2H+. The inverted selenophene β-CH protons resonate around 0.037 ppm confirming the inversion of the central selenophene rings. The thiophene ring protons (b and c) resonate as two doublets at 8.91 ppm and 9.25 ppm respectively, while the pyrrole β-CH protons appear as a singlet at 8.2 ppm. The mesityl –CH protons are at 7.22 ppm, while the methyl protons are in the expected region. Protonation of the pyrrole nitrogens leads to large shielding and deshielding of ring protons depending on the nature of the rings (Fig. S45 and S46, ESI†). The inverted selenophene β-CH protons experience a significant shielding of 4 ppm and appear at −4.04 ppm, suggesting that the selenophene rings come into the plane exposing the β-CH protons to the ring current of the macrocycle upon protonation.
 | ||
| Fig. 5 1H NMR spectrum: (a) 15A (298 K) and (b) 15A·2H+(298 K) in toluene D8. | ||
The thiophene ring β-CH protons (b and c) experience deshielding (2.0 ppm for ‘c’ protons and 1.1 ppm for ‘b’ protons) and appear at 10.28 ppm and 10.93 ppm respectively. The pyrrole NH protons appear as a broad signal at −4.59 ppm, suggesting the aromatic nature of the macrocycle.
The difference in chemical shifts of most deshielded and most shielded ring protons (represented by Δδ) for various octaphyrin isomers listed in Table 1 gives some insight into the aromaticity of the macrocycles. The results are as follows: (i) A isomers have lower aromatic character relative to B isomers (Δδ = 8.97 vs. 12.11); (ii) protonated derivatives exhibit significantly larger Δδ values relative to freebase forms. The chemical shifts of inverted ring β-CH protons depend on the orientation of the inverted ring relative to the mean macrocyclic plane. In the A isomer, the centrally inverted heterocyclic rings are not completely planar due to which these protons experience only partial ring current. The terminally inverted rings in the B isomer are oriented in the plane of the macrocycle and hence these protons are more shielded relative to A isomer protons accounting for larger Δδ values. Upon protonation of pyrrole nitrogens, the inverted rings come into the plane of the macrocycle and the β-CH protons are completely exposed to the ring current of the macrocycle, which results in larger shielding of these protons. Hence, the protonated derivatives show larger aromaticity. Support for such a conclusion comes from the single crystal X-ray analysis of freebase and protonated derivatives of 10A and 10B (vide infra).
| Octaphyrin isomer | Δδ (ppm) | Octaphyrin isomer | Δδ (ppm) |
|---|---|---|---|
| 10A | 9.73 | 13A | 9.06 |
| 10B | 12.11 | 13B | 11.25 |
| 10B·2H+ | 15.98 | 13A·2H+ | 17.36 |
| 13B·2H+ | 18.14 | ||
| 11A | 8.97 | ||
| 11B | 11.12 | 15A | 9.21 |
| 11B·2H+ | 15.15 | 15A·2H+ | 15.52 |
2.3. NICS(0) calculations and AICD plots
The NICS(0) values were calculated for all the individual heterocyclic rings of octaphyrins (10, 13 and 15) in freebase and protonated forms (Charts S1–S3, ESI†).31 It is observed in all the cases that the NICS(0) value for the pyrrole rings in the freebase form is 0.1 ppm, and those for inverted heterocyclic rings are from −3.3 ppm to −4.4 ppm, whereas those for the normal heterocyclic rings are between −18.1 ppm and −25.8 ppm, respectively, and the overall NICS(0) values are from −9.95 ppm to −12.04 ppm, suggesting the aromatic character in the freebase form. Upon protonation, the pyrrole rings have a higher NICS value as compared to the freebase form (−13.4 ppm to −15.2 ppm), whereas a nominal increase in that of the inverted heterocyclic rings (−4.2 ppm to −5.6 ppm) and marginal shift in that of the normal heterocyclic rings (−17.5 ppm to −20.6 ppm) with an overall increase in NICS values from −13.66 ppm to −15.18 ppm reveal that the protonated forms are more aromatic than the freebase form, justifying the conclusion drawn from 1H NMR chemical shift data. For example, the NICS value of 10B and 10B·2H+ is shown in Fig. 6 and found to be −11.4 ppm in 10B and −15.17 ppm in 10B·2H+, justifying that protonation enhances the aromatic character as compared to the freebase form. | ||
| Fig. 6 NICS(0) values (ppm) of (a) 10B and (b) 10B·2H+. | ||
Furthermore, the anisotropy induced current density plots (AICD plots) obtained at an isosurface value of 0.026 clearly suggest clockwise orientation of current density vectors indicating the presence of diatropic ring current in the octaphyrins (Charts S1–S3, ESI†).
2.4. Single crystal X-ray analysis
The proposed structures of octaphyrin isomers were confirmed through single crystal X-ray analysis. The single crystal 10 obtained by slow diffusion of CH2Cl2 solution in n-hexane crystalizes in a monoclinic crystal lattice with the P21/n space group.32 (Fig. S47–S49, Table S1, ESI†). As reflected in 1H NMR spectral analysis, both the isomers with the same empirical formula exist in the crystal lattice. Both the molecules contain two terthiophene units connected with two pyrrole units via meso carbon bridges. The only difference is in the inversion of one of the thiophene rings in the terthiophene units. The middle thiophene is inverted in 10A, whereas in 10B, one of the terminal rings is inverted (Fig. 7). The difference in the dihedral angle between the two planes (C5, C10, C5′ and C10′ vs. C45, C50, C45′ and C50′) is 38.84° (Fig. S47, ESI†). As reflected by the Δδ values (9.73 vs. 12.11) in Table 1, the terminally inverted thiophene rings (10B) are deviated by 16.27°, whereas the middle inverted thiophene rings are deviated by 21.23° (10A), confirming that the terminal rings are moving towards the plane and experiencing an effective ring current as compared to the middle thiophene rings. The maximum deviation of the middle thiophene ring is reflected in the steric repulsion between the inner core hydrogen atoms (H39⋯H38′). The meso aryl rings in 10 are almost perpendicular to the mean plane of the macrocycle (for 10A 79.89°, 83.03°, 79.89°, and 83.03° and for 10B 86.37°, 81.10°, 86.37°, and 81.10°) as observed in other meso-aryl expanded porphyrinoids.33 The crystal analyses of 10 revealed three different intermolecular hydrogen bonding interactions between (i) π-clouds of the inner thiophene (S2′) of 10B and C43–H43 of 10A, (ii) π-clouds of the meso-mesityl unit of 10A and C17′–H17′ of 10B and (iii) π-clouds of the meso-mesityl unit of 10B and C71–H71B of 10A and the bond distances and angles of S2′(π)⋯C43–H43, Mes(π)⋯C17′–H17′ and Mes(π)⋯C71–H71B are 2.83 Å & 136.14° and 2.71 Å & 137.01° and 3.15 Å & 150.91° respectively (Fig. S50 and S51, ESI†).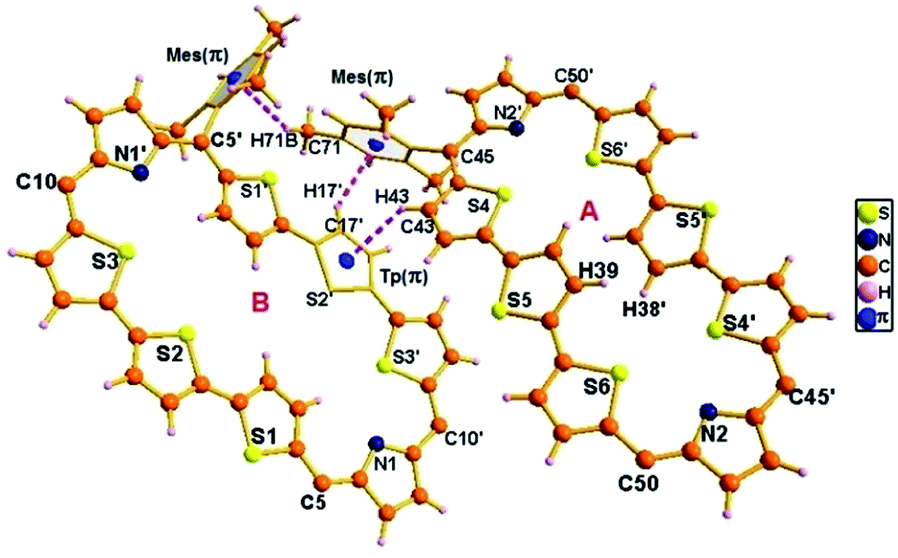 | ||
| Fig. 7 Single-crystal X-ray structure of 10. The dotted line indicates intermolecular hydrogen bonding interactions. The meso mesityl substituents which are not involved in hydrogen bonding interactions are omitted for clarity. | ||
The exclusively stabilized 10B·2H+ isomer upon protonation was unambiguously confirmed by single crystal X-ray analysis (Fig. 8). The molecule crystallizes in a triclinic lattice with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Fig. S52–S54, Table S1, ESI†).31 Two perchlorate anions (O1/O1′) are involved in intermolecular hydrogen bonding interaction with the protonated imine NH (N1–H1/N1′–H1′) and the bond distance and angle of N1–H1⋯O1/N1′–H1′⋯O1′ are 2.33 Å and 118.41° respectively. These N–H⋯O bond length and bond angles compare well with those of a previously reported octaphyrin–TFA complex.34 The hydrogen bonding interactions generate a zig-zag one dimensional array in the solid state (Fig. S55, ESI†). The terminal thiophene unit (S1/S1′) is hardly deviated (12.99°) from the mean macrocyclic plane (C5, C10, C5′, and C10′) and maintains the planarity as observed in the freebase form of 10. The deviation is lower as compared to 10B (16.27°), proving the higher aromatic ring current in the terminal thiophene unit of the protonated form compared to the freebase form, as reflected by the higher Δδ value (15.98 vs. 12.11) in Table 1. By replacing counter anion from ClO4− to SO42− ion, the crystal lattice contains 10B·2H+ with one SO42− ion, where the SO42− ion generates zig-zag 1d array with intermolecular hydrogen bonding interaction with distance and angle of 2.17 Å and 129.55° (Fig. S56 and S57, ESI†).
space group (Fig. S52–S54, Table S1, ESI†).31 Two perchlorate anions (O1/O1′) are involved in intermolecular hydrogen bonding interaction with the protonated imine NH (N1–H1/N1′–H1′) and the bond distance and angle of N1–H1⋯O1/N1′–H1′⋯O1′ are 2.33 Å and 118.41° respectively. These N–H⋯O bond length and bond angles compare well with those of a previously reported octaphyrin–TFA complex.34 The hydrogen bonding interactions generate a zig-zag one dimensional array in the solid state (Fig. S55, ESI†). The terminal thiophene unit (S1/S1′) is hardly deviated (12.99°) from the mean macrocyclic plane (C5, C10, C5′, and C10′) and maintains the planarity as observed in the freebase form of 10. The deviation is lower as compared to 10B (16.27°), proving the higher aromatic ring current in the terminal thiophene unit of the protonated form compared to the freebase form, as reflected by the higher Δδ value (15.98 vs. 12.11) in Table 1. By replacing counter anion from ClO4− to SO42− ion, the crystal lattice contains 10B·2H+ with one SO42− ion, where the SO42− ion generates zig-zag 1d array with intermolecular hydrogen bonding interaction with distance and angle of 2.17 Å and 129.55° (Fig. S56 and S57, ESI†).
 | ||
| Fig. 8 Single-crystal X-ray structure of 10B·2H+. The dotted line indicates intermolecular hydrogen bonding interactions with ClO4− ions. Meso mesityl substituents are omitted for clarity. | ||
Repeated attempts to crystallize 13 failed in our hands. Hence, the structures of 13A and 13B were optimized at the M06L/6-31G** level. Fig. 9a and b show the optimized structures of the isomers of 13. For 13A, the middle selenophene and thiophene rings are inverted, where in 13B the terminal thiophene rings are inverted, which are consistent with the 1H NMR data (Fig. S37–S40 in the ESI†), where similar ring inversions were observed. Upon protonation, both 13A·2H+ and 13B·2H+ were formed and the optimized structures of the protonated derivatives are given in Chart S2 in the ESI.†
 | ||
| Fig. 9 M06L/6-31G** level optimized geometry of (a) 13A and (b) 13B. | ||
Octaphyrin 15 crystalizes in a monoclinic crystal lattice with the P21/c space group (Fig. S58 and S59, Table S2, ESI†). As observed in 10, two molecules of 15 are present in the single unit cell; however both are identical isomers in which only the middle selenophene ring is inverted (Fig. 10). The terminally inverted isomer B was not formed in the reaction. The difference in the dihedral angle between the two molecular planes (C7, C12, C7′, and C12′ vs. C7ʺ, C12′′, C7′′′, and C12′′′) is 21.41°. The middle selenophene unit in 15A is tilted by around 20.10° from the mean macrocyclic plane, whereas, the rest thiophene and pyrrolic rings are more or less planar with the mean molecular plane. The marginal difference in deviation (20.10° vs. 21.23°) of the middle selenophene unit over the middle thiophene unit (10A) adopts a similar trend to that observed in Δδ values (9.21 vs. 9.73) as shown in Table 1. Like 10A, here also the repulsion between the inverted selenophene β-hydrogens (H1⋯H18) is responsible for maximum deviation. The meso mesityl groups are nearly perpendicular to the mean macrocyclic plane (78.30°, 76.37°, 78.30° and 76.37°). The structural analysis of 15A revealed two weak intermolecular hydrogen bonding interactions between the two molecules, (i) Mes(π)⋯C4′–H4′ and (ii) Mes(π)⋯C44′′′–H44′′′ and the bond distances and angles were (i) 3.30 Å and 132.35° and (ii) 3.19 Å and 123.57° respectively (Fig. S60 and S61, ESI†).
 | ||
| Fig. 10 Single-crystal X-ray structure of 15A with intermolecular hydrogen bonding interactions. The meso mesityl substituents which are not involved in hydrogen bonding interactions are omitted for clarity. | ||
The single crystal X-ray structure of 15A·2H+ is shown in Fig. 11. The crystal was grown by slow evaporation of toluene in methanol at room temperature. The compound crystallizes in a triclinic crystal lattice with the P space group and contains two ClO4− ions (Fig. S62 and S63, Table S2, ESI†). The structural analysis reveals that the middle selenophene rings of 2,5-di(thiophen-2-yl)selenophene are inverted as in the freebase form. The inverted selenophene units deviate by 17.92° from the mean macrocyclic plane (C7, C12, C7′, and C12′) due to the steric repulsion between the β-CH protons (H1⋯H18) of the inverted selenophene moieties. The deviation in 15A·2H+ is 3.48° less as compared to its freebase 15A, further confirming the 4 ppm upfield shift of the inverted β-CH protons of 15A·2H+ as revealed in 1H NMR spectroscopy. As reflected in 15A the remaining heterocyclic rings are nearly coplanar (9.96° and 14.55°), whereas the meso mesityl substituents are nearly perpendicular (63.50°, 77.99°, 63.50° and 77.99°) to the mean macrocyclic plane. The two ClO4− ions are located above and below the plane of the macrocycle with intermolecular hydrogen bonding interaction with the protonated imine NH's (N1–H1/N1′–H1′) and the bond distance and angle of N1–H1⋯O1/N1′–H1′⋯O1′ are 2.54 Å and 107.83° respectively.
 | ||
| Fig. 11 Single-crystal X-ray structure of 15A·2H+. The dotted line indicates intermolecular hydrogen bonding interactions with two ClO4− ions. Meso mesityl substituents are omitted for clarity. | ||
2.5. DFT calculations
The exclusive formation of a particular isomer relative to the other suggests a small energy difference between the isomers. Any small external perturbation can alter this difference in energy and favour one isomer over the other. Keeping this in mind, we have calculated single point energies on the optimized structures of isomers in freebase and protonated forms using the M06L/CC-pVTZ//M06L/6-31G** level of theory. Table S3† summarizes the energies calculated. For example, the table shows that 10A is more stable than 10B by 2 kcal mol−1. However, 10B·2H+ is found to be more stable than 10A·2H+ by 0.5 kcal mol−1. Furthermore the protonation is found to have a more stabilizing effect on 10B·2H+ relative to 10A·2H+ due to increased aromaticity. In addition, the calculations do not take into account the effect of counter anions and crystal packing forces. Taken together these observations explain the stabilization of 10B·2H+ relative to 10A·2H+. In the case of 13, 13A is more stable by 3.4 kcal mol−1 relative to 13B. Upon protonation, these energy differences between the two isomers remain the same and protonation effects are similar in both A and B isomers, explaining the formation of both isomers 13A·2H+ and 13B·2H+ upon protonation. In the case of 15, the calculations showed that 15A is more stable than 15B by 4.8 kcal mol−1, supporting the formation of only isomer 15A. Even in the protonated state 15A·2H+ is found to be more stable than 15B·2H+ by 5.9 kcal mol−1, justifying the formation of 15A·2H+ upon protonation.3. Conclusions
In conclusion, syntheses and spectral and structural characterization of four 34π core modified octaphyrins have been described. Spectral and X-ray structural studies indicate that octaphyrins exhibit rotational isomers and the structure of the isomer depends on the nature of the heteroatom present in the core of the macrocycle. The octaphyrins are aromatic both in freebase and protonated forms. In the protonated form, two counter anions (ClO4− and SO42−) bind to the macrocycle through N–H⋯O hydrogen bonding interaction and anions are found above and below the macrocyclic plane. The NICS(0) values and AICD plots satisfactorily explain the aromaticity and the presence of diatropic ring current in both freebase and protonated forms. DFT calculations support the exclusive formation of a particular isomer upon protonation in terms of stabilization energies. Further studies on their excited state properties and nonlinear optical behaviour by the two photon absorption technique are in progress to exploit their diverse chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. K. C. thanks the National Institute of Science Education and Research (NISER), Department of Atomic Energy (DAE) for funding. A. G. and S. D. thank NISER for the fellowship. A. S. thanks the Research and Development (R&D) for the project (NISER/R&D-158/SERB/CH1802/2018/1116). We thank Mr Pankaj Kalita and Mr Subhyan Chakraborty (NISER) for useful discussion regarding XRD and NMR.Notes and references
- (a) N. Shivran, S. C. Gadekar and V. G. Anand, Chem.–Asian J., 2017, 12, 6–20 CrossRef CAS PubMed; (b) T. Sarma, G. Kim, S. Sen, W.-Y. Cha, Z. Duan, M. D. Moore, V. M. Lynch, Z. Zhang, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2018, 140, 12111–12119 CrossRef CAS PubMed; (c) S.-i. Ishida, J. Kim, D. Shimizu, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2018, 57, 5876–5880 CrossRef CAS PubMed; (d) N. Sprutta and L. Latos-Grażyński, Chem.–Eur. J., 2001, 7, 5099–5112 CrossRef CAS PubMed; (e) D. Seidel, V. Lynch and J. L. Sessler, Angew. Chem., Int. Ed., 2002, 41, 1422–1425 CrossRef CAS.
- (a) E. Vogel, M. Bröring, J. Fink, D. Rosen, H. Schmickler, J. Lex, K. W. K. Chan, Y.-D. Wu, D. A. Plattner, M. Nendel and K. N. Houk, Angew. Chem., Int. Ed. Engl., 1995, 34, 2511–2514 CrossRef CAS; (b) M. Bröring, J. Jendrny, L. Zander, H. Schmickler, J. Lex, Y.-D. Wu, M. Nendel, J. Chen, D. A. Plattner, K. N. Houk and E. Vogel, Angew. Chem., Int. Ed. Engl., 1995, 34, 2515–2517 CrossRef.
- K. Mitsuno, T. Yoshino, I. Gupta, S. Mori, S. Karasawa, M. Ishida and H. Furuta, Angew. Chem., Int. Ed., 2017, 56, 14252–14256 CrossRef CAS PubMed.
- V. G. Anand, S. K. Pushpan, S. Venkatraman, A. Dey, T. K. Chandrashekar, B. S. Joshi, R. Roy, W. Teng and K. R. Senge, J. Am. Chem. Soc., 2001, 123, 8620–8621 CrossRef CAS PubMed.
- J. S. Reddy, S. Mandal and V. G. Anand, Org. Lett., 2006, 8, 5541–5543 CrossRef CAS PubMed.
- H. Rath, J. Sankar, V. Prabhuraja, T. K. Chandrashekar, B. S. Joshi and R. Roy, Chem. Commun., 2005, 3343–3345 RSC.
- V. G. Anand, S. Saito, S. Shimizu and A. Osuka, Angew. Chem., Int. Ed., 2005, 44, 7244–7248 CrossRef CAS PubMed.
- G. Karthik, W.-Y. Cha, A. Ghosh, T. Kim, A. Srinivasan, D. Kim and T. K. Chandrashekar, Chem.–Asian J., 2016, 11, 1447–1453 CrossRef CAS PubMed.
- E. Vogel, M. Köcher, H. Schmickler and J. Lex, Angew. Chem., Int. Ed. Engl., 1986, 25, 257–259 CrossRef.
- J. L. Sessler, E. A. Brucker, S. J. Weghorn, M. Kisters, M. Schäfer, J. Lex and E. Vogel, Angew. Chem., Int. Ed. Engl., 1994, 33, 2308–2312 CrossRef.
- E. Vogel, M. Bröring, S. J. Weghorn, P. Scholz, R. Deponte, J. Lex, H. Schmickler, K. Schaffner, S. E. Braslavsky, M. Müller, S. Pörting, C. J. Fowler and J. L. Sessler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1651–1654 CrossRef CAS.
- (a) H. Furuta, T. Asano and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 767–768 CrossRef CAS; (b) P. J. Chmielewski, L. Latos-Grażyński, K. Rachlewicz and T. Głowiak, Angew. Chem., Int. Ed. Engl., 1994, 33, 779–781 CrossRef.
- V. V. Roznyatovskiy, C.-H. Lee and J. L. Sessler, Chem. Soc. Rev., 2013, 42, 1921–2204 RSC.
- T. Chatterjee, A. Srinivasan, M. Ravikanth and T. K. Chandrashekar, Chem. Rev., 2017, 117, 3329–3376 CrossRef CAS.
- T. Sharma and P. K. Panda, Chem. Rev., 2017, 117, 2785–2838 CrossRef PubMed.
- (a) K. Rachlewicz, N. Sprutta, P. J. Chmielewski and L. Latos-Grażyński, J. Chem. Soc., Perkin Trans. 2, 1998, 969–976 RSC; (b) S. J. Narayanan, B. Sridevi, T. K. Chandrashekar, U. Englich and K. Ruhlandt-Senge, Inorg. Chem., 2001, 40, 1637–1645 CrossRef CAS PubMed.
- S. K. Pushpan, A. Srinivasan, V. G. Anand, S. Venkatraman, T. K. Chandrashekar, B. S. Joshi, R. Roy and H. Furuta, J. Am. Chem. Soc., 2001, 123, 5138–5139 CrossRef CAS.
- L. Simkhovich, S. Rosenberg and Z. Gross, Tetrahedron Lett., 2001, 42, 4929–4931 CrossRef CAS.
- I. Gupta, A. Srinivasan, T. Morimoto, M. Toganoh and H. Furuta, Angew. Chem., Int. Ed., 2008, 47, 4563–4567 CrossRef CAS PubMed.
- S. Gokulnath, K. Yamaguchi, M. Toganoh, S. Mori, H. Uno and H. Furuta, Angew. Chem., Int. Ed., 2011, 50, 2302–2306 CrossRef CAS PubMed.
- A. Srinivasan, T. Ishizuka, A. Osuka and H. Furuta, J. Am. Chem. Soc., 2003, 125, 878–879 CrossRef CAS.
- Y.-S. Xie, K. Yamaguchi, M. Toganoh, H. Uno, M. Suzuki, S. Mori, S. Saito, A. Osuka and H. Furuta, Angew. Chem., Int. Ed., 2009, 48, 5496–5499 CrossRef CAS PubMed.
- V. G. Anand, S. K. Pushpan, S. Venkatraman, S. J. Narayanan, A. Dey, T. K. Chandrashekar, R. Roy, B. S. Joshi, S. Deepa and G. N. Sastry, J. Org. Chem., 2002, 67, 6309–6319 CrossRef CAS PubMed.
- K. Zhang, J. Zhang, X. Li, R. Guo, H. Ågren, Z. Ou, M. Ishida, H. Furuta and Y. Xie, Org. Lett., 2015, 17, 4806–4809 CrossRef CAS.
- R. Misra and T. K. Chandrashekar, Acc. Chem. Res., 2008, 41, 265–279 CrossRef CAS PubMed.
- (a) A. Chaudhary, A. Srinivasan, T. K. Chandrashekar, The Handbook of Porphyrin Science, K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Ltd, 2014, 32, pp. 271–366 Search PubMed; (b) J.-Y. Shin, K. S. Kim, M.-C. Yoon, J. M. Lim, Z. S. Yoon, A. Osuka and D. Kim, Chem. Soc. Rev., 2010, 39, 2751–2767 RSC; (c) J. M. Lim, Z. S. Yoon, J.-Y. Shin, K. S. Kim, M.-C. Yoon and D. Kim, Chem. Commun., 2008, 261–273 RSC; (d) R. Kumar, R. Misra, T. K. Chandrashekar, A. Nag, D. Goswami, E. Suresh and C. H. Suresh, Eur. J. Org. Chem., 2007, 4552–4562 CrossRef CAS; (e) H. Rath, J. Sankar, V. Prabhuraja, T. K. Chandrashekar, A. Nag and D. Goswami, J. Am. Chem. Soc., 2005, 127, 11608–11609 CrossRef CAS.
- W.-Y. Cha, T. Kim, A. Ghosh, Z. Zhang, X.-S. Ke, R. Ali, V. M. Lynch, J. Jung, W. Kim, S. Lee, S. Fukuzumi, J. S. Park, J. L. Sessler, T. K. Chandrashekar and D. Kim, Nat. Chem., 2017, 9, 1243–1248 CrossRef CAS.
- H. Rath, V. Prabhuraja, T. K. Chandrashekar, A. Nag, D. Goswami and B. S. Joshi, Org. Lett., 2006, 8, 2325–2328 CrossRef CAS.
- S. Kumar and M. Ravikanth, J. Org. Chem., 2017, 82, 12359–12365 CrossRef CAS.
- R. Kumar, R. Misra, T. K. Chandrashekar and E. Suresh, Chem. Commun., 2007, 43–45 RSC.
- The NICS(0) values at the centre of the macrocycle were not calculated because of the following. From the X-ray structure, the centre position of macrocycle is 1.85 Å away from the nearest carbon atom. The orientation of the inverted thiophene/selenophene rings w.r.t the centre point lies in the middle of the π bonded area of the C–C bond of thiophene/selenophene rings. The overall aromaticity of the centre point of the macrocycle is dominated by the π-character of this C–C bond which affects the precise NICS(0) calculation. Thus the respective NICS(0) values are calculated at the centre point of each heterocyclic ring both in freebase and protonated forms of all octaphyrins.
- CCDC 1857151 (10), 1857152 (10·2HClO4), 1857153 (10·H2SO4), 1887399 (15) and 1903728 (15·2HClO4) contains the supplementary crystallographic data for this paper.†.
- T. K. Chandrashekar, V. Prabhuraja, S. Gokulnath, R. Sabarinathan and A. Srinivasan, Chem. Commun., 2010, 46, 5915–5917 RSC.
- V. G. Anand, S. Venkatraman, H. Rath, T. K. Chandrashekar, W. Teng and K. Ruhlandt-Senge, Chem.–Eur. J., 2003, 9, 2282–2290 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of all new compounds and crystallographic data for compound 10 (1857151), 10·2HClO4 (1857152), 10·H2SO4 (1857153), 15 (1887399) and 15·2HClO4 (1903728). CCDC 1857151–1857153, 1887399 and 1903728. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01633c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |