
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure of formylglycine-generating enzyme in complex with copper and a substrate reveals an acidic pocket for binding and activation of molecular oxygen†
Dzmitry A.
Miarzlou
 ,
Florian
Leisinger
,
Daniel
Joss
,
Daniel
Häussinger
and
Florian P.
Seebeck
*
,
Florian
Leisinger
,
Daniel
Joss
,
Daniel
Häussinger
and
Florian P.
Seebeck
*
Department of Chemistry, University of Basel, Mattenstrasse 24a, Basel 4002, Switzerland. E-mail: florian.seebeck@unibas.ch; Tel: +41 612071143
First published on 18th June 2019
Abstract
The formylglycine generating enzyme (FGE) catalyzes oxidative conversion of specific peptidyl-cysteine residues to formylglycine. FGE mediates O2-activation and hydrogen-atom abstraction in an active site that contains Cu(I) coordinated to two cysteine residues. Similar coordination geometries are common among copper-sensing transcription factors and copper-chaperone but are unprecedented among copper-dependent oxidases. To examine the mechanism of this unusual catalyst we determined the 1.04 Å structure of FGE from Thermomonospora curvata in complex with copper and a cysteine-containing peptide substrate. This structure unveils a network of four crystallographic waters and two active site residues that form a highly acidic O2-binding pocket juxtaposed to the trigonal planar tris-cysteine coordinated Cu(I) center. Comparison with structures of FGE in complex with Ag(I) and Cd(II) combined with evidence from NMR spectroscopy and kinetic observations highlight several structural changes that are induced by substrate binding and prime the enzyme for O2-binding and subsequent activation.
Introduction
Copper is a versatile catalyst for oxygen-dependent reactions. Combined with appropriate ligands, copper can cycle between the oxidation states I, II and III to activate molecular oxygen (O2) and to form reactive oxygen species that can initiate very difficult reactions.1–3 Mechanistic appreciation of this reactivity is an important prerequisite to understand the fundamental role of copper in biology and to exploit its potential in chemical synthesis.4 There are two groups of copper-proteins that are most relevant for the subject of the present report: mononuclear copper enzymes that utilize O2 as an oxidant, and copper-binding proteins that suppress unspecific and cytotoxic activities of copper in the cell.5The group of known mononuclear copper enzymes includes lytic polysaccharide monooxygenases (LPMO),6,7 particulate methane monooxygenase (pMMO),8 peptidylglycine-α-hydroxylating monooxygenase (PHM),9,10 copper amine oxidase (CAO),11 and dopamine-β-hydroxylase (DBH).12 These enzymes bind copper in histidine-dominated tetrahedral or square planar coordination spheres. The structure of these enzymes have inspired the development of abiotic copper-ligands in order to examine specific aspects of copper-catalysis, but also with an eye on application in organic synthesis.2,13 Not surprisingly, most of these compounds also contain nitrogen-rich ligand sets.
The mononuclear copper proteins of the second group do not necessarily catalyze reactions but rather contribute to cellular copper-transport and sensing. Representative examples are the Cu-responsive transcriptional activator CueR,14 and the copper chaperone Atox1.15 These proteins bind Cu(I) in a linear bis-cysteine coordination sphere with extremely high affinity.16 This coordination-type provides an ideal solution to form redox inert protein:copper complexes with maximal thermodynamic stability, while enabling reversible transfer of Cu(I) from one protein to another.15,17
The formylglycine generating enzyme (FGE) is unique, since it cannot be attributed to either the first or the second group of copper-binding proteins. FGE binds a single Cu(I) by linear bis-cysteine coordination and catalyzes copper-dependent O2 activation.18,19 FGE catalyzes the oxidative conversion of specific peptidyl-cysteines to formylglycine via abstraction of the pro-(R)-β-hydrogen atom (fGly, Fig. 1). This posttranslational modification is important for sulfatases which use the hydrated form of fGly as catalytic nucleophile.20 FGE has attracted scientific interest for several reasons. Initially, this enzyme was discovered in the pursuit of finding the molecular cause for the storage disease multiple sulfatase deficiency.20,21 The subsequent realization that FGE can be used as a tool to introduce electrophilic aldehyde functions into recombinant proteins highlighted the considerable biotechnological potential of this enzyme.22–24 Finally, the recent discovery that FGE is a copper-dependent enzyme opened the door for mechanistic studies on this unique enzyme.18,19,25,26 The crystal structure of FGE from Thermomonospora curvata (tcFGE) in complex with Ag(I) (tcFGE_Ag, PDB: 5NXL), and most recently, the crystal structure of FGE from Streptomyces coelicolor in complex with Cu(I) (scFGE_Cu, PDB: 6MUJ), combined with biochemical characterization showed that two active site cysteines are the only metal ligands and that the Cu(I) bound state is the catalytic resting state.25–27 In this way, FGE represents an entirely new type of copper-enzyme. Structural and mechanistic studies on this enzyme may inspire new directions in the design of abiotic copper-catalysts.2 Conversely, the similarities of the primary metal coordination sphere in FGE and non-catalytic Cu-binding proteins raise the questions as to what secondary features may enable O2-activation by FGE. Identification of these features may help to identify redox enzymes, masquerading as copper-trafficking proteins, or could provide guidelines to engineer novel redox-activity into existing copper–proteins.
 | ||
| Fig. 1 FGE-catalyzed oxidation of peptidyl-cysteine to formylglycine (fGly) is initiated by abstraction of the pro-(R)-β-hydrogen atom from the substrate (red). | ||
In this report we describe the crystal structure of tcFGE in complex with Cu(I) and a 17-residue substrate analog. Based on this structure together with NMR spectroscopy and kinetic characterization, we identify an acidic O2-binding pocked juxtaposed to the copper center as key determinant for efficient O2-activation.
Results and discussion
Crystallization of tcFGE in complex with Cu(I) and a substrate-analog
Recombinant tcFGE was produced in Escherichia coli (see ESI†).19 The purified enzyme was crystallized in the presence of 1.2 equivalents of Cu(I) and 2.8 equivalents of a synthetic 17-residue substrate-analog. To stabilize the enzyme:substrate complex, crystallization and room-temperature handling of the crystals were executed under anoxic conditions. The sequence of the substrate-analog (FGE-27: Abz-ATTPLCGPSRASILSGR, Abz = o-aminobenzoic acid) was derived from the active site segment of a putative arylsulfatase (WP_012850446) from T. curvata. Using isothermal titration calorimetry (ITC), we established that this peptide binds to an inactive variant of tcFGE (tcFGEW228F, see below) with a Kd = 40 ± 10 nM (Fig. S2†), indicating that the wild type enzyme would also form a stable complex with this peptide. Single crystals were isolated from solutions containing purified tcFGE (21.5 mg mL−1), CuCl, peptide, PEG 8000 (10%), Tris–HCl (0.1 M, pH 7.0) and MgCl2 (0.2 M). Diffraction data could be processed up to a resolution of 1.04 Å (Fig. 2A). At this resolution conservative estimates of standard deviations (r.m.s.) are ±0.05 Å for bond lengths and ±2° for bond angles.28 The unbiased electron density revealed a continuous polypeptide chain from −1 to 302 for tcFGE and from Abz (−6) to Gly (11) for the substrate FGE-27. Two Ca(II) ions were identified in two conserved calcium binding sites (Fig. S3†).26,29,30 The high resolution of this data set allowed placement of 494 crystallographic waters, in particular four waters in the active site (H2O_1–4) with high confidence (Fig. S4†). The overall structure of tcFGE and its similarity to human FGE (hsFGE) and scFGE has been discussed previously.26,29,30 Therefore, the main focus here will be on the structure of the substrate binding pocket and the metal binding site. For this discussion we compare tcFGE in complex with Cu(I) and substrate (tcFGE_Cu_S) with the structure of tcFGE in complex Ag(I) (tcFGE_Ag) as a model for tcFGE in complex with Cu(I) (tcFGE_Cu). The structure of scFGE in complex with Cu (scFGE_Cu) has also become available very recently.27 The metal binding sites in scFGE_Cu and tcFGE_Ag were found to be essentially isostructural. | ||
| Fig. 2 (A) Structure of tcFGE in complex with Cu(I) and substrate (tcFGE_Cu_S). The 17-residue peptide substrate (violet). (B) The m|Fo| − D|Fc| omit map for the substrate model is contoured at σ-level = 3.0. (C) The C-terminus of the substrate folds into a short 310-helix and interacts with neighboring tcFGE chains in the crystal (D and E). The dashed lines in (E) indicate nearest contacts, not necessarily attractive interactions. | ||
Position of the substrate
The unbiased electron density for the peptide reveals ordered positions for all residues except for the C-terminal Arg10 (Fig. 2B). The residues in the N-terminal part (Ala-5–Ser4), except for Cys1, adopt extended backbone conformations (ϕ = −50° to −136°; ψ = 120–170°). The conformation of Cys1 (ϕ = −108°; ψ = −20°) induces a sharp turn allowing this residue to access the copper localized at the deepest point of the substrate binding pocket (Fig. 2). Pro-2 and Pro3 make extensive hydrophobic interactions with Phe38/Tyr273 and Phe86/Trp109 respectively. In contrast, Ala-5, Thr-4, Thr-3 and Leu-1 make little or no contact with the protein surface. The C-terminal residues Arg3–Ile6 adopt dihedral angles consistent with a helical conformation (ϕ = −90 to 62; ψ = −30 to −5). Indeed, the network of intramolecular backbone–backbone hydrogen bonds is reminiscent of a short 310-helix (Fig. 2C). The side chain of Ser4 hydrogen bonds to the backbone amide of Ser7 (3.0 Å), serving as a N-terminal helix cap.31 In return, the side chain of Ser7 hydrogen bonds with the backbone carbonyl of Ser4 (2.7 Å). The guanidinium side chain of Arg5 makes a π–cation interaction with the indole ring of Trp84, in addition to hydrogen-bonds with Ser290 (2.8 Å), and the backbone carbonyl of Trp84 (2.9 Å). The side chain of Ile8 binds to a hydrophobic dell lined by the side chains of Trp84, Phe86, Met99 and Ala101. The side chains of Ala6, Leu9 and the following residues (Ser10, Gly11 and Arg12) make no interaction with the protein surface. The helical structure of the substrate may be further stabilized by weak interactions with neighboring tcFGE chains (Fig. 2D and E).hsFGE has been crystallized with a seven-residue substrate (LCTPSRA, hsFGE_S, PDB: 2AIK).32 This structure contains no metal in the active site and Cys366 (Cys274 in tcFGE) was mutated to Ser. Instead, the substrate cysteine is linked via disulfide bond to Cys341 (Cys269 in tcFGE). Despite these damages, the equivalent substrate residues in tcFGE_Cu_S and hsFGE_S adopt almost identical positions in the substrate binding pocket (Fig. S5†). Apparently, residues 0–5, the conserved residues Pro3 and Arg5 in particular (numbering according to tcFGE_Cu_S) make sufficient interactions with the substrate binding pocket to steer the targeted cysteine residue into the active site – even in the absence of Cu(I).
Indeed, the biotechnological potential of FGE depends on its ability to target cysteine residues embedded in the minimal LCXPXR motif. The limited length of this motif can be inserted into N- or C-termini or surface exposed loops of unrelated proteins without significant disturbance of the structure and stability of the parent protein.22,23,33,34 On the other hand, comparison of kinetic parameters from in vitro studies with 9-, 14- and 23-residue substrates showed that longer substrates are recognized with significantly higher affinity.18,19,35 The structure of tcFGE_Cu_S provides a convincing explanation for this observation. The C-terminal residues Arg5–Ile8 adopt a defined secondary structure that positions Ile8 for additional hydrophobic contact with FGE. Likewise, Pro-2 also makes a specific contact with the enzyme that has not been observed in the structure of hsFGE_S. The more comprehensive picture of the enzyme:substrate interaction seen in the structure of tcFGE_Cu_S could provide a valuable starting point for the design of FGE variants that accept alternative motifs with applications in orthogonal bifunctionalization of recombinant proteins.36
Primary and secondary coordination sphere
The structure of FGE_Cu_S demonstrates that the peptidyl cysteine (Cys1) from the substrate is a direct ligand of the catalytic Cu(I). This finding corroborates XAS observations suggesting trigonal coordination of Cu(I) by scFGE and its substrate.27 The three thiol side chains of Cys269 (Cu–SCys269: 2.2 Å), Cys274 (Cu–SCys274: 2.3 Å) and the Cys1 (Cu–SCys1: 2.2 Å) coordinate Cu(I) in an irregular trigonal planar complex with significant deviations from the expected 120° S–Cu–S bond angles (SCys269–Cu–SCys274: 108°; SCys274–Cu–SCys1: 104°; SCys269–Cu–SCys1: 147°). The angle between the two FGE-derived cysteines and Cu(I) is significantly smaller than the value (134°) derived from DFT-based modelling based on the structure of scFGE_Cu.27 Instead, the much larger SCys269–Cu–SCys1 angle opens the triangle enabling two crystallographic waters (H2O_1, 3.3 Å; H2O_2, 3.5 Å) to form near van der Waals contact with Cu(I). Notably, these two waters are also immobilized by planar trigonal solvation spheres. H2O_2 is in contact with H2O_1, H2O_3 and H2O_4 (all 2.8 Å, O1–O2–O4: 111°; O1–O2–O3: 124°; O3–O2–O4: 119°;). H2O_1 also binds to the indole side chain of Trp228 (2.9 Å) and Ser266 (2.8 Å, OSer266–O1–NTrp228: 89°; NTrp228–O1–O2: 129°; O2–O1–OSer266: 140°). In addition, H2O_1 and H2O_2 make weak contacts with the thiolates of Cys269 (3.1 Å) and Cys-1 (3.3 Å). This solvation geometry deviates significantly from the more stable tetrahedral solvation spheres observed in bulk water,37 suggesting that the two crystallographic waters are held in unstable positions. Release of H2O_1 and H2O_2 from this pocket may provide important enthalpic driving force to enhance O2-binding (see below).38–41Inspection of the next neighbors to Ser266, H2O_3 and H2O_4 provides information about the position of the protons in this hydrogen-bonding network. In addition to contacting H2O_1, the side chain of Ser266 also hydrogen bonds with the backbone amide of Leu268 (3.0 Å). This interaction suggests that Ser266 is a hydrogen-bond donor with respect to H2O_1. H2O_3 is in hydrogen-bonding contact with Trp108 (2.9 Å), Trp109 (2.9 Å) and the backbone carbonyl of Leu-1 of the substrate (2.7 Å). This tetrahedral system suggests that H2O_3 is a hydrogen bond donor with respect to H2O_2. H2O_4 is in contact with the backbone carbonyl of Leu268 (2.8 Å) and is otherwise engulfed by hydrophobic moieties of Phe194, Tyr273 and Cys269. This environment too suggests that H2O_4 is a hydrogen bond donor with respect to H2O_2. Hence, the pocket that hosts H2O_1 and H2O_2 in the tcFGE_Cu_S structure appears as a highly acidic microenvironment.
Hydrogen bonding to metal-coordinated thiolates are known to affect the nucleophilicity and redox potential of metal–thiolate complexes.42–45 Such interactions are indeed observable in tcFGE_Cu_S. Cys1 makes two weak hydrogen bonds to H2O_2 and Arg276, 3.3 Å, Cys269 hydrogen bonds to H2O_1 and the backbone amide of Ser266 (3.3 Å), whereas Cys274 only points to the backbone amide of His270 (3.6 Å). This network may play an important role in stabilizing the high electron density of the tris-thiolate Cu(I) complex. Incidentally, Cys274 appears as the least stabilized thiolate in the complex suggesting that this residue may be most vulnerable to oxidation during catalysis or as a result of abortive side reactions (see below).
Comparison of Ag, Cu and Cd-bound FGE reveals a sequence of coordination changes
The structure of tcFGE_Cu_S (resolution: 1.04 Å) complements the structures of the same enzyme in complex with Ag(I) (resolution: 1.66 Å) or with Cd(II) (tcFGE_Cd, PDB: 5NYY, resolution: 1.28 Å). Comparison of the three structures provide a high-resolution picture of the structural changes that take place as the coordination sphere expands from linear to trigonal to tetrahedral in order to allow inner-sphere coordination of substrate and O2.Superposition of the tcFGE_Ag and tcFGE_Cu_S structures highlights three significant changes that are induced by substrate binding. First, the metal center moves by 1.1 Å as it changes from linear to trigonal coordination (Fig. 3). This movement reduces the distance from the metal to H2O_1 and H2O_2 by about 0.3 Å and 0.8 Å respectively. As a second change, H2O_1 and H2O_2 are displaced by 0.6 Å from predominantly tetrahedral to purely trigonal planar hydrogen bonding environments. In addition, the interaction partners of H2O_2 change. In tcFGE_Ag H2O_2 hydrogen bonds with four partners: H2O_1 (2.6 Å) H2O_50 (equivalent to H2O_3, 2.8 Å), H2O_105 (2.7 Å) and Tyr273 (2.8 Å, Fig. S7†). Substrate binding displaces H2O_105, leaving H2O_2 with only three nearest neighbors in tcFGE_Cu_S. In addition, the side chain of Tyr273 is rotated out of the active site to establish a hydrogen-bond to the backbone amide of Leu-1 of the substrate. Instead, H2O_4 is recruited as third neighbor of H2O_2 in tcFGE_Cu_S. This exchange is likely accompanied with a change in hydrogen bond polarity. In tcFGE_Ag Tyr273 hydrogen bonds with the backbone carbonyl of Leu268 (2.7 Å) (Fig. S7†), indicating that Tyr273 is a hydrogen bond acceptor with respect to H2O_2. In contrast, H2O_4 in tcFGE_Cu_S likely acts as a hydrogen bond donor (see above). These changes suggest that substrate binding induces destabilization of H2O_1 and H2O_2. A third substrate-induced change occurs at Cys274. The dihedral angle along Cα–Cβ of this residue changed by 58° (Nα–Cα–Cβ–S) and the amide function between Cys274 and Asn275 rotated by 135° presumably as a result of the change from linear to trigonal Cu(I)-coordination (Fig. S6†). A similar rotation was observed in the tcFGE_Cd structure (135°). Hence, this reorganization also seems to be driven by substrate binding.
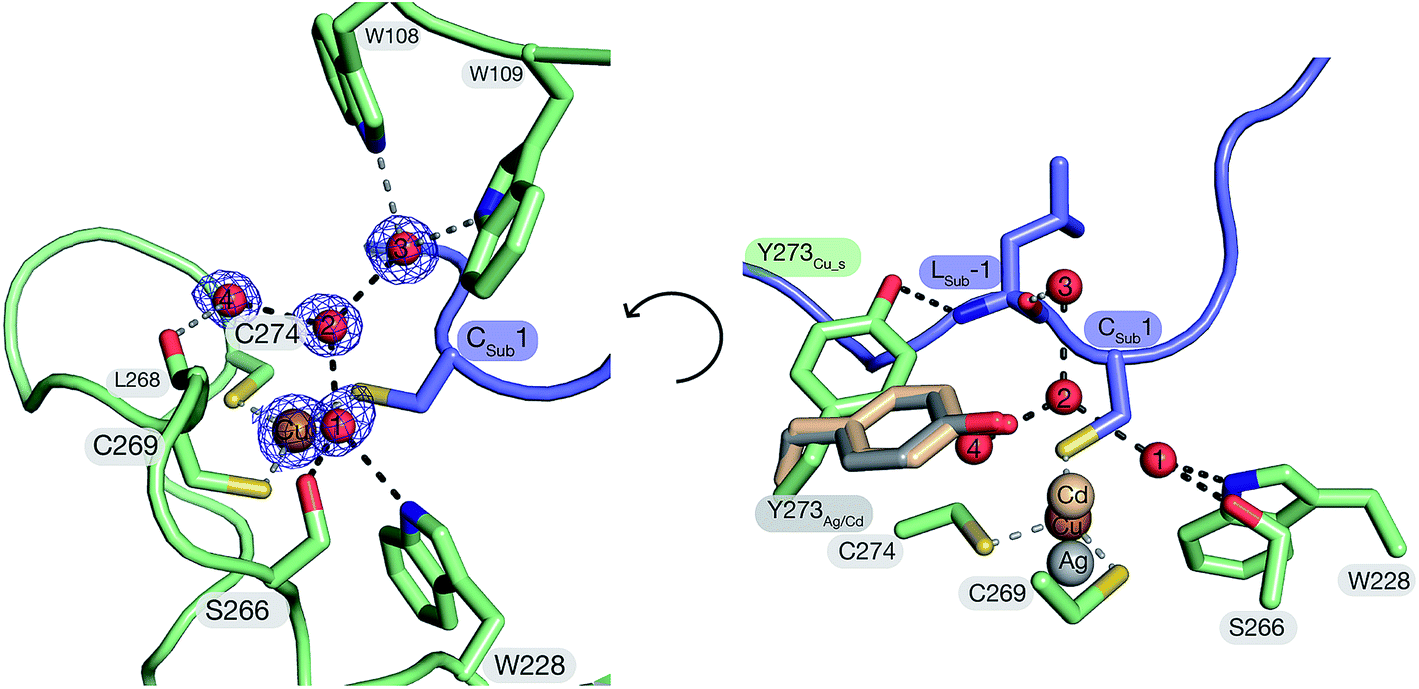 | ||
| Fig. 3 Left: The primary and secondary coordination sphere of Cu(I) in the crystal structure of tcFGE_Cu_S (protein: green, substrate: violet). The 2m|Fo| − D|Fc| omit map of Cu(I) and the four crystallographic water molecules (H2O_1–4) is contoured at σ-level = 1.0. Right: Active site of tcFGE_Cu_S including the metals and the side chain of Tyr273 from the structures of tcFGE_Ag (gray) and tcFGE_Cd (beige). | ||
The structure of tcFGE_Cd provides a model for the enzyme with a tetrahedral coordination site which may be required simultaneous coordination of the substrate and a reactive oxygen species.27 Superposition of the structures of tcFGE_Cu_S and tcFGE_Cd shows that the transition from trigonal to tetrahedral coordination moves the metal again by 0.8 Å and reduces the distance between the metal and the position of H2O_1 and H2O_2 down to 2.8 Å and 2.7 Å (Fig. 3). Tyr273 adopts the same position as in tcFGE_Ag, confirming that the orientation of this residue is dependent on the presence of substrate, rather than on the geometry of the meal complex (Fig. 3). Otherwise, the tcFGE_Cu_S and tcFGE_Cd structures are remarkably similar, suggesting that most active site reorganization that occurs en route from the Cu(I) resting state to the reactive complex may be induced by substrate-binding.
Trp228 and Ser266 form an anion binding site
The structure of tcFGE_Cu_S identifies the position of H2O_1 and H2O_2 as the only available pocket for O2 to initiate an interaction with the copper center. The opposite face of the trigonal complex is blocked by the side chains of Trp228 and Arg276. Hence, further scrutiny of the subtle changes that occur at this site in response to substrate binding may provide additional insight as to how this unusual metal center can activate O2. The apo structures of hsFGE (PDB: 1Y1E)29 and scFGE (PDB: 2Q17),30 the metalated forms tcFGE_Ag,26scFGE_Cu,27 and tcFGE_Cu_S all contain one crystallographic water (H2O_1) in hydrogen-bonding distance to Ser266 (2.6–2.8 Å) and Trp228 (2.9–3.2 Å, Fig. S7†). In the apo structures the hydrogen bonds with Trp228 are likely weak due to a small bond angle (N–H–O < 130°, Table S4†). Nevertheless, several crystal structures of hsFGE with halides bound to the approximate position of H2O_1 indicate that this pocket is endowed with significant affinity for anionic species.32,46 Most interestingly, addition of metal and substrate to tcFGE increased the bond angle between H2O_1 and the indole of Trp228 to 148° (tcFGE_Ag) and 157° (tcFGE_Cu_S) (Table S4†), suggesting that the presence of metal and substrate also increases bonding interactions at the O2-binding site. Also, based on the observed anion-affinity of this pocket we have proposed previously that H2O_1 in tcFGE_Ag might be a hydroxide that serves as the base that deprotonates the thiol of the incoming peptidyl cysteine.26Observation of Trp228 by NMR
To examine the hydrogen bonding interaction between H2O_1 and Trp228 in more detail we used NMR spectroscopy. In addition to directly interrogate the chemical environment of specific protons NMR spectroscopy has the key advantage over X-ray measurements (at low temperatures) that the enzyme is examined in solution and at room temperature. 1H–15N TROSY HSQC spectra (transverse relaxation-optimized spectroscopy heteronuclear single quantum correlation) were measured using uniformly 15N-labelled tcFGE and tcFGEW228F in apo form and in complex with Cu(I) on a 600 MHz Bruker NMR instrument.47 Superposition of these spectra, in combination with a triple resonance HNCO experiment that distinguishes amide-specific resonances from indole-specific resonances (Fig. S8 and S9†),48 allowed unambiguous identification of the Hε1 and Nε1 signals relating to the indole side chain of Trp228 (Fig. S10–S12†). The chemical shifts of indole Hε1 and Nε1 signals are highly sensitive to their environment, reporting on the solvation of the N–H function and the aromatic ring system.49,50 Therefore, we reasoned that the Trp288 signal could be used to monitor the protonation state and movements of H2O_1. The recorded spectra show that addition of Cu(I) to apo tcFGE induced a significant downfield shift of the Hε1 signal (Table 1 and Fig. 4). Addition of the substrate to tcFGE_Cu induced a further Hε1 shift of similar magnitude. Addition of metal also caused slight deshielding of Nε1, consistent with increased strength of the hydrogen bond between H2O_1 and Trp228. In contrast, addition of substrate caused a marked shielding effect. Inspection of the crystal structures shows, that the indole ring of Trp228 is solvent exposed in tcFGE_Ag and is buried in tcFGE_Cu_S (Fig. S13†). Hence, addition of the substrate reduces the polarity of the medium above the indole ring consistent with the observed shielding effect. The observed deshielding of the indole proton is fully consistent with the proposition that metal and substrate binding increase bonding interactions at the O2-binding site.| Enzyme | Metal | δ Hε1/Ne1 Trp228, ppm | Δ δ Hε1/Ne1 (Trp228), ppm | ||||
|---|---|---|---|---|---|---|---|
| E | E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M :M |
E:M:S |
E → E:M |
E:M → E:M:S |
E → E:M:S |
||
| a The errors of the given values are estimated to be <0.02 ppm. | |||||||
| tcFGE | Ag(I) | 9.69/128.93 | 9.88/129.95 | 10.26/126.81 | 0.19/1.02 | 0.38/−3.14 | 0.57/−2.12 |
| tcFGE | Cu(I) | 9.69/128.93 | 9.98/130.75 | 10.22/126.66 | 0.29/1.82 | 0.24/−4.09 | 0.53/−2.27 |
| tcFGES266A | Cu(I) | 9.75/129.29 | 10.04/130.78 | 10.22/126.77 | 0.29/1.49 | 0.18/−4.01 | 0.47/−2.52 |
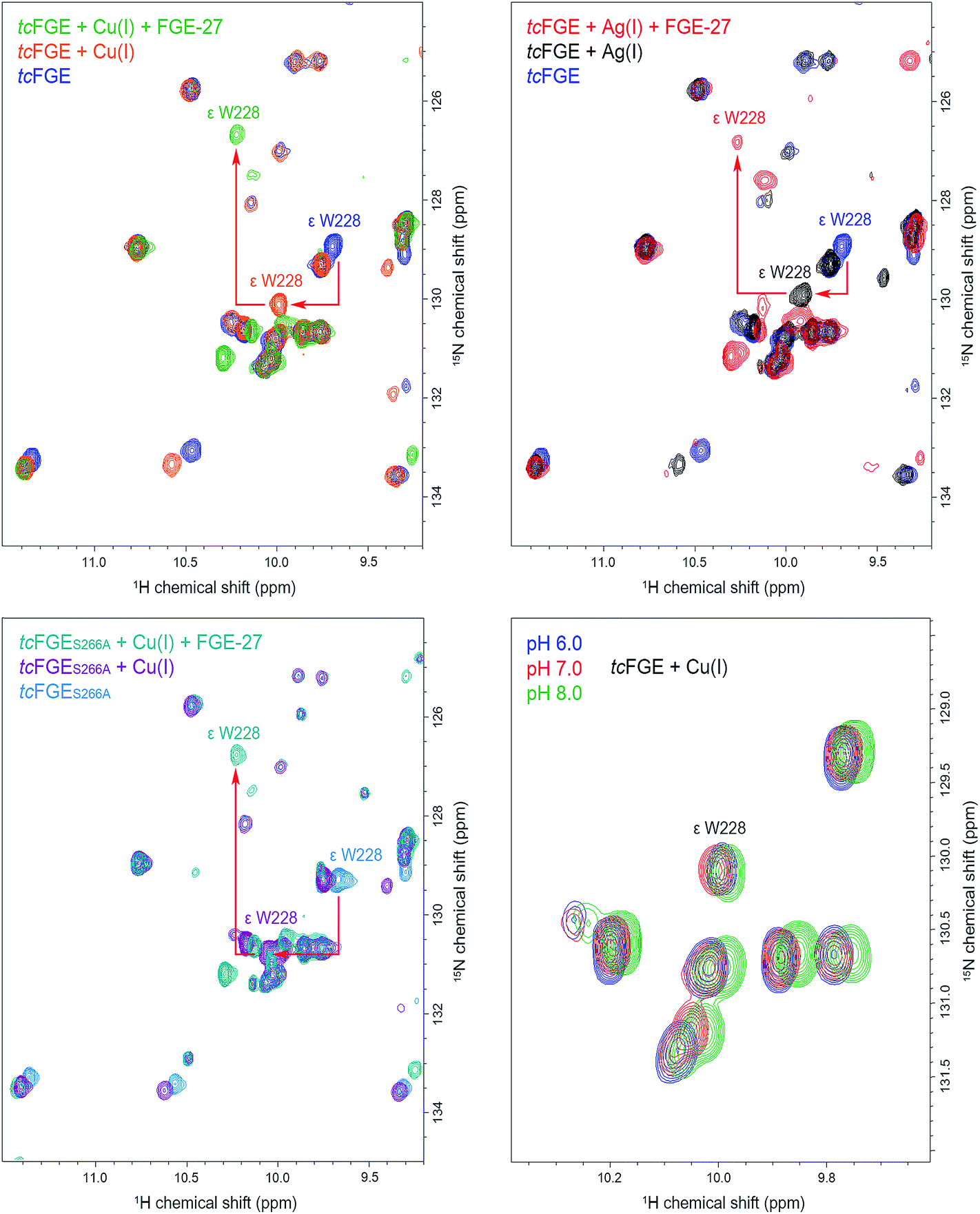 | ||
| Fig. 4 1H–15N TROSY HSQC spectra of uniformly 15N-labelled 300 μM tcFGE in 20 mM phosphate buffer, 50 mM NaCl, 2 mM DTT, 25 C. The downfield area of the amide region is presented. Top left: The overlay of the spectra of apo-form tcFGE, pH 8.0 (dark blue), tcFGE in the presence of 450 μM CuSO4, pH 8.0 (orange), tcFGE in the presence of 450 μM CuSO4 and 1 mM FGE-27, 2 mM glucose, 2 units (U) glucose oxidase (GO), 200 U catalase (CAT), pH 8.1 (green). Top right: The overlay of the spectra of apo-form tcFGE, pH 8.0 (dark blue), tcFGE in the presence of 450 μM AgNO3, pH 8.0 (black), tcFGE in the presence of 450 μM AgNO3 and 1 mM FGE-27, 2 mM glucose, 2 U GO, 200 U CAT, pH 8.1 (red). Bottom left: The overlay of the spectra of apo-form tcFGES266A, pH 8.0 (blue), tcFGES266A in the presence of 450 μM CuSO4, pH 8.0 (magenta), tcFGES266A in the presence of 450 μM CuSO4 and 1 mM FGE-27, 2 mM glucose, 2 U GO, 200 U CAT, pH 8.1 (dark cyan). Bottom right: The overlay of the spectra tcFGE in the presence of 450 μM CuSO4 measured at different pH. | ||
Complexation of apo tcFGE with Ag(I) induced a significantly smaller downfield shift of the Hε1/Nε1 signal than observed for Cu(I) (Table 1 and Fig. 4). A likely cause for this small but significant difference is that the bond angle between the sulfur ligands and the metal is larger for Ag(I) than for Cu(I).27 This slight movement of the metal would push H2O_1 closer to Trp228, and therefore strengthen the interaction between the two. The structures of tcFGE_Ag (S–Ag–S: 178°) and scFGE_Cu (S–Cu–S: 171°) indeed document a small structural difference.27 Addition of the substrate to tcFGE_Ag shifted the Hε1/Nε1 signal of tcFGE_Ag_S to exactly the same position as measured with tcFGE_Cu_S (Table 1), suggesting that the two complexes adopt very similar structures. The crystallographic and spectroscopic findings validate Ag(I) as a reliable structural mimic of Cu(I) in FGE.
Using this sensitive NMR methodology, we examined the proposition that H2O_1 in the tcFGE_Cu is stabilized as a hydroxide. Even though H2O_1 is not a metal ligand, it is possible that the proximity of an additional cationic charge, compounded by the interactions with Ser266 and Trp228 could stabilize a hydroxide at physiological pH. Since the pKa of water closely matches that of indole rings, a hydrogen bond between indole and hydroxide is likely stronger than between indole and water. Superposition of three 1H–15N TROSY HSQC spectra for tcFGE_Cu measured at pH 6, 7 and 8 showed no change of the Trp228 signal relative to other indole-specific resonances (Fig. 4). Based on this observation we concluded that either the pKa of H2O_1 is far below 6, or that H2O_1 is neutral at physiological pH. The second option is far more likely.
Role of Ser266 and Trp266 in the resting state and in catalysis
Finally, we examined the effect of the Ser266 to Ala mutation on the interaction of H2O_1 with Trp228. 1H–15N TROSY HSQC spectra recorded for tcFGES266,tcFGES266A_Cu and tcFGES266A_Cu_S revealed only small or no deshielding effects on the Hε1/Nε1 signal, suggesting that the absence of the Ser266 side chain does not affect the interaction between H2O_1 and Trp228. Hence, the role of Ser266 in immobilizing H2O_1 in the tcFGE_Cu_S complex is limited. In stark contrast, the same mutation causes a 50-fold reduction in catalytic turnover (kcat) without affecting substrate binding (Km, Table 2).25 The kinetic and spectroscopic properties of tcFGES266A suggest that Ser266 provides a hydrogen bond that become important after substrate-binding. Similarly, mutation of Trp228 to Phe (tcFGEW288F) also specifically affects kcat without hurting substrate binding. The lack of this hydrogen bond donor in the O2-binding site is likely to contribute to the demise of catalysis by this variant.| Enzyme | k cat, [min−1] | K m, [μM] | k cat/Km, [min−1 M−1] |
|---|---|---|---|
| a Kinetic parameters were determined in the presence of 2 μM CuSO4, 5 mM DTT, 50 mM NaCl and 50 mM Tris buffer pH 8.0, at 25C. Rate determined by monitoring the formation of fGly. b From ref. 25. | |||
| tcFGE | 1.4 ± 0.07 | 530 ± 40 | 2700 ± 200 |
| tcFGES266Ab | 0.025 ± 0.002 | 520 ± 240 | 49 ± 8 |
| tcFGEW228F | 0.0073 ± 0.0002 | 390 ± 20 | 19 ± 1 |
The unusual resting state of FGE
The active site of FGE is unusual for several reasons. First, catalysts that use a bis-cysteine coordinated Cu(I) as a starting point for O2-dependent C–H cleavage is unexplored territory. A large number of studies have shown quite conclusively that imidazole, amine, amide and thioether ligands are particularly well suited to facilitate copper-mediated O2-activation, not the least because these ligands stabilize high-valent copper species.1–3,51,52 From this perspective, bis- or tris-thiolate coordination spheres seem odd choices for designing copper-dependent oxidation catalysts. Thiolate ligands stabilize Cu(I) which should render electron transfer to O2 more difficult. On the other hand, thiolates are intrinsically vulnerable to oxidation which may open efficient paths for catalyst destruction. A specific complication of the FGE-catalyzed reaction is that the two active site cysteines and the substrate contain altogether six chemically equivalent cysteinyl Cβ–H bonds. Hence, the geometry of the active site must ensure that the reactive oxygen species exclusively attacks the pro-(R)-β-hydrogen on the substrate.19 The structure of tcFGE_Cu_S visualizes how the enzyme achieves regiospecificity. All three cysteines approach Cu(I) in a similar angle (Cβ–S–Cu: 103–120°) and place their Cβ at a similar distance to the copper center (3.2–3.5 Å). However, only the pro-(R)-β-hydrogen of the substrate points towards the presumed O2-binding site (Fig. 5).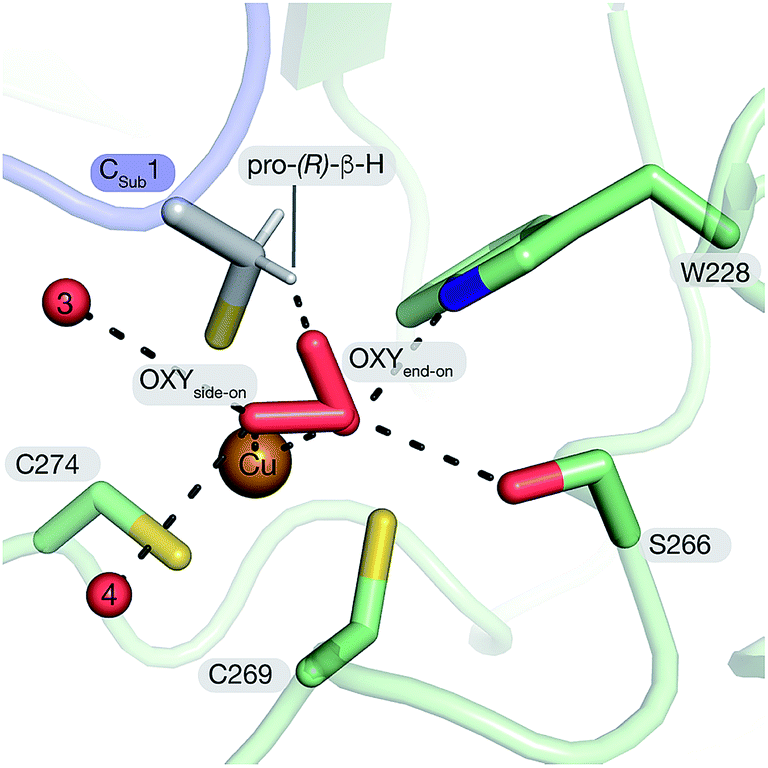 | ||
| Fig. 5 Structure-based model of tcFGE_Cu_S with superoxide coordinated to Cu(II) in side-on mode (η1) or end-on mode (η2). | ||
The second unusual feature of FGE is that the substrate and the reactive oxygen species both coordinate to the metal.1,27 Metal-dependent oxidases often bind O2via displacement of a weakly bound metal ligand, such as water, at a pre-formed coordination site. Formation of the coordination bond is accompanied by reduction of O2 to superoxide via inner-sphere electron transfer. For example, PHM coordinates Cu(I) via the side chains of two histidines and one methionine in a near tetrahedral geometry. The fourth ligand is a water molecule that is displaced by O2.9,10 In LPMO Cu(I) is coordinated by a histidine brace (Nα/Nπ) and the side chain of a second histidine.7 The square planar coordination sphere is completed by a chloride or a water molecule filling the dedicated O2 binding site. CAO coordinates copper with three histidines and a loosely bound water. Displacement by O2 occurs without change in the coordination geometry.11 We note that the path leading to the cupric superoxide in CAO is controversial. In the conventional mechanism O2 forms a coordination bond with Cu(I).53 An alternative proposal suggests that prebound O2 is first reduced by a protein-derived redox cofactor, and then combines as superoxide with Cu(II).54
In contrast to these enzymes, the substrate complex of tcFGE contains no preformed coordination site for a fourth ligand. The tris-thiolate Cu(I) complex is completely planar and the closest crystallographic waters (H2O_1 and H2O_2) are too distant for attractive interactions. Hence, direct binding of O2 would require a change in coordination geometry. The structure of tcFGE in complex with Cd(II) showed a tetrahedral coordination sphere around this bivalent metal. Therefore it is plausible that oxidation of Cu(I) to Cu(II) may trigger a change from trigonal planar to tetrahedral coordination.26 Importantly, if O2-coordination requires structural change, and this change requires oxidation of Cu(I), it is unlikely that this redox reaction occurs via inner-sphere electron transfer as in other mononuclear copper enzymes. A more plausible scenario is that O2 first binds to the active site without direct contact to the metal (C, Fig. 6). Outer sphere electron transfer from Cu(I) to prebound O2 followed by coordination would produce the predicted cupric superoxide complex (D). The efficiency of outer sphere electron transfer would critically rely on a binding site that increases the redox potential of pre-bound O2.
 | ||
| Fig. 6 Proposed catalytic mechanism of FGE. The reaction starts with sequential binding of substrate and O2 forming first complex B (crystallized) and then C. Outer sphere electron transfer from Cu(I) to O2 could produce the Cu(II) superoxide species D (modelled in Fig. 5). Homolytic abstraction of the pro-(R)-β-hydrogen atom from the substrate (E), followed by release and hydrolysis of the oxidized peptide product and reduction of the oxidized enzyme form (F) return the enzyme to the catalytically active resting state A. | ||
This requirement highlights the third unusual feature of FGE. The presumed O2-binding site in tcFGE is lined with an extensive array of hydrogen bond donors. Hydrogen-bonding has been recognized as important modulator of the stability and reactivity of copper-coordinated oxygen species in enzymes and in abiotic catalysts.1,2,55–57 For example, hydrogen-bonding to the proximal oxygen atom of a synthetic cupric superoxide complex has been shown to increase its stability and its propensity for hydrogen atom abstraction in direct correlation with hydrogen-bonding strength.55 Furthermore, specific hydrogen-bonding to the distal or to the proximal oxygen has been shown to affect the activity of copper-oxygen adducts in very different ways.56 On the other hand, oxygen-binding pockets lined by four well-positioned hydrogen bond donors are rare among metal-dependent catalysts. Studies on synthetic cages showed that solvation of O2 with multiple amide- or amine-donated hydrogen bonds can significantly stabilize superoxide and peroxide anions.58,59 The acidic O2-binding pocket in FGE may play a similar stabilizing role and thereby facilitate electron transfer from the electron-rich tris-thiolate Cu(I) to O2. The observation that mutation of Ser266 or Trp228 dramatically reduce the activity of tcFGE is consistent with this interpretation.
Conclusions
In this report we describe the crystal structure of tcFGE in complex with Cu(I) and a 17-residue peptide. Comparisons of this complex with the structures of tcFGE bound to Ag(I) or Cd(II) highlight substrate-induced changes that may prime the active site for O2-binding. On the other hand, we also found that substrate binding does not induce a preformed coordination site for O2 at the metal center. These observations raise the possibility that FGE activates O2 in a different way than other metallo-enzymes. Specifically, we propose that O2 first extracts an electron from Cu(I) via outer sphere electron transfer before combining to a cupric superoxide species. This and other testable hypotheses emanating from our structural investigating set the stage for deciphering the detailed mechanism of this unusual oxidation catalyst. We also hope to inspire the synthesis and characterization of abiotic catalysts that exploit this reactivity of bis- and tris-thiolate coordinated copper species.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Dr Oliver Einsle (Univ. Freiburg) for the access to the equipment used for anaerobic crystallization, the Swiss Lightsource (Villigen, Switzerland) for access to the beamline facilities and beamline staff for support. This project was supported by a starting grant from the European Research Council (ERC-2013-StG 336559), the NCCR for Molecular Systems Engineering and by the “Professur für Molekulare Bionik”.References
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059–2107 CrossRef CAS PubMed.
- W. Keown, J. B. Gary and T. D. Stack, J. Biol. Chem., 2017, 22, 289–305 CAS.
- S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed.
- J. T. Rubino and K. J. Franz, J. Inorg. Biochem., 2012, 107, 129–143 CrossRef CAS PubMed.
- R. J. Quinlan, M. D. Sweeney, L. L. Leggio, H. Otten, J. C. N. Poulsen, K. S. Johansen, K. B. R. M. Krogh, C. I. Jorgensen, M. Tovborg, A. Anthonsen, T. Tryfona, C. P. Walter, P. Dupree, F. Xu, G. J. Davies and P. H. Walton, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15079–15084 CrossRef CAS PubMed.
- K. E. Frandsen, T. J. Simmons, P. Dupree, J. C. Poulsen, G. R. Hemsworth, L. Ciano, E. M. Johnston, M. Tovborg, K. S. Johansen, P. von Freiesleben, L. Marmuse, S. Fort, S. Cottaz, H. Driguez, B. Henrissat, N. Lenfant, F. Tuna, A. Baldansuren, G. J. Davies, L. Lo Leggio and P. H. Walton, Nat. Chem. Biol., 2016, 12, 298–303 CrossRef CAS PubMed.
- L. Cao, O. Caldararu, A. C. Rosenzweig and U. Ryde, Angew. Chem., Int. Ed., 2018, 57, 162–166 CrossRef CAS PubMed.
- S. T. Prigge, B. A. Eipper, R. E. Mains and L. M. MAmzel, Science, 2004, 304, 864–867 CrossRef CAS PubMed.
- S. T. Prigge, A. S. Kolhekar, B. A. Eipper, R. E. Mains and M. Amzel, Nat. Struct. Biol., 1999, 6, 976–983 CrossRef CAS PubMed.
- C. M. Wilmot, J. Hajdu, M. J. McPherson, P. F. Knowles and S. E. Phillips, Science, 1999, 286, 1724–1728 CrossRef CAS PubMed.
- T. V. Vendelboe, P. Harris, Y. Zhao, T. S. Walter, K. Harlos, K. El Omari and H. E. M. Christensen, Sci. Adv., 2016, 2, e1500980 CrossRef PubMed.
- J. Y. Lee and K. D. Karlin, Curr. Opin. Chem. Biol., 2015, 25, 184–193 CrossRef CAS PubMed.
- A. Changela, K. Chen, Y. Xue, J. Holschen, C. E. Outten, T. V. OHalloran and A. Mondragon, Science, 2003, 301, 1383–1387 CrossRef CAS PubMed.
- A. K. Wernimont, D. L. Huffman, A. L. Lamb, T. V. OHalloran and A. C. Rosenzweig, Nat. Struct. Biol., 2000, 7, 766–771 CrossRef CAS PubMed.
- A. K. Boal and A. C. Rosenzweig, Chem. Rev., 2009, 109, 4760–4779 CrossRef CAS PubMed.
- L. Banci, I. Bertini, F. Cantini, I. C. Felli, L. Gonnelli, N. Hadjiliadis, R. Pierattelli, A. Rosato and P. Voulgaris, Nat. Chem. Biol., 2006, 2, 367–368 CrossRef CAS PubMed.
- P. G. Holder, L. C. Jones, P. M. Drake, R. M. Barfield, S. Banas, G. W. de Hart, J. Baker and D. Rabuka, J. Biol. Chem., 2015, 290, 15730–15745 CrossRef CAS PubMed.
- M. Knop, P. Engi, R. Lemnaru and F. P. Seebeck, ChemBioChem, 2015, 16, 2147–2150 CrossRef CAS PubMed.
- T. Dierks, B. Schmidt, L. V. Borissenko, J. H. Peng, A. Preusser, M. Mariappan and K. von Figura, Cell, 2003, 113, 435–444 CrossRef CAS.
- M. P. Cosma, S. Pepe, I. Annunziata, R. F. Newbold, M. Grompe, G. Parenti and A. Ballabio, Cell, 2003, 113, 445–456 CrossRef CAS PubMed.
- I. S. Carrico, B. L. Carlson and C. R. Bertozzi, Nat. Chem. Biol., 2007, 3, 321–322 CrossRef CAS PubMed.
- M. J. Appel and C. R. Bertozzi, ACS Chem. Biol., 2015, 10, 72–84 CrossRef CAS PubMed.
- T. Krüger, T. Dierks and N. Sewald, Biol. Chem., 2019, 4000, 289–297 Search PubMed.
- M. Knop, T. Q. Dang, G. Jeschke and F. P. Seebeck, ChemBioChem, 2017, 18, 161–165 CrossRef CAS PubMed.
- M. Meury, M. Knop and F. P. Seebeck, Angew. Chem., Int. Ed., 2017, 56, 8115–8119 CrossRef CAS PubMed.
- M. J. Appel, K. K. Meier, J. Lafrance-Vanasse, H. Lim, C. L. Tsai, B. Hedman, K. O. Hodgson, J. A. Tainer, E. I. Solomon and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(12), 5370–5375 CrossRef CAS PubMed.
- M. Jaskolski, M. Gilski, Z. Dauter and A. Wlodawer, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 611–620 CrossRef CAS PubMed.
- T. Dierks, A. Dickmanns, A. Preusser-Kunze, B. Schmidt, M. Mariappan, K. von Figura, R. Ficner and M. G. Rudolph, Cell, 2005, 121, 541–552 CrossRef CAS PubMed.
- B. L. Carlson, E. R. Ballister, E. Skordalakes, D. S. King, M. A. Breidenbach, S. A. Gilmore, J. M. Berger and C. R. Bertozzi, J. Biol. Chem., 2008, 283, 20117–20125 CrossRef CAS PubMed.
- A. J. Doig, M. W. MacArthur, B. J. Stapley and J. M. Thornton, Protein Sci., 1997, 6, 147–155 CrossRef CAS PubMed.
- D. Roeser, A. Preusser-Kunze, B. Schmidt, K. Gasow, J. G. Wittmann, T. Dierks, K. von Figura and M. G. Rudolph, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 81–86 CrossRef CAS PubMed.
- M. Knop, R. Lemnaru and F. P. Seebeck, ChemBioChem, 2017, 18, 1755–1761 CrossRef CAS PubMed.
- E. L. Smith, J. P. Giddens, A. T. Iavarone, K. Godula, L. X. Wang and C. R. Bertozzi, Bioconjugate Chem., 2014, 25, 788–795 CrossRef CAS PubMed.
- J. H. Peng, S. Alam, K. Radhakrishnan, M. Mariappan, M. G. Rudolph, C. May, T. Dierks, K. von Figura and B. Schmidt, FEBS J., 2015, 282, 3262–3274 CrossRef CAS PubMed.
- T. Krüger, S. Weiland, G. Falck, M. Gerlach, M. Boschanski, S. Alam, K. M. Müller, T. Dierks and N. Sewald, Angew. Chem., Int. Ed., 2018, 57, 7245–7249 CrossRef PubMed.
- T. Urbic and K. A. Dill, J. Am. Chem. Soc., 2018, 140, 17106–17113 CrossRef CAS PubMed.
- A. Biela, N. N. Nasief, M. Betz, A. Heine, D. Hangauer and G. Klebe, Angew. Chem., Int. Ed., 2013, 52, 1822–1828 CrossRef CAS PubMed.
- M. C. Chervenak and E. J. Toone, J. Am. Chem. Soc., 1994, 116, 10533–10539 CrossRef CAS.
- P. W. Snyder, J. Mecinovic, D. T. Moustakas, S. W. r. Thomas, M. Harder, E. T. Mack, M. R. Lockett, A. Héroux, W. Sherman and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17889–17894 CrossRef CAS PubMed.
- F. Biedermann, W. M. Nau and H. J. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11158–11171 CrossRef CAS PubMed.
- P. C. Roehm and J. M. Berg, J. Am. Chem. Soc., 1998, 120, 13083–13087 CrossRef CAS.
- S. J. Chiou, C. G. Riordan and A. L. Rheingold, Proc. Natl. Acad. Sci. U. S. A., 2003, 1000, 3695–3700 CrossRef PubMed.
- A. Dey, T. A. Okamura, N. Ueyama, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2005, 127, 12046–12053 CrossRef CAS PubMed.
- X. Yang, S. Niu, T. Ichiye and L. S. Wang, J. Am. Chem. Soc., 2004, 126, 15790–15794 CrossRef CAS PubMed.
- D. Roeser, B. Schmidt, A. Preusser-Kunze and M. G. Rudolph, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 621–627 CrossRef CAS PubMed.
- K. Pervushin, R. Riek, G. Wider and K. Wüthrich, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12366–12371 CrossRef CAS PubMed.
- S. Grzesiek and A. Bax, J. Magn. Reson., 1992, 96, 432–440 CAS.
- W. Sicinska, W. M. Westler and H. F. DeLuca, Proteins, 2005, 61, 461–467 CrossRef CAS PubMed.
- E. T. Mollova, D. E. Metzler, A. Kintanar, H. Kagamiyama, H. Hayashi, K. Hirotsu and I. Miyahara, Biochemistry, 1997, 36, 615–625 CrossRef CAS PubMed.
- S. M. Adam, G. B. Wijeratne, P. J. Rogler, D. E. Diaz, D. A. Quist, J. J. Liu and K. D. Karlin, Chem. Rev., 2018, 118, 10840–11022 CrossRef CAS PubMed.
- L. Ciano, G. J. Davies, W. B. Tolman and P. H. Walton, Nat. Catal., 2018, 1, 571–577 CrossRef CAS.
- E. M. Shepard, K. M. Okonski and D. M. Dooley, Biochemistry, 2008, 47, 13907–13920 CrossRef CAS PubMed.
- S. A. Mills, Y. Goto, Q. Su, J. Plastino and J. P. Klinman, Biochemistry, 2002, 41, 10577–10584 CrossRef CAS PubMed.
- M. Bhadra, J. Y. C. Lee, R. E. Cowley, S. Kim, M. A. Siegler, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2018, 140, 9042–9045 CrossRef CAS PubMed.
- S. I. Mann, T. Heinisch, T. R. Ward and A. S. Borovik, J. Am. Chem. Soc., 2017, 139, 17289–17292 CrossRef CAS PubMed.
- S. Hong, Y.-M. Lee, K. Ray and W. Nam, Coord. Chem. Rev., 2017, 334, 25–42 CrossRef CAS.
- N. Lopez, D. J. Graham, R. J. McGuire, G. E. Alliger, Y. Shao-Horn, C. C. Cummins and D. G. Nocera, Science, 2012, 335, 450–453 CrossRef CAS PubMed.
- E. W. Dahl, J. J. Kiernicki, M. Zeller and N. K. Szymczak, J. Am. Chem. Soc., 2018, 140, 10075–10079 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed descriptions of all experiments, supporting Fig. (S1–S15) and Tables (S1–S4) are shown in the ESI. See DOI: 10.1039/c9sc01723b |
| This journal is © The Royal Society of Chemistry 2019 |