
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceβ-Strand inspired bifacial π-conjugated polymers†
Saikat
Chaudhuri
,
Manikandan
Mohanan‡
,
Andreas V.
Willems‡
,
Jeffery A.
Bertke
 and
Nagarjuna
Gavvalapalli
*
and
Nagarjuna
Gavvalapalli
*
Department of Chemistry, Institute for Soft Matter Synthesis and Metrology, Georgetown University, 3700 O st NW, Washington, D.C. 20057, USA. E-mail: ng554@georgetown.edu
First published on 9th May 2019
Abstract
Access to diverse, relatively high molecular weight soluble linear polymers without pendant solubilizing chains is the key to solution state synthesis of structurally diverse nanoribbons of conjugated materials. However, realizing soluble 1D-π-conjugated polymers without pendant solubilizing chains is a daunting task. Herein, inspired from the polypeptide β-strand architecture, we have designed and developed novel bifacial π-conjugated polymers (Mn: ca. 24 kDa) that are soluble (ca. 70 to >250 mM) despite the absence of pendant solubilizing chains. The impact of varying the bifacial monomer height on polymer solubility, optical properties, and interactions with small molecules is reported.
Introduction
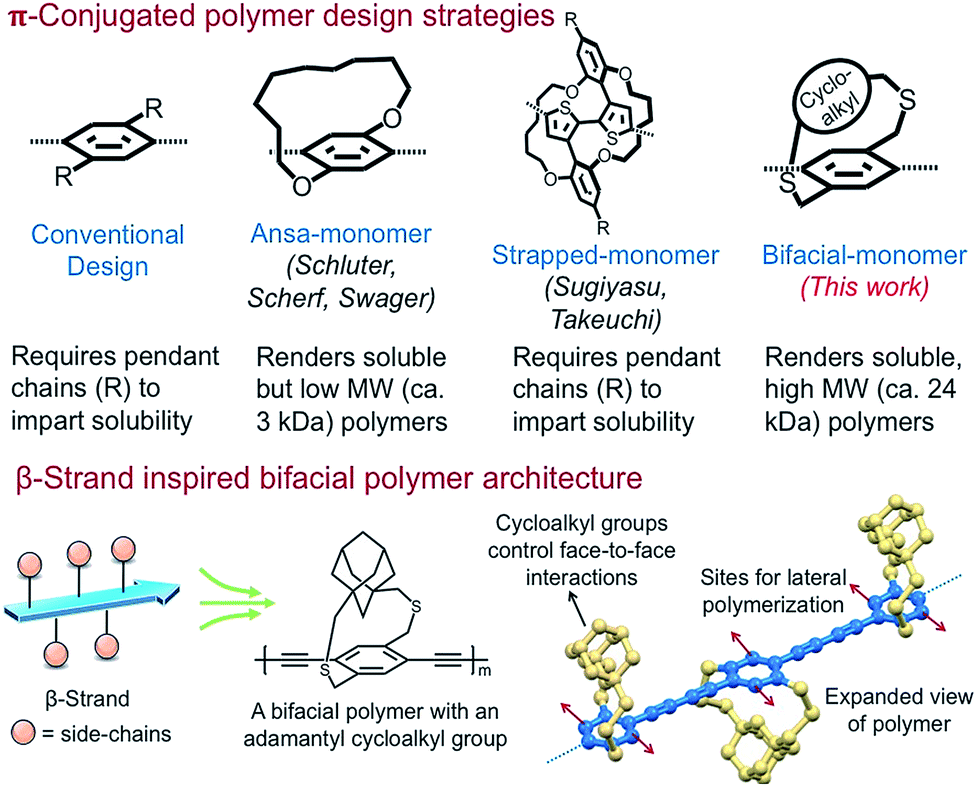
Extended π-conjugation in conformationally rigid planar polymers including ladder polymers, nanoribbons, 2D-grids, and 2D-π-conjugated polymers (2D-π-CoPs) provides intriguing optical and electronic properties compared to 1D-π-conjugated polymers (1D-π-CoPs).1–17 However, the lack of solution state synthesis, assembly, and solution processability of the 2D-π-conjugated materials limits their structural diversification and use in various applications.10,11,18–22 Although appending pendant solubilizing chains is a successful strategy in overcoming the above-mentioned challenges in the conformationally flexible 1D-π-CoPs, it is not effective for the 2D-π-conjugated materials of larger width due to strong van der Waals interactions.10,11,18–22Solution state synthesis of nanoribbons requires linear polymers that are soluble without pendant solubilizing chains so that monomers can be added laterally to extend the ribbon width (Scheme 1). In addition, for the resultant nanoribbons to be soluble, the π-surface of the ribbons should be masked efficiently to disrupt inter-ribbon interactions.12–16 Realizing these critical requirements, Schluter has shown two decades ago in a seminal paper that incorporating macrocycle containing monomers without pendant solubilizing chains, known as ansa-monomers, disrupts interchain (π–π) interactions and enables soluble low-molecular weight linear polymers as well as the corresponding ladder polymers of molecular weight 2.5 kDa (Scheme 1).11,20,23,24 Soluble polymers of high molecular weight are needed to generate nanoribbons of larger dimensions and harness the advantages of electronic conjugation. Thus, access to diverse, relatively high molecular weight soluble linear polymers without pendant solubilizing chains is the key to realizing solution state synthesis of structurally diverse nanoribbons.
 | ||
| Scheme 1 β-Strand inspired bifacial polymer architecture. | ||
Nature routinely forms 2D-sheets i.e., β-sheets through lateral polymerization of polypeptide β-strands.25 The peptide β-strands are designed such that the side-chains on alternate repeat units are positioned above and below the strand to allow the strands to come close and polymerize laterally through hydrogen-bonding interactions. Since the β-strand design renders soluble β-strands, allows lateral polymerization of the strands to generate sheets, and also controls face-to-face aggregation of the β-sheets,26,27 we envisioned designing molecular building blocks that emulate the β-strand architecture for the solution state synthesis of ladders, nanoribbons, and 2D-grids.
Herein, inspired from the β-strand design, we have designed and shown that the bifacial cyclophane monomers, despite not having pendant solubilizing chains, render soluble, high molecular weight (24 kDa) π-conjugated linear polymers. The bifacial polymers are made of bifacial cyclophane building blocks consisting of a polymerizable aryl group (aryl face) and a π-surface masking cycloalkyl group (cycloalkyl face) (Schemes 1 and 2). The aryl group of the bifacial monomer generates a π-conjugated polymer upon polymerization whereas the cycloalkyl groups positioned above and below the π-conjugation plane, similar to the side chains in the β-strands, mask the π-surface and control face-to-face intersheet interactions, and hence their solubility and assembly (Schemes 1 and 2).
 | ||
| Scheme 2 Chemical structures of bifacial monomers and polymers synthesized in this study. | ||
Importantly, the bifacial monomer design allows to easily vary the monomer height (vide infra) by changing the cycloalkyl group (e.g., from cyclohexyl to adamantyl) without significantly altering the monomer width (Scheme 2). The bifacial polymer solubility increased by changing the monomer height as well as incorporating a co-monomer of different height. The solubility of the bifacial polymers (BiPs) (ca. 70 to >250 mM) exceeded the solubility limit (ca. 70 mM) of the conventional poly(p-phenylenebutadiynylenes) (PPB) based π-conjugated polymer with pendant solubilizing chains. The impact of varying the bifacial monomer height on optical properties, assembly and polymer interactions with small molecules is also studied.
The proposed bifacial monomer design differs from strapped-polymers and insulated molecular wires28–30 in which the polymer backbone is completely sheathed and therefore lateral polymerization to generate the 2D-polymers is not possible. Also, since only the π-surface is masked in the bifacial polymers, their interchain charge transport is expected to be comparable or better than the completely insulated molecular wires,28,29 making the BiPs useful for optical and electronic applications. Importantly, the cycloalkyl groups can be cleaved post-polymerization or post-processing, if needed, using established desulfurization protocols to enhance intersheet interactions.31
Results and discussion
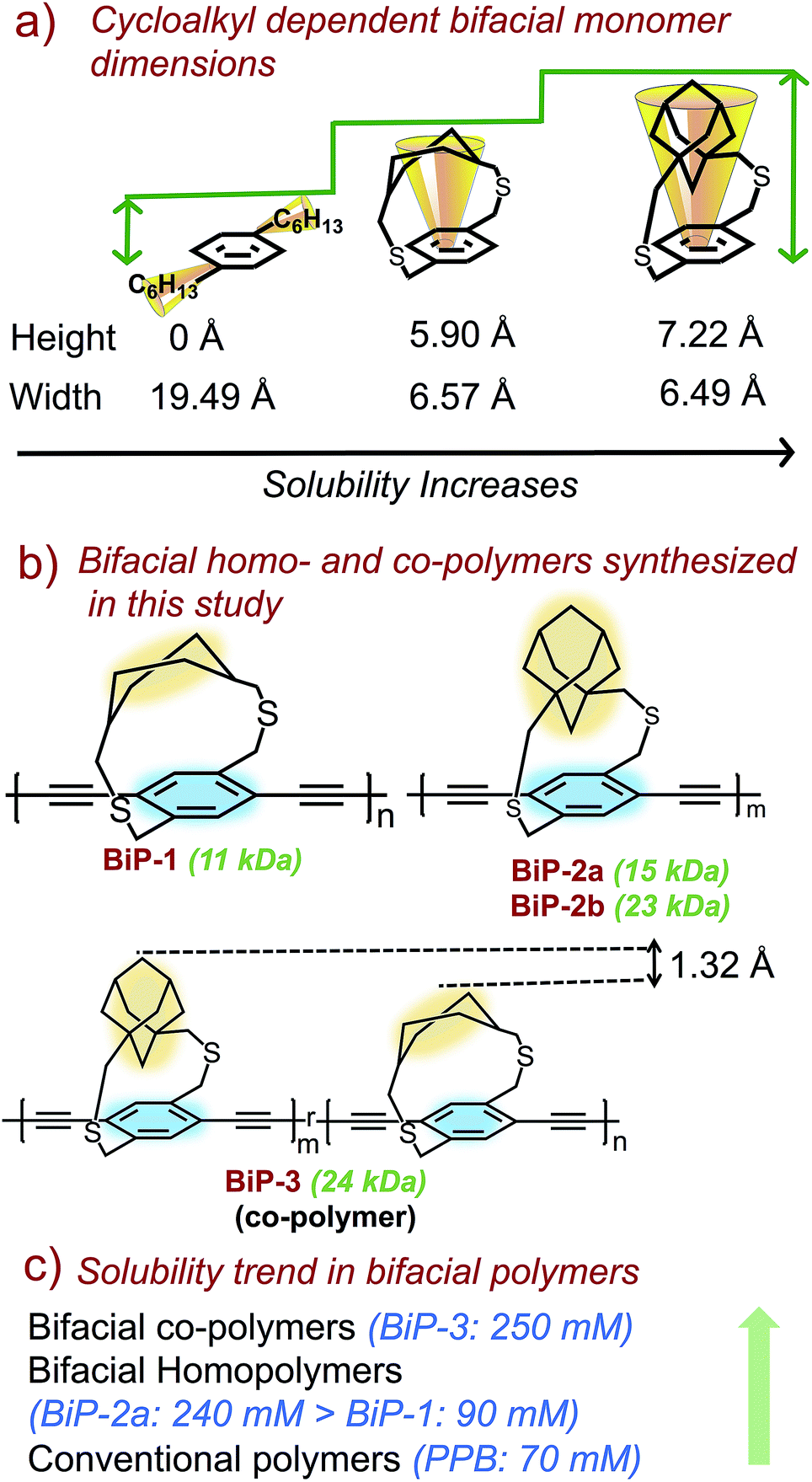
Bifacial molecules have been known since the 1990's32–34 but surprisingly they have not been used as building blocks for polymers yet. The bifacial monomers were synthesized using the modified reported protocols (Scheme 3 and ESI†). 2,5-Dibromo p-xylene was brominated using NBS (N-bromosuccinimide) to generate 2,5-dibromo-p-xylylene dibromide. 1,4-Cyclohexyl dimethanol (1) was converted into 1,4-cyclohexyl dimethanethioacetate (2) and reduced to generate 1,4-cyclohexyl dimethanethiol (3). 2,5-Dibromo-p-xylylene dibromide and the dithiol compound (3) were reacted under dilute conditions to generate 1,4-dibromo cyclohexanocyclophane (4). Sonogashira coupling of (4) with TMS-acetylene followed by desilylation resulted in 1,4-diethynyl cyclohexanocyclophane monomer (6). 1,4-Diethynyl adamantanocyclophane bifacial monomer (12) was generated from 1,3-adamantane dimethanol (7) following the similar protocols. 1,3-Adamantane dimethanol was prepared by esterifying and reducing 1,3-adamantane dicarboxylic acid. The height of compounds 4 and 10i.e., the distance from normal to the phenyl plane to the farthest atom in the cycloalkyl group was determined from single crystal X-ray structures (Fig. 1, S1 and S2†). As expected, the height of 10 (7.22 Å) with an adamantyl cycloalkyl group is greater than that of 4 (5.90 Å) with a cyclohexyl group, and the heights of both these monomers are higher than the conventional 1,4-dihexyl benzene (0 Å), which lacks the cycloalkyl groups on top of the π-surface. | ||
| Scheme 3 Synthesis of bifacial polymers. Reaction conditions: (a) thioacetic acid, DIAD, PPh3, THF; (b) LiAlH4, THF; (c) 2,5-dibromo-p-xylylene dibromide, KOH, C6H6, EtOH; (d) (i) TMS-acetylene, Pd(PPh3)4, CuI, toluene, triethylamine; (e) TBAF, THF; (f) CuCl, TMEDA, O2, solvent; (g) TMS-acetylene, Pd(PPh3)4, CuI, piperidine. | ||
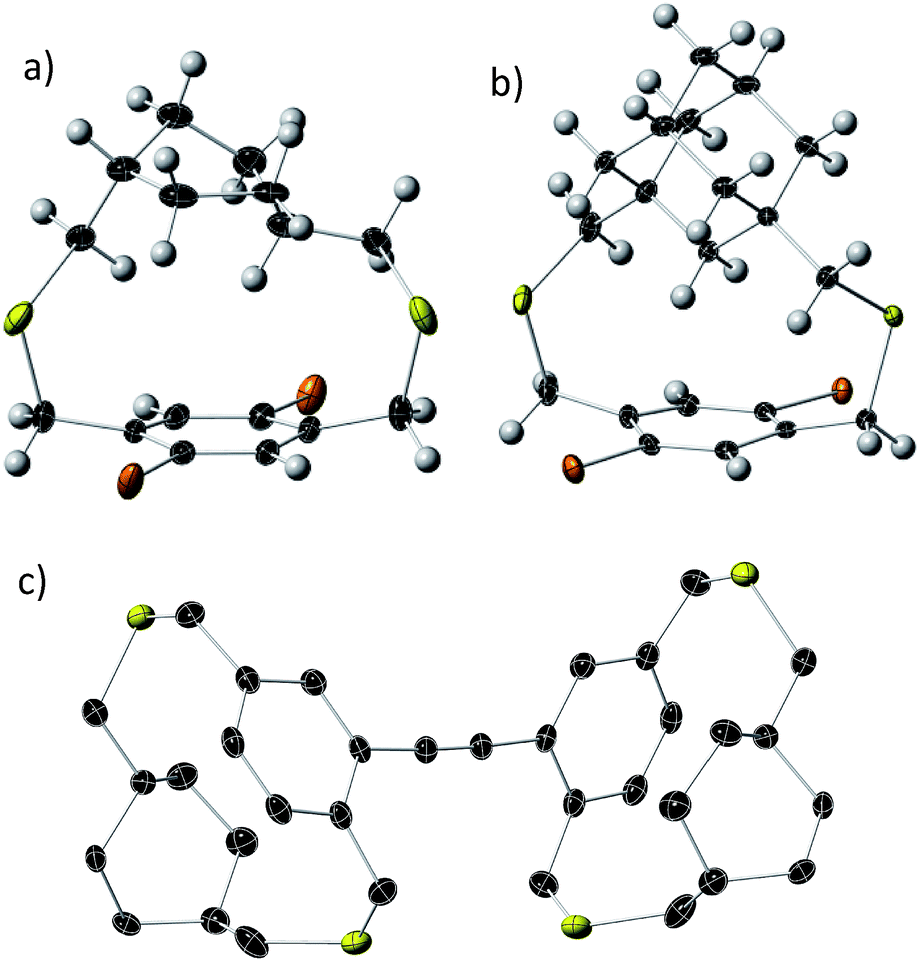 | ||
| Fig. 1 Single crystal X-ray thermal ellipsoid plots of (a) compound 4; (b) compound 10; and (c) compound 15 (dimer) with thermal ellipsoids at 50% probability. Black-C, yellow-S, orange-Br, white-H. | ||
Monomers 6 and 12 were subjected to Glaser–Hay polymerization to obtain the BiP polymers (Schemes 2, 3 and Fig. S3†). The polymerization mixture was precipitated in methanol and the crude polymer was subjected to soxhlation in ethyl acetate and chloroform. The polymer collected in chloroform was re-precipitated in ether, dried and used for characterization. Three bifacial homopolymers (BiP-1, BiP-2a and BiP-2b), one bifacial co-polymer (BiP-3), and a conventional polymer with dihexyl pendant solubilizing chains (PPB) were synthesized (Scheme 3). Stereoisomers of the monomers as synthesized were used for polymerization. The BiP-1 was synthesized from both the diastereomeric (arising due to cis and trans cyclohexyl substitution) and racemic mixture (formed during cyclophane formation) of monomer 6. The BiP-2a and BiP-2b were synthesized from the racemic mixture (formed during cyclophane formation) of monomer 12. Thus, all the synthesized bifacial polymers are atactic in nature i.e., there is no control over orientation of the repeat units along the polymer backbone.
The synthesized BiPs are soluble in several typical organic solvents (e.g., THF, CHCl3, DMF and chlorobenzene) that are used to solubilize π-conjugated polymers. In order to determine the solubilization power of the bifacial monomer compared to the conventional monomer, solubility limit of the polymers were determined in chloroform from multiple trials. The solubility limits were determined by dissolving polymers in chloroform at elevated temperature (55 °C) for about 20 minutes and then bringing the solution to room temperature.
The amount of polymer dissolved in chloroform was determined using the Beer–Lambert law (see ESI Fig. S4–S8†). The polymer solubility limit follows the order: PPB (16 kDa) < BiP-1 (11 kDa) ⋘ BiP-2a (15 kDa) (see Table 1). During the solubility limit test, an insoluble polymer was observed in solution in the case of both BiP-1 and PPB indicating that the solutions are saturated. Unlike the PPB and BiP-1, to our pleasant surprise, the BiP-2a is completely soluble in chloroform even at three times the amount of the PPB polymer and the polymer solution turned highly viscous making it difficult to determine the exact solubility limit. In order to avoid the discrepancy due to high molecular weight of the adamantyl repeat unit, the solubility limits are also determined in mM with respect to the repeat unit molecular weight, and the trend still holds well (Table 1). Solubility limit of the adamantyl bifacial polymer reduced when the polymer weight increased to 23 kDa (BiP-2b) indicating that as the molecular weight increases the solubility limit reduces for a given bifacial repeat unit (Table 1). Nonetheless, the solubility limit of BiP-2b is still higher than the conventional polymer (PPB) despite the molecular weight of BiP-2b (23 kDa) being higher than the PPB (16 kDa).
| Polymers | M n (kDa) | M w (kDa) | Absorption maximumb (Δλmax nm) | Molar extinction coefficient (M−1 cm−1) | Emission maximumb,c (λmax nm) | Solubility limit in CHCl3 | K sv (× 104) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Solution | Thin film | Δλmax | mg mL−1 | mM | ||||||
| a Tetrahydrofuran gel permeation chromatography with polystyrene standards. b Recorded in CHCl3. c Excited at absorption maximum. d Polymer fluorescence quenching studies with TCNQ. | ||||||||||
| BiP-1 | 11.0 | 29.3 | 421 | 433 | 12 | 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 ± 300 500 ± 300 |
437 | 29 ± 5 | 90 ± 15 | 2.6 ± 0.3 |
| BiP-2a | 15.0 | 31.8 | 423 | — | — | 27100 ± 700 |
— | >92 | >240 | — |
| BiP-2b | 23.0 | 48.8 | 424 | 433 | 9 | 25200 ± 100 |
448 | 26 ± 3 | 70 ± 8 | 1.3 ± 0.1 |
| BiP-3 | 24.0 | 51.4 | 424 | 430 | 6 | 32200 ± 300 |
445 | >87 | >250 | 1.7 ± 0.3 |
| PPB | 16.1 | 35.6 | 415 | 443 | 28 | 29900 ± 300 |
433 | 21 ± 2 | 70 ± 7 | 11 ± 1 |
To test whether fractionation of polymers is happening during the solubility limit tests i.e., if the low molecular weight polymer preferentially dissolves in chloroform while the high molecular weight polymer stays insoluble, GPC traces of the soluble and insoluble portions of the BiP-1 were recorded and compared with the BiP-1 polymer sample before the solubility limit test (Fig. S3f†). There is no significant difference in the polymer molecular weights (within 0.5 kDa) and polydispersities (within 0.1) of these three samples, which indicates that there is no fractionation of the polymer during the solubility limit tests. It is worth noting that during the purification stage all the BiPs went through fractionation via soxhlet extraction. Ethylacetate was used to extract the low molecular weight polymer while chloroform was used to obtain the chloroform soluble polymer fraction of the as synthesized polymers, and only the chloroform soluble fraction is used for all characterization. Even though the BiP-1 has a higher polydispersity compared to the BiP-2a, the absence of fractionation confirms that the insoluble polymer is indeed the excess BiP-1 polymer (beyond the solubility limit) and not just the high molecular weight polymer fraction of the BiP-1. The high molecular weight of BiP-2a along with the absence of fractionation in BiP-1 makes it reasonable to compare the solubility limits of these two polymers. Within the adamantyl polymer series, the BiP-2b has higher polymer molecular weight than the BiP-2a but both have similar polydispersities. Thus, the solubility limit comparison within these two polymers clearly highlights the impact of polymer molecular weight on solubility. Similar to the conventional polymers, the bifacial polymers with higher molecular weight have lower solubility limit.
In order to understand the impact of repeat units of dissimilar height on polymer solubility and interchain interactions, a random co-polymer of 6 and 12 (BiP-3) was synthesized (Schemes 2 and 3). We are thrilled to find that the solubility limit of the co-polymer (BiP-3, 24 kDa) is more than three times higher than the adamantyl bifacial homopolymer (BiP-2b, 23 kDa) of similar molecular weight (Table 1). Even at three times the amount of BiP-2b, the co-polymer is completely soluble in chloroform and the polymer solution turned highly viscous making it difficult to determine the exact solubility limit. Since the cycloalkyl groups are far from the ethynes, they have negligible steric and electronic effect on the kinetics of the Glaser–Hay coupling. This is evident from the 1:1 ratio of repeat units in the co-polymers, which reflects the feed ratio of the monomers as determined from 1H NMR. Thus, both the bifacial monomers of different height (1.328 Å height difference) are randomly positioned along the polymer backbone, which makes the height profile of the π-surface rugged along the polymer backbone (Scheme 2). Since both the BiP-2b and BiP-3 polymers have similar molecular weight and polydispersities, we attribute the enhanced solubility limit of the co-polymer compared to the homopolymers to the ruggedness of the π-surface, which leads to inefficient polymer chain packing and hence weaker interchain interactions.
In the case of ansa-poly(para-phenylene)s made using the ansa-monomers (Scheme 1), it has been shown that the isotactic and syndiotactic polymers (polymers synthesized from enantiomerically pure monomers) are less soluble compared to the atactic polymer (polymer synthesized from a racemic mixture).23 Thus, the presence of different stereoisomers along the polymer backbone also helps to weaken interchain interactions and improve polymer solubility. In the case of BiPs also, since a stereoisomeric mixture of monomers is used to synthesize the polymers, the generated bifacial polymers contain stereoisomers associated with the monomer along the polymer backbone resulting in atactic polymers. In addition to the presence of the cycloalkyl groups, the atactic nature of the polymer backbone also helped to further weaken the interchain interactions and generate soluble polymers without pendant solubilizing chains. Since all the BiPs are synthesized from a stereoisomeric mixture of monomers, improved solubility of the BiP-2a compared to BiP-1 is attributed to a change in the height of the monomer. Improved solubility of the BiP-3 compared to BiP-2b is attributed to a random arrangement of the repeat units of different height along the polymer backbone.
In order to understand the impact of the bifacial monomer height on the strength of interchain interactions, change in absorption maximum of the polymers from solution to thin film are compared (Fig. 2). The absorption maxima of all the bifacial homopolymers as well as the BiP-3 co-polymer in chloroform are ca. 424 nm, which is 9 nm higher than the PPB (Table 1). Similar absorption maxima of the BiPs and PPB indicate that the bifacial architecture has minimal effect on the solution state absorption spectrum. The absorption maxima of the BiPs in thin films are ca. 430 nm, which is 6–10 nm red-shifted compared to the solution state maxima. On the other hand, absorption maximum of the PPB in thin film is 442 nm, which is 28 nm red-shifted compared to the solution state maximum (Table 1). The higher red shift in PPB is due to strong interchain π–π interactions between the unhindered π-surface of the polymers. The lower red shift (6–12 nm) in the case of BiPs support that the cycloalkyl groups weaken interchain interactions between the polymers. The emission spectra of the BiPs in chloroform are shown in Fig. 3. The difference in solution state absorption maxima to emission maxima for the BiPs are between 16–24 nm, which indicates that there is not much difference in the conformational changes during emission as the repeat unit size is increased by changing the cycloalkyl group from cyclohexyl to adamantyl.
 | ||
| Fig. 2 UV-vis absorption spectra of BiP polymers. (Top) In solution state; (bottom) thin film. | ||
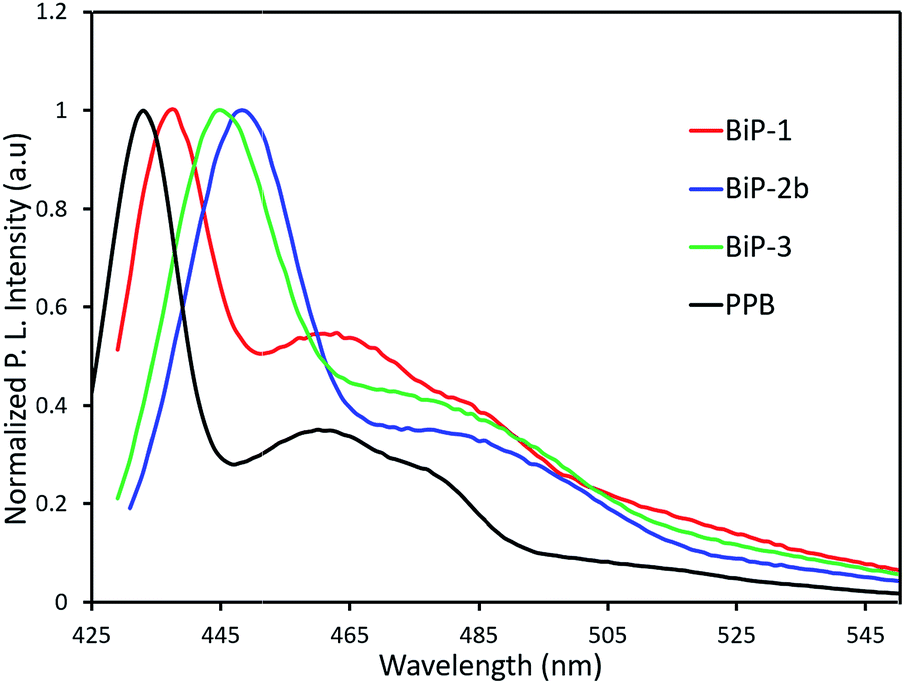 | ||
| Fig. 3 Fluorescence spectra of BiP polymers. | ||
The powder X-ray diffraction data of all the BiPs shows two peaks below 25° 2θ (Fig. 4). For the BiP-1 the two peaks correspond to 7.18 Å and 4.12 Å. Since the BiPs lack pendant solubilizing chains their assembly is expected to be different from the conventional conjugated polymers with solubilizing chains. To gain insights into polymer assembly in the BiPs, a model-dimer molecule (15) is synthesized (see ESI†). The dimer crystal structure (Fig. 1 and ESI†) suggests existence of two different layers of polymer chains in the solid state in which one of the layers is offset with respect to the other and there is an interdigitation of cycloalkyl groups between the layers. This type of assembly pattern is reminiscent of the assembly in polyacetylenes, which also lack the pendant solubilizing chains.35 The crystal structure of the model dimer molecule indicates presence of ABA type stacking in the BiPs as shown in Fig. 4. The interlayer separation distance in BiP-1 was found to be 7.18 Å from PXRD, which corresponds to the peak at lower 2θ. The interchain separation within the layer along the lateral direction is 8.24 Å but in the PXRD the corresponding overtone peak at higher 2θ (4.12 Å) is appearing. In the case of BiP-2b and BiP-3 due to the increased height of the adamantyl moiety the interlayer distance from PXRD is found to be greater than the BiP-1 and corresponds to 7.5 Å. Only a small increase in the interlayer separation for BiP-2b and BiP-3 indicates strong interdigitation of the adamantyl cycloalkyl groups in these polymers. Similar interchain distance for both the BiP-2b and BiP-3 copolymer confirms that the improved solubility of BiP-3 is due to weaker interactions in the co-polymer and not due to a difference in polymer assembly in the solid state.
 | ||
| Fig. 4 (Top) PXRD of BiPs and (bottom) cartoon depicting the assembly of polymers in solid state. | ||
To further elucidate how the bifacial monomer height impacts the BiP interactions with other molecules, fluorescence quenching studies of the BiPs with TCNQ (tetracyanoquinodimethane) acceptor were carried out and compared with that of the conventional polymer (PPB) (Fig. 5 and S9–S11†). As the TCNQ concentration increased the fluorescence intensity of all the polymers reduced resulting in an upward curvature of the Stern–Volmer (SV) plot. Thus, the data was fitted to a reported non-linear SV equation36 to obtain the SV constant (Ksv) (Fig. 5 and S12†). The Ksv decreased with an increase in the monomer height and follows the order PPB ≫ BiP-1 > BiP-3 > BiP-2b. Moreover, there is almost an order of magnitude reduction in the Ksv from PPB to BiP-2b (Table 1). To the best of our knowledge varying the Ksv of poly(p-phenyleneethynylene),36 which is similar to the PPB backbone structure, up to an order of magnitude by varying the pendant side chains has not been demonstrated so far. All the BiPs have a similar polymer backbone; thus, the quenching constant primarily depends on how close a TCNQ molecule can approach the polymer backbone. In the case of bifacial polymers, the polymer backbone mimics a cylinder wherein the radius of the cylinder is determined by the height of the monomer. The larger the radius of the cylinder (radius of BiP-2b > BiP-3 > BiP-1), the farther the TCNQ is from the polymer backbone, thereby resulting in a lower quenching constant. BiPs have lower quenching constant than the PPB even though the width of the PPB repeat unit and hence the PPB cylinder diameter is larger because in the case of BiPs the cycloalkyl groups are directly positioned above and below the π-surface and thus are more effective in weakening the BiP–TCNQ interactions and reducing the Ksv by an order of magnitude.
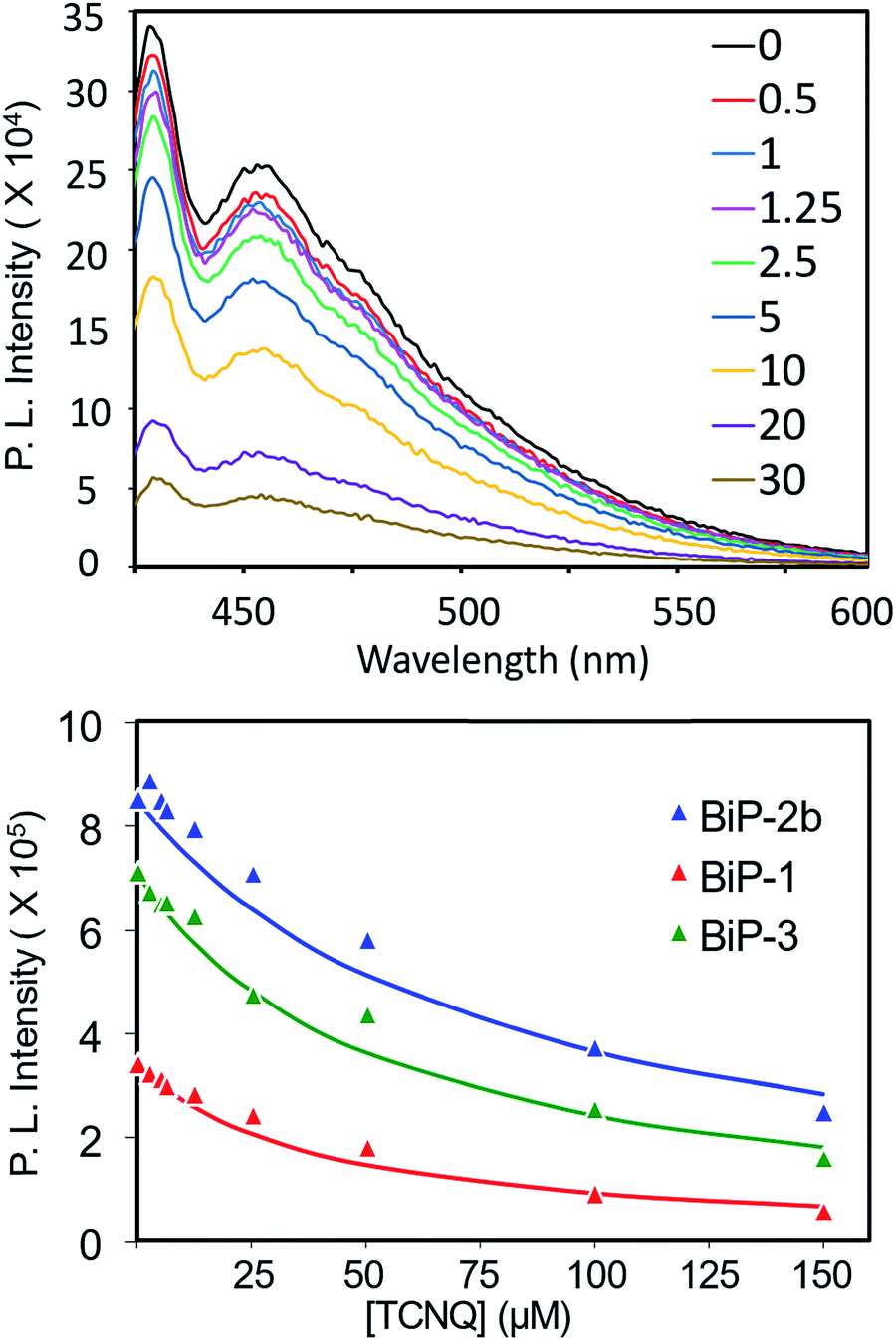 | ||
| Fig. 5 (Top) TCNQ concentration dependent BiP-1 emission spectra in chloroform, TCNQ to polymer repeat unit concentration is shown in the legend; (bottom) Stern–Volmer plot and fit of bifacial polymers. | ||
Conclusions
In conclusion, novel bifacial π-conjugated polymers that mimic the β-strand architecture are developed. The lack of pendant solubilizing chains, and the ability to easily vary monomer height without significantly altering the monomer width and to generate relatively high molecular weight soluble polymers make the bifacial polymers potential building blocks to control growth, assembly, and solution processability of the 2D-π-conjugated materials. By proper design i.e., by incorporating functional moieties that undergo lateral polymerization, the high molecular weight, soluble bifacial polymers can be converted into ladder polymers and nanoribbons. Also, the lack of pendant solubilizing chains and higher solubility will help to surpass the current limitation on the number of strands used for 2D-grid growth. Finally, similar to the β-strands in β-fibrils, the bifacial architecture provides control over face-to-face interactions in 2D-π-CoPs, which enables control of the growth, assembly and solution processability of the 2D-π-CoPs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Donors of the American Chemical Society Petroleum Research Fund (#58444-DNI7) for partial support of this research.Notes and references
- B. A. G. Hammer and K. Mullen, Chem. Rev., 2016, 116, 2103–2140 CrossRef CAS PubMed.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453–465 CrossRef CAS PubMed.
- L. Ma, S. Wang, X. Feng and B. Wang, Chin. Chem. Lett., 2016, 27, 1383–1394 CrossRef CAS.
- L. Yang and D.-C. Wei, Chin. Chem. Lett., 2016, 27, 1395–1404 CrossRef CAS.
- A. Narita, X. L. Feng, Y. Hernandez, S. A. Jensen, M. Bonn, H. F. Yang, I. A. Verzhbitskiy, C. Casiraghi, M. R. Hansen, A. H. R. Koch, G. Fytas, O. Ivasenko, B. Li, K. S. Mali, T. Balandina, S. Mahesh, S. De Feyter and K. Mullen, Nat. Chem., 2014, 6, 126–132 CrossRef CAS PubMed.
- J. Lee, A. J. Kalin, T. Y. Yuan, M. Al-Hashimi and L. Fang, Chem. Sci., 2017, 8, 2503–2521 RSC.
- P. M. Byers and I. V. Alabugin, J. Am. Chem. Soc., 2012, 134, 9609–9614 CrossRef CAS PubMed.
- D. Hertel, U. Scherf and H. Bassler, Adv. Mater., 1998, 10, 1119–1122 CrossRef CAS.
- S. R. Bheemireddy, M. P. Hautzinger, T. Li, B. Lee and K. N. Plunkett, J. Am. Chem. Soc., 2017, 139, 5801–5807 CrossRef CAS PubMed.
- C. S. Hartley, E. L. Elliott and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 4512–4513 CrossRef CAS PubMed.
- I. C.-Y. Hou, Y. Hu, A. Narita and K. Müllen, Polym. J, 2017, 50, 3–20 CrossRef.
- D. Lehnherr, C. Chen, Z. Pedramrazi, C. R. DeBlase, J. M. Alzola, I. Keresztes, E. B. Lobkovsky, M. F. Crommie and W. R. Dichtel, Chem. Sci., 2016, 7, 6357–6364 RSC.
- Y. J. Li, Z. Y. Jia, S. Q. Xiao, H. B. Liu and Y. L. Li, Nat. Commun., 2016, 7, 11637 CrossRef PubMed.
- F. Schlutter, T. Nishiuchi, V. Enkelmann and K. Mullen, Angew. Chem., Int. Ed., 2014, 53, 1538–1542 CrossRef PubMed.
- T. Y. Zheng, Z. X. Cai, R. Ho-Wu, S. H. Yau, V. Shaparov, T. Goodson and L. P. Yu, J. Am. Chem. Soc., 2016, 138, 868–875 CrossRef CAS PubMed.
- C. D. Zhu, D. Wang, D. Y. Wang, Y. Zhao, W. Y. Sun and Z. Z. Shi, Angew. Chem., Int. Ed., 2018, 57, 8848–8853 CrossRef CAS PubMed.
- Y. Morisaki, T. Ishida and Y. Chujo, Macromolecules, 2002, 35, 7872–7877 CrossRef CAS.
- A. D. Schluter, P. Payamyar and H. C. Ottinger, Macromol. Rapid Commun., 2016, 37, 1638–1650 CrossRef PubMed.
- J. Sakamoto, J. van Heijst, O. Lukin and A. D. Schluter, Angew. Chem., Int. Ed., 2009, 48, 1030–1069 CrossRef CAS PubMed.
- A. D. Schluter, M. Loffler and V. Enkelmann, Nature, 1994, 368, 831–834 CrossRef.
- S. Gratz, D. Beyer, V. Tkachova, S. Hellmann, R. Berger, X. L. Feng and L. Borchardt, Chem. Commun., 2018, 54, 5307–5310 RSC.
- A. Narita, X. Y. Wang, X. L. Feng and K. Mullen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- R. Fiesel, J. Huber, U. Apel, V. Enkelmann, R. Hentschke, U. Scherf and K. Cabrera, Macromol. Chem. Phys., 1997, 198, 2623–2650 CrossRef CAS.
- D. K. Fu, B. Xu and T. M. Swager, Tetrahedron, 1997, 53, 15487–15494 CrossRef CAS.
- P. N. Cheng, J. D. Pham and J. S. Nowick, J. Am. Chem. Soc., 2013, 135, 5477–5492 CrossRef CAS.
- M. S. Lamm, K. Rajagopal, J. P. Schneider and D. J. Pochan, J. Am. Chem. Soc., 2005, 127, 16692–16700 CrossRef CAS PubMed.
- C. J. Bowerman and B. L. Nilsson, Biopolymers, 2012, 98, 169–184 CrossRef CAS PubMed.
- F. Cacialli, J. S. Wilson, J. J. Michels, C. Daniel, C. Silva, R. H. Friend, N. Severin, P. Samori, J. P. Rabe, M. J. O'Connell, P. N. Taylor and H. L. Anderson, Nat. Mater., 2002, 1, 160–164 CrossRef CAS PubMed.
- M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028–1064 CrossRef CAS PubMed.
- C. J. Pan, C. H. Zhao, M. Takeuchi and K. Sugiyasu, Chem.–Asian J., 2015, 10, 1820–1835 CrossRef CAS PubMed.
- J. Rentner, M. Kljajic, L. Offner and R. Breinbauer, Tetrahedron, 2014, 70, 8983–9027 CrossRef CAS.
- R. Lemmerz, M. Nieger and F. Vogtle, J. Chem. Soc., Chem. Commun., 1993, 1168–1170, 10.1039/c39930001168.
- S. T. Lin, F. M. Yang and L. H. Lin, Tetrahedron, 1997, 53, 16123–16130 CrossRef CAS.
- F. M. Yang and S. T. Lin, J. Org. Chem., 1997, 62, 2727–2731 CrossRef CAS PubMed.
- S. Flandrois and A. Boukhari, Chemical Physics of Intercalation, ed. A. P. Legrand and A. Flandrois, Springer Science + Business Media, LLC, 1987, pp. 311–317 Search PubMed.
- K. Campbell, A. Zappas, U. Bunz, Y. S. Thio and D. G. Bucknall, J. Photochem. Photobiol., A, 2012, 249, 41–46 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of small molecules and polymers. CCDC 1885228–1885230. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01724k |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |