
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed enantioselective decarboxylative allylic alkylation of fully substituted N-acyl indole-derived enol carbonates†
Eric J.
Alexy‡
 a,
Tyler J.
Fulton‡
a,
Haiming
Zhang
*b and
Brian M.
Stoltz
*a
a,
Tyler J.
Fulton‡
a,
Haiming
Zhang
*b and
Brian M.
Stoltz
*a
aWarren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: stoltz@caltech.edu
bSmall Molecule Process Chemistry, Genentech, Inc., 1 DNA Way, South San Francisco, CA 94080, USA. E-mail: zhang.haiming@gene.com
First published on 17th May 2019
Abstract
The first enantioselective palladium-catalyzed decarboxylative allylic alkylation of fully substituted N-acyl indole-derived enol carbonates forming acyclic all-carbon quaternary stereocenters is reported. Excellent yields up to 99% and enantioselectivities up to 98% ee are obtained through the use of a new electron-deficient phosphinoxazoline (PHOX) ligand. Control of substrate enolate geometry is crucial for high selectivity. The obtained α-quaternary N-acyl indoles are formal ester equivalents, and represent a useful handle for further synthetic transformations.
Introduction
The catalytic enantioselective preparation of all-carbon quaternary stereocenters has become an important area of research owing to the prominence of such structures in natural products and their potential utility in medicinal chemistry.1 Toward this end, numerous strategies have been developed for the synthesis of this challenging motif in cyclic systems.1,2 Meanwhile, approaches to quaternary stereocenters in acyclic systems have proven more challenging, largely due to the need for stereodefined olefins and enolates typically necessary for high selectivities.3 Recently, our lab has investigated the utility of palladium-catalyzed decarboxylative allylic alkylation for preparing acyclic all-carbon quaternary stereocenters.4 In particular, we identified a unique transformation of fully-substituted acyclic enol carbonates to afford the corresponding α-quaternary ketones in an enantioselective fashion (Fig. 1A).4c While the enol carbonates could be prepared with high stereochemical fidelity utilizing recently reported conditions,5 we were surprised to find that the enolate geometry of the substrate was inconsequential, with the same enantiomer of product obtained in the same enantioselectivity regardless of the ratio of starting enolates. This result runs contrary to reports in the literature highlighting the central importance of controlling enolate geometry in acyclic palladium-catalyzed allylic alkylation.4a,6 To extend the scope of the transformation, we sought to examine ester enolates as possible substrates, allowing for the synthetically more versatile α-quaternary carboxylic acid derivatives to be accessed. As background, the Trost group has previously reported the enantioselective palladium-catalyzed allylic alkylation of two different ester enolate equivalents; 2-acylimidazoles7a and N-acyl oxazolidinones (Fig. 1B).7b However, both of these reports were limited to trisubstituted enolates, highlighting the difficulty of forming acyclic quaternary stereocenters via enolate chemistry. Unique strategies have emerged as a means of bypassing this issue. For example, Shimizu and Kanai reported a dual boron–palladium catalytic system, which allowed access to α-quaternary carboxylic acids through a symmetrical, chiral carboxylic acid dianion (Fig. 1C).8 Additionally, both rhodium9 and iridium-catalyzed10 allylic alkylation have previously been shown to provide acyclic quaternary stereocenters. With this in mind, additional general methods addressing the synthesis of this challenging and useful structural motif warrant further investigation. Toward this end, we sought to prepare stereodefined acyclic ester enolates, or enolate equivalents, investigate the E/Z selectivity of their synthesis, and determine the importance of this geometry by demonstration of their application in enantioselective palladium-catalyzed allylic alkylation (Fig. 1D).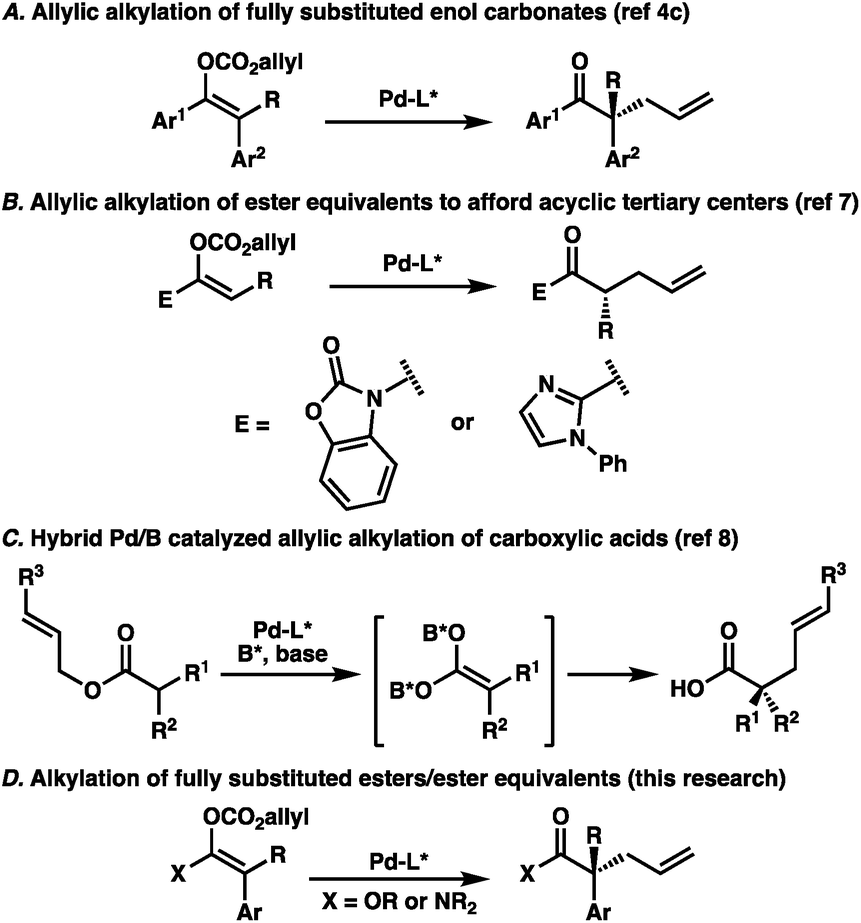 | ||
| Fig. 1 Enantioselective Pd-catalyzed allylic alkylation of acyclic substrates. | ||
Results and discussion
We began by investigating the selective enolization of sterically varied α-alkyl/aryl substituted esters and were intrigued to find that treatment with LiHMDS and dimethylethylamine at 25 °C,5 followed by enolate trapping with allyl chloroformate, provides the desired ester-derived enol carbonates in high E/Z selectivity, albeit in modest yield (eqn (1)). Given that previous examples of enolization with this protocol had been performed on aryl ketones, this unexpected selectivity was highly exciting. | (1) |
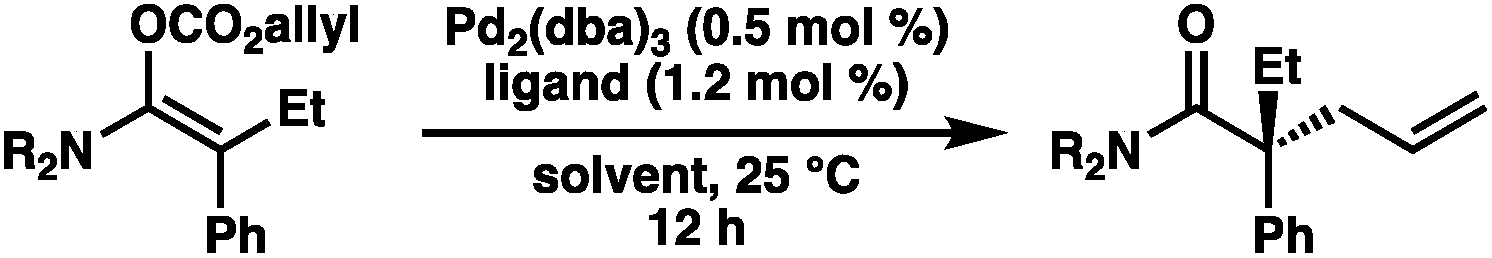
Our excitement was quickly tempered, however, as the resulting allyl carboxy ketene acetals demonstrated surprisingly poor reactivity under standard palladium-catalyzed allylic alkylation conditions, requiring elevated temperatures to reach appreciable levels of conversion. Despite optimization efforts, the best result obtained utilized the ethyl ester-derived substrate and provided the alkylation product in only 43% yield as a racemate using (S)-CF3-t-BuPHOX (L1) as the ligand (eqn (2)). Both phenyl and t-Bu ester-derived substrates failed to provide significant levels of the alkylation product.
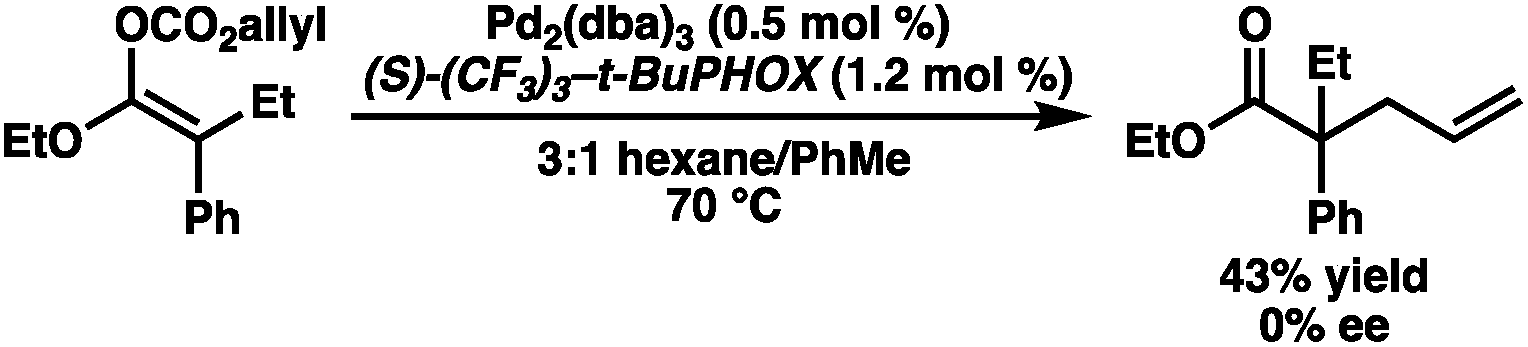 | (2) |
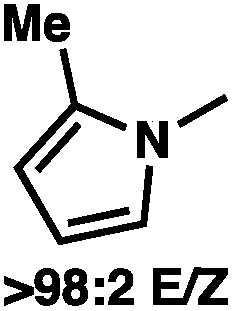
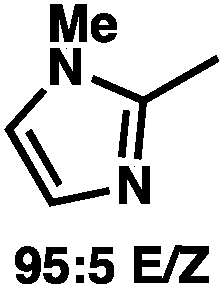
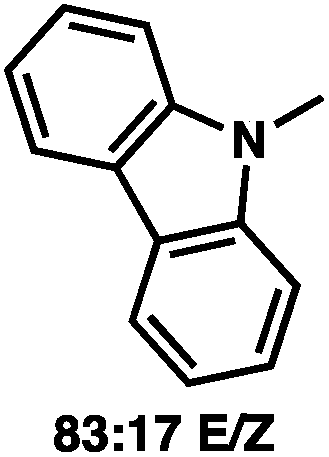
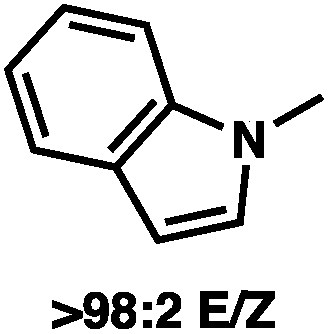
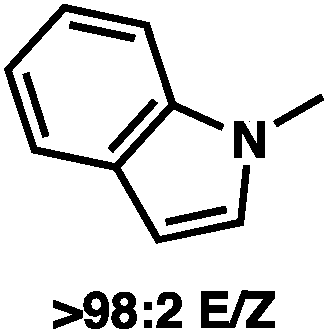
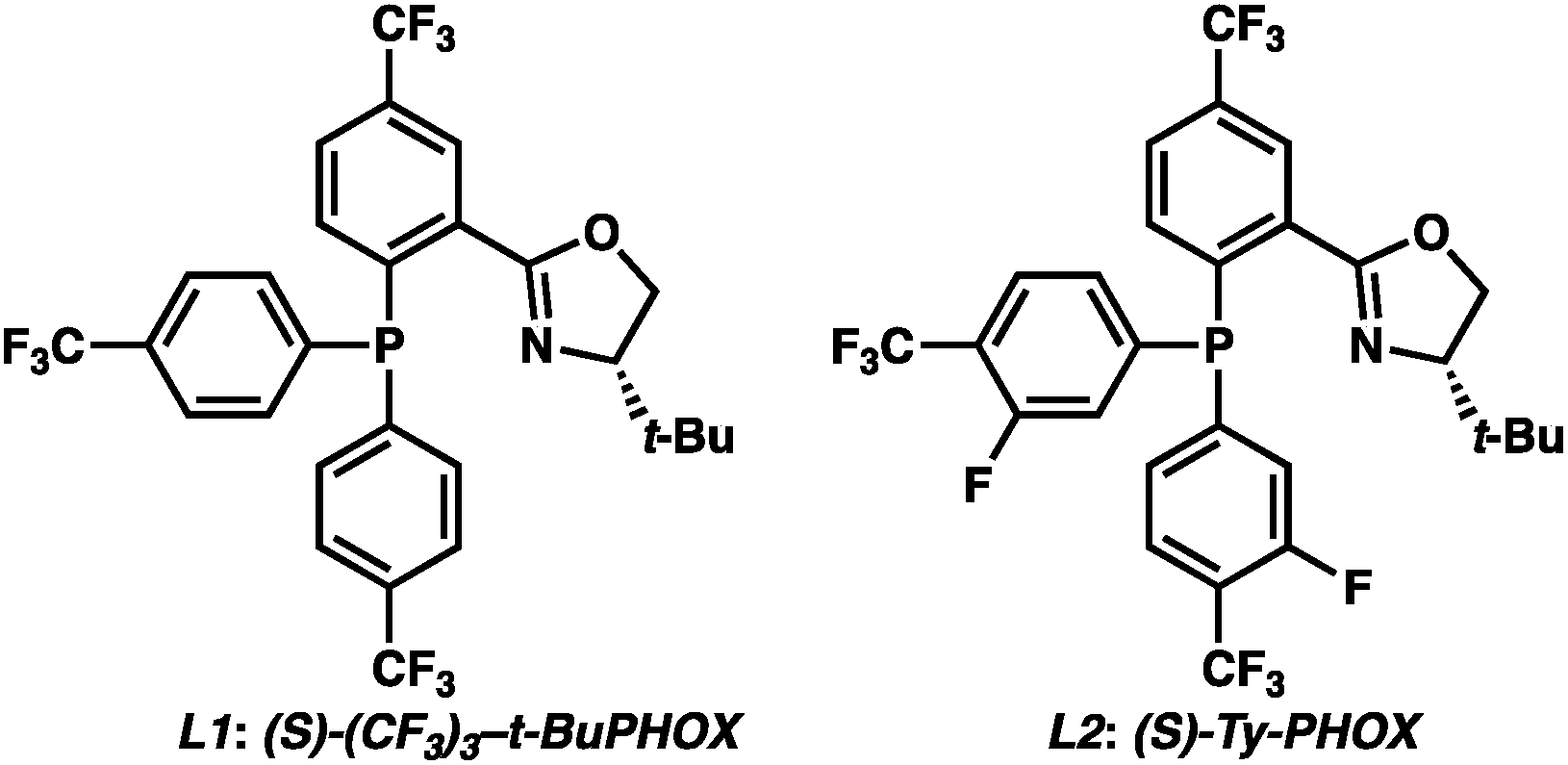
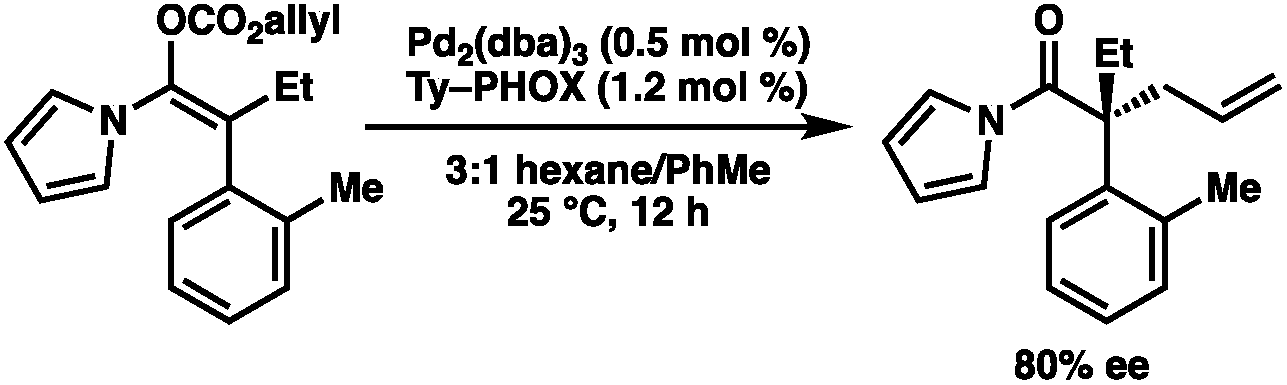
To circumvent the issues with ester-derived enol carbonates, we turned instead toward ester enolate equivalents. Based on prior precedent and our own recent findings in the area of cyclic enolate derivatives,6b,11 we investigated N-acyl-heterocycles as potential substrates. We were pleased to find that a variety of N-acyl-heterocycle derived enol carbonates can be prepared in good to excellent E/Z selectivity under analogous selective-enolization conditions, and provided more promising reactivity in a subsequent enantioselective alkylation (Table 1). Utilizing conditions our group identified for the enantioselective allylic alkylation of acyclic ketones as a starting point, we found that the N-acyl pyrrole-derived enol carbonate affords the desired allylic alkylation product in a high 92% yield and a moderate 73% ee with electron-deficient phosphinoxazoline L1 as the ligand (Table 1, entry 1). The ee was improved to 83% by switching to a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methylcyclohexane/PhMe solvent mixture (entry 2), however, further experimentation led to no additional increase in selectivity for this substrate. By increasing the steric bulk on the pyrrole through the inclusion of an ortho-methyl group (entry 3), the high enolization selectivity is retained, however, the resultant alkylation product is obtained in only 65% ee. An N-methylimidazole-derived substrate (entry 4) was found to give no conversion under the alkylation conditions. Further increasing the steric bulk by moving to carbazole (entry 5) results in a drop in enolization selectivity, and again, no reaction is observed in the allylic alkylation. The optimal substrate was found to be an N-acyl indole (entry 6), which both enolizes selectively and affords the desired alkylation product in an excellent 95% yield and 90% ee. After additional experimentation, we found that the use of a novel electron-deficient PHOX ligand (S)-Ty-PHOX (L2) provides the desired product in an improved 99% yield and 95% ee when a slightly modified 3:1 hexane/toluene solvent mixture is used. Notably, the reaction is performed using only 0.5 mol% Pd2(dba)3 and 1.2 mol% ligand and is complete in 12 h at 25 °C.
:1 methylcyclohexane/PhMe solvent mixture (entry 2), however, further experimentation led to no additional increase in selectivity for this substrate. By increasing the steric bulk on the pyrrole through the inclusion of an ortho-methyl group (entry 3), the high enolization selectivity is retained, however, the resultant alkylation product is obtained in only 65% ee. An N-methylimidazole-derived substrate (entry 4) was found to give no conversion under the alkylation conditions. Further increasing the steric bulk by moving to carbazole (entry 5) results in a drop in enolization selectivity, and again, no reaction is observed in the allylic alkylation. The optimal substrate was found to be an N-acyl indole (entry 6), which both enolizes selectively and affords the desired alkylation product in an excellent 95% yield and 90% ee. After additional experimentation, we found that the use of a novel electron-deficient PHOX ligand (S)-Ty-PHOX (L2) provides the desired product in an improved 99% yield and 95% ee when a slightly modified 3:1 hexane/toluene solvent mixture is used. Notably, the reaction is performed using only 0.5 mol% Pd2(dba)3 and 1.2 mol% ligand and is complete in 12 h at 25 °C.
|
|
||||
|---|---|---|---|---|
| Entry | R2N– | Ligand | Solvent | eeb,c (yield) |
| a Conditions: 0.1 mmol substrate, 0.5 mol% Pd2(dba)3, 1.2 mol% ligand, 1.0 mL solvent, 25 °C. b Determined by chiral SFC analysis. c Yield of isolated product. d MeCy = methylcyclohexane. | ||||
| 1 |
|
L1 | 3:1 hexane/PhMe |
73 (92) |
| 2 |
|
L1 | 3:1 MeCyd/PhMe |
83 (97) |
| 3 |
|
L1 | 3:1 MeCy/PhMe |
65 (ND) |
| 4 |
|
L1 | 3:1 MeCy/PhMe |
NR |
| 5 |
|
L1 | 3:1 MeCy/PhMe |
NR |
| 6 |
|
L1 | 3:1 MeCy/PhMe |
90 (95) |
| 7 |
|
L2 | 3:1 hexane/PhMe |
95 (99) |
|
||||
With optimal reaction conditions identified, we sought to examine the impact of substrate enolate geometry. Contrary to our prior report on ketone enolates,4c in which the starting material enolate ratio was found to be inconsequential to reaction selectivity, we found that the geometry of the N-acyl indole-derived enol carbonate (1a) has a significant impact on the selectivity of the reaction (eqn (3)). Specifically, the use of a 21:79 E/Z mixture of enol carbonates provides the alkylation product (2a) in a significantly diminished 66% ee with L2 as the ligand, albeit favoring the same enantiomer of product that is observed using a 98:2 E/Z mixture. This result hints at some degree of enolate geometry equilibration in the catalytic process, but to a lesser degree than our ketone system.4c An analogous effect was observed with L1, as well as with the other N-acyl substrates examined during optimization.
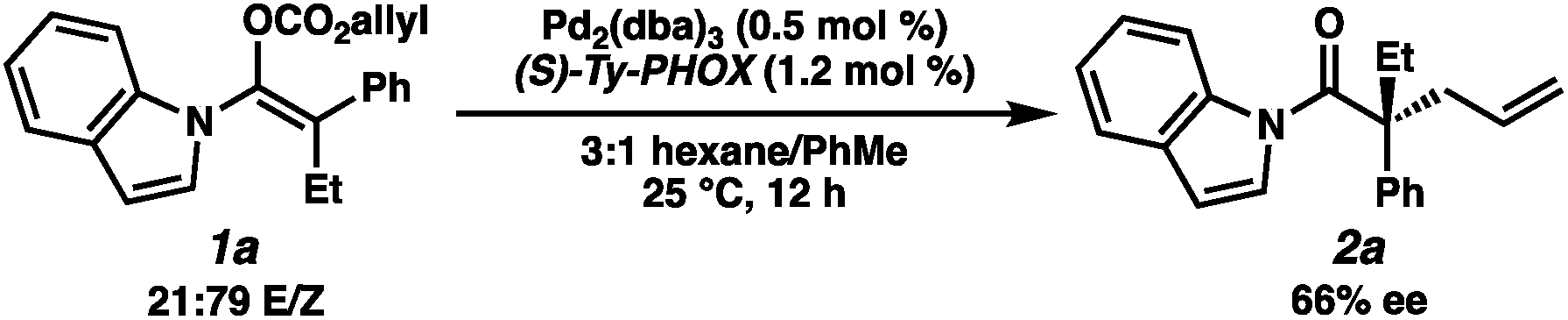 | (3) |
Having optimized conditions in hand, and now understanding the critical importance of olefin geometry in this allylic alkylation, we examined the scope of the transformation for accessing various acyclic all-carbon quaternary stereocenters (Table 2). All enol carbonates were prepared in excellent E/Z ratios (see ESI† for details) and the corresponding alkylation products were obtained in high yields. Longer alkyl chains at the α-position such as n-pentyl (2b) and those with branching (2c) provide the alkylation products in excellent enantioselectivity (96% ee). Benzyl-containing substrate 2d was also well tolerated, albeit with a slightly lower ee (90%). The presence of electron-rich aryl groups resulted in a slight increase in selectivity, with both p-Me (2e) and p-OMe (2f) substrates yielding the desired products in 98% ee each, and the bis-OMe substrate (2g) in 95% ee. Notably, both methoxy-containing substrates 2f and 2g required longer reaction times to reach full conversion (i.e., 24 h). An examination of electron-poor aryl groups demonstrated that, while p-Cl (2h), p-Br (2i), and p-F (2j) groups were well tolerated (94–96% ee), a p-CF3 (2k) containing substrate provided the product in a diminished 72% ee, believed to result from increased stabilization of the resultant enolate.2f Additionally, substitution on the indole was tolerated, with methyl-containing substrate (2l) providing the product in 98% ee and the bromo substrate (2m) in 93% ee. While ortho substitutions on the α-aryl ring provided low selectivities with N-acyl indole-derived substrates, employing the smaller N-acyl 3-methyl pyrrole allowed o-Me (2n) and o-Br (2o) alkylation products to be obtained in moderate 89% and 80% ee respectively, allowing formation of an exceedingly hindered quaternary stereocenter. The use of a more electron-rich N-acyl heterocycle (i.e. 3-methyl pyrrole vs. pyrrole) provides a slight boost in enantioselectivity.12
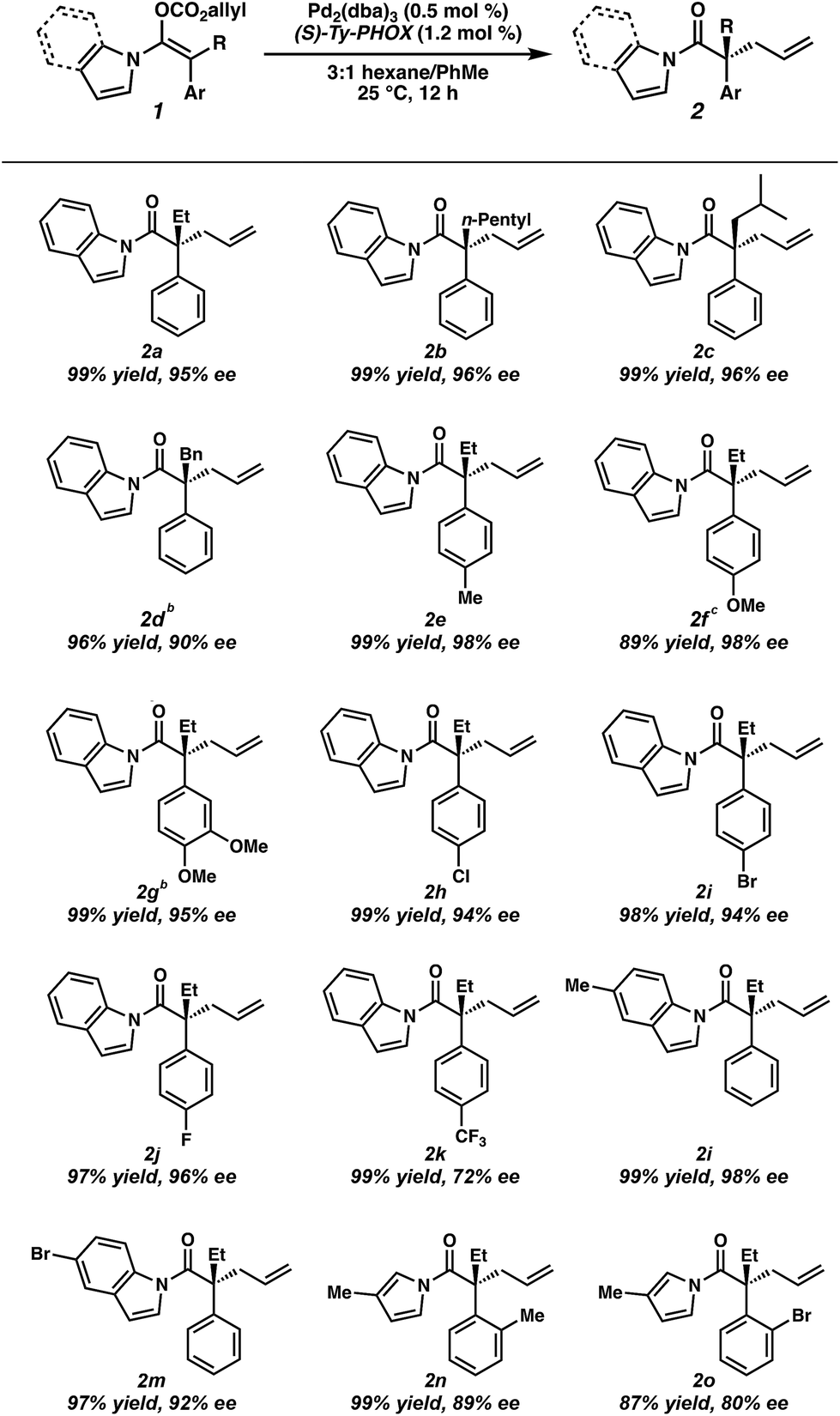
The broad scope of α-aryl substituents tolerated in this transformation (and the related ketone allylation) is surprising given that previous examples of α-aryl stabilized and other weakly basic enolates had demonstrated them to be poor substrates for enantioselective allylic alkylation using PHOX ligands, with near racemic alkylation products.2f To rationalize this new observation, as well as explain the necessity for a flat aromatic western portion of the substrate, we propose that the α-aryl group is likely rotated out of plane with respect to the enolate and that the indole is planar and in coordination with the enolate to avoid steric clashing between both aromatic groups (Fig. 2). An attractive edge-to-face interaction between the two aromatic substituents may also play a role. As a result, no significant resonance stabilization is afforded to the enolate from the α-aryl substituent and high enantioselectivities can be achieved, although inductive stabilization through electron-withdrawing functionality may still result in lower enantioselectivities (e.g.2k).
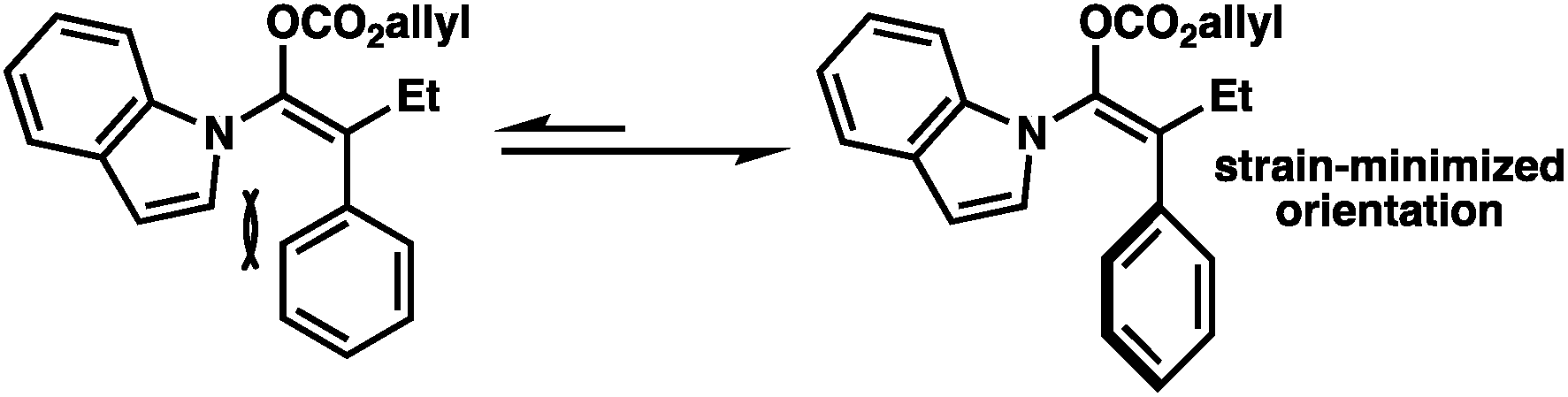 | ||
| Fig. 2 Proposed rationale for tolerance of α-aryl substituents. | ||
To further understand our hypothesis, we prepared α-pyrrolo substrate 1p and subjected it to the optimized palladium-catalyzed conditions (Fig. 3). We reasoned that an α-pyrrole would mimic an α-phenyl substituent in the same way that the acyl-indole mimics a phenyl ketone. This is indeed the case, with alkylation product 2p being obtained in an excellent 97% yield and 99% enantioselectivity, providing acess to an interesting amino-acid derivative.
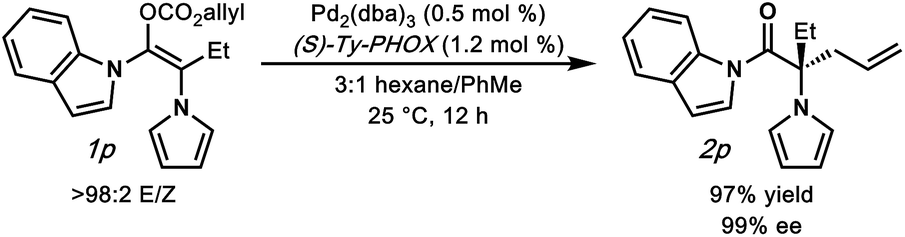 | ||
| Fig. 3 Investigation of an α-pyrrole substituted N-acyl indole substrate. | ||
To highlight the utility of the prepared α-quaternary N-acyl indoles we next turned toward examining derivatizations (Fig. 4). First, treatment of alkylation product 2a with KOTMS smoothly affords the carboxylic acid 3, which can be isolated in 99% yield following a chromatography-free procedure utilizing an acid–base extraction. Ethyl ester 4 was obtained in 75% yield by the addition of KOEt at 25 °C. Full reduction of 2a to the corresponding primary alcohol 5 could be achieved with LiAlH4 in 84% yield. Lastly, Wacker oxidation conditions affords 1,4-dicarbonyl compound 6 in 95% yield as a 5:1 mixture of ketone/aldehyde. Surprisingly, 2a was found to be unreactive to Grignard reagents as well as direct amidation conditions, even at elevated temperatures, suggesting the potential to employ this moiety as a robust and orthogonal carboxylic acid masking group in synthesis.
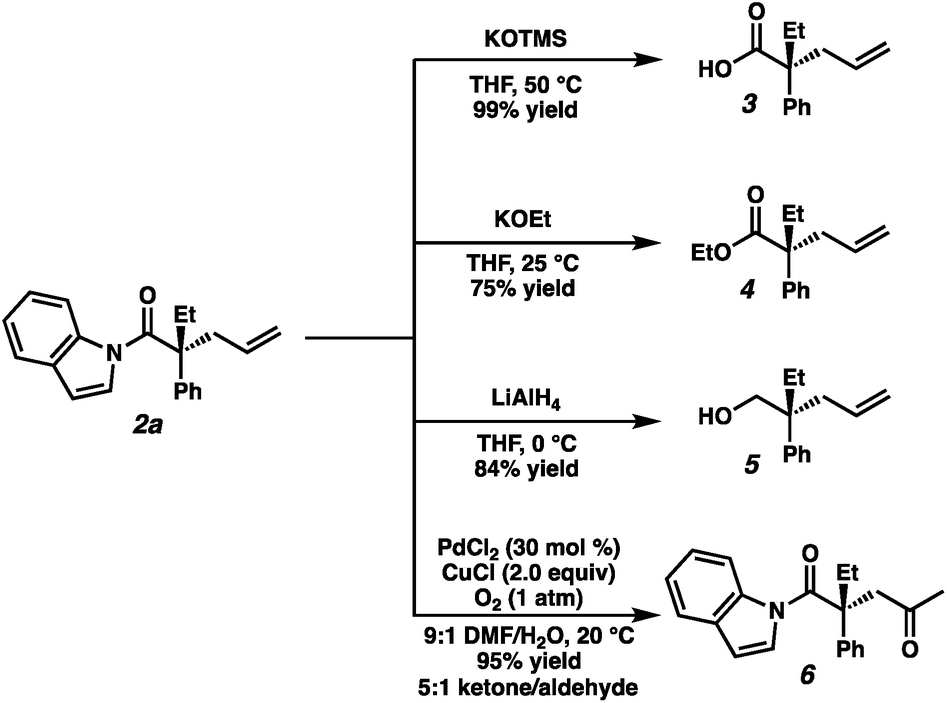 | ||
| Fig. 4 Derivatization of alkylation products. | ||
Conclusions
In summary, the first enantioselective palladium-catalyzed allylic alkylation of N-acyl indole-derived enol carbonates furnishing acyclic all-carbon quaternary stereocenters has been developed. Crucial to the excellent selectivities obtained is the preparation of the enol carbonates as single geometrical enolate isomers as well as the use of a new electron-deficient PHOX ligand, (S)-Ty-PHOX (L2). Additional studies addressing the scope and applications of this transformation as well as the subtle mechanistic distinctions associated with this and other recently described acyclic systems are currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank NIH-NIGMS (R01GM080269), the Gordon and Betty Moore Foundation, and Caltech for financial support. E. J. A. thanks the National Science Foundation for a predoctoral fellowship. We thank Dr David VanderVelde (Caltech) for NMR expertise. Dr Scott Virgil (Caltech) is thanked for instrumentation and SFC assistance. Dr Michael Takase and Lawrene Henling are acknowledged for assistance with X-ray analysis.Notes and references
- Y. Liu, S. J. Han, W. B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed.
- (a) S. F. Martin, Tetrahedron, 1980, 36, 419–460 CrossRef CAS; (b) E. J. Corey and A. Guzman-Perez, Angew. Chem. Int. Ed., 1998, 37, 388–401 ( Angew. Chem. , 1998 , 110 , 402–415 ) CrossRef; (c) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363–5367 CrossRef CAS PubMed; (d) I. Marek and G. Sklute, Chem. Commun., 2007, 1683–1691 RSC; (e) B. M. Trost and C. Jiang, Synthesis, 2006, 369–396 CrossRef CAS; (f) D. C. Behenna, J. T. Mohr, N. H. Sherden, S. C. Marunescu, A. M. Harned, K. Tani, M. Seto, S. Ma, Z. Novak, M. R. Krout, R. M. McFadden, J. L. Roizen, J. A. Enquist Jr, D. E. White, S. R. Levine, K. V. Petrova, A. Iwashita, S. C. Virgil and B. M. Stoltz, Chem.–Eur. J., 2011, 17, 14199–14223 CrossRef CAS PubMed; (g) B. M. Trost, J. Xu and T. Schmidt, J. Am. Chem. Soc., 2009, 131, 18343–18357 CrossRef CAS.
- (a) J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564–12580 CrossRef CAS PubMed; (b) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593–4623 RSC; (c) Y. Minko and I. Marek, Chem. Commun., 2014, 50, 12597–12611 RSC; (d) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, J. Am. Chem. Soc., 2014, 136, 2682–2694 CrossRef CAS PubMed; (e) T. S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340–344 CrossRef CAS PubMed; (f) K. Zhang, Q. Peng, X. L. Hou and D. Y. Wu, Angew. Chem., Int. Ed., 2008, 47, 1741–1744 ( Angew. Chem. , 2008 , 120 , 1765–1768 ) CrossRef CAS PubMed.
- (a) P. Starkov, J. T. Moore, D. C. Duquette, B. M. Stoltz and I. Marek, J. Am. Chem. Soc., 2017, 139, 9615–9620 CrossRef CAS PubMed; (b) E. J. Alexy, S. C. Virgil, M. D. Bartberger and B. M. Stoltz, Org. Lett., 2017, 19, 5007–5009 CrossRef CAS PubMed; (c) E. J. Alexy, H. Zhang and B. M. Stoltz, J. Am. Chem. Soc., 2018, 140, 10109–10112 CrossRef CAS PubMed.
- (a) B. X. Li, D. N. Le, K. A. Mack, A. McClory, N.-K. Lim, T. Cravillion, S. Savage, C. Han, D. B. Collum, H. Zhang and F. Gosselin, J. Am. Chem. Soc., 2017, 139, 10777–10783 CrossRef CAS PubMed; (b) K. A. Mack, A. McClory, H. Zhang, F. Gosselin and D. B. Collum, J. Am. Chem. Soc., 2017, 139, 12182–12189 CrossRef CAS PubMed.
- (a) B. M. Trost, J. Xu and T. Schmidt, J. Am. Chem. Soc., 2009, 131, 18343–18357 CrossRef CAS PubMed; (b) Y. Ariyarathna and J. A. Tunge, Org. Biomol. Chem., 2014, 12, 8386–8389 RSC.
- (a) B. M. Trost, K. Lehr, D. J. Michaelis, J. Xu and A. K. Buckl, J. Am. Chem. Soc., 2010, 132, 8915–8917 CrossRef CAS PubMed; (b) B. M. Trost, D. J. Michaelis, J. Charpentier and J. Xu, Angew. Chem. Int. Ed., 2012, 51, 204–208 ( Angew. Chem. , 2012 , 124 , 208–212 ) CrossRef CAS PubMed.
- T. Fujita, T. Yamamoto, Y. Morita, H. Chen, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2018, 140, 5899–5903 CrossRef CAS PubMed.
- (a) B. W. H. Turnbull and P. A. Evans, J. Am. Chem. Soc., 2015, 137, 6156–6159 CrossRef CAS PubMed; (b) T. B. Wright and P. A. Evans, J. Am. Chem. Soc., 2016, 138, 15303–15306 CrossRef CAS PubMed; see also: (c) P. A. Evans, S. Oliver and J. Chae, J. Am. Chem. Soc., 2012, 134, 19314–19317 CrossRef CAS PubMed; (d) P. A. Evans and S. Oliver, Org. Lett., 2013, 15, 5626–5629 CrossRef CAS PubMed; (e) B. W. H. Turnbull, S. Oliver and P. A. Evans, J. Am. Chem. Soc., 2015, 137, 15374–15377 CrossRef CAS PubMed.
- (a) W.-B. Liu, C. M. Reeves and B. M. Stoltz, J. Am. Chem. Soc., 2013, 135, 17298–17301 CrossRef CAS PubMed; (b) S. Shockley, J. C. Hethcox and B. M. Stoltz, Angew. Chem., Int. Ed., 2017, 56, 11545–11548 ( Angew. Chem. , 2017 , 129 , 11703–11706 ) CrossRef CAS PubMed; (c) J. C. Hethcox, S. Shockley and B. M. Stoltz, Angew. Chem., Int. Ed., 2018, 57, 8664–8667 ( Angew. Chem. , 2018 , 130 , 8800–8803 ) CrossRef CAS PubMed.
- (a) B. P. Pritchett, E. J. Donckele and B. M. Stoltz, Angew. Chem., Int. Ed., 2017, 56, 12624–12627 ( Angew. Chem. , 2017 , 129 , 12798–12801 ) CrossRef CAS PubMed; (b) B. P. Pritchett, J. Kikuchi, Y. Numajiri and B. M. Stoltz, Angew. Chem., Int. Ed., 2016, 55, 13529–13532 ( Angew. Chem. , 2016 , 128 , 13727–13730 ) CrossRef CAS PubMed.
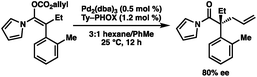
- The standard N-acyl pyrrole substrate provides the o-Me product in 80% ee.
 .
.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR and IR spectra, SFC traces, X-ray crystallographic data. CCDC 1908785. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01726g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |