
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesymmetrization of cyclic 1,3-diketones via Ir-catalyzed hydrogenation: an efficient approach to cyclic hydroxy ketones with a chiral quaternary carbon†
Quan
Gong
a,
Jialin
Wen
 *ab and
Xumu
Zhang
*ac
*ab and
Xumu
Zhang
*ac
aDepartment of Chemistry, Southern University of Science and Technology, 1088 Xueyuan Road, Shenzhen, 518055, China. E-mail: wenjl@sustech.edu.cn; zhangxm@sustech.edu.cn
bAcademy of Advanced Interdisciplinary Studies, Southern University of Science and Technology, 1088 Xueyuan Road, Shenzhen, 518055, China
cShenzhen Grubbs Institute, Southern University of Science and Technology, 1088 Xueyuan Road, Shenzhen, 518055, China
First published on 21st May 2019
Abstract
We herein report an efficient method to synthesize cyclic hydroxy ketones with a chiral quaternary center. Catalyzed by an Ir/f-ampha complex, cyclic α,α-disubstituted 1,3-diketones were hydrogenated, giving mono-reduced products with both high enantioselectivities and diastereoselectivities. In addition, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C
C and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds could survive in this catalytic system. This method was applied in the preparation of (+)-estrone. No diols were observed in this chemical transformation. The enantiomeric and diastereomeric induction were achieved as a result of steric hindrance.
C bonds could survive in this catalytic system. This method was applied in the preparation of (+)-estrone. No diols were observed in this chemical transformation. The enantiomeric and diastereomeric induction were achieved as a result of steric hindrance.
Introduction
Desymmetrization reactions have been proved to be an efficient method to generate compounds bearing chiral quaternary carbons1 which have historically been problematic in synthetic chemistry.2,3 A successful example of this strategy is mono-reduction of cyclic α,α-disubstituted 1,3-diketones, which typically gives hydroxy ketones with two vicinal stereogenic centers.1 This product has been demonstrated to be a versatile synthon that attracts synthetic chemists. Many synthetic studies were documented to use this 5- or 6-member ring synthon to construct complex molecules with multiple stereogenic centers. Successful examples include coriolin,4 anguidine,5 (+)-crotogoudin,6 (+)-paspaline,7,8 (+)-estrone,9 cortistatins10 and aplysiasecosterol A (Fig. 1).11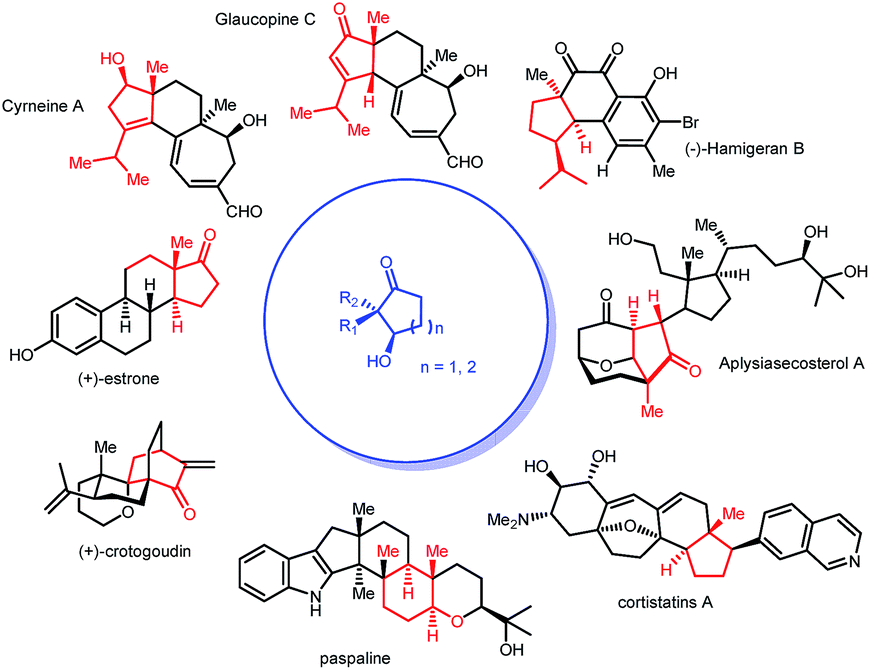 | ||
| Fig. 1 Chiral cyclic hydroxy ketones in total synthesis. | ||
The construction of this important synthon, however, is limited to enzyme catalyzed reduction,12,13 Ru-catalyzed transfer hydrogenation14,15 and Corey–Bakshi–Shibata9 reduction with borane. These methods suffer from the drawbacks of narrow substrate scope, moderate selectivity and high catalyst loading. The challenges of mono-reduction of cyclic α,α-disubstituted 1,3-diketones lie in areas such as (1) enantioselectivity and diastereoselectivity being realized in one step and (2) the prevention of over-reduction to diols. In the hydride transfer step, different facial approaches towards the substrate lead to two pairs of diastereomers (marked with blue and red arrows in Fig. 2). Enantioselectivity originates in the differentiation of a quaternary carbon from a methylene group (Fig. 2).
 | ||
| Fig. 2 Origin of stereoselectivities in mono-reduction of 1,3-diketones. | ||
Our group has been dedicated to transition metal catalyzed ketone reduction during the recent two decades and has developed a series of ferrocene-based tridentate ligands for iridium catalyzed hydrogenation.16–18 A variety of simple or functionalized ketones can be hydrogenated to chiral alcohols with remarkably high ees and turnover numbers (TONs). To the best of knowledge, direct hydrogenation has not been applied in the preparation of the aforementioned synthon. Due to our continuous interest in construction of chiral molecules via transition metal catalyzed asymmetric hydrogenation, we envisioned that this efficient catalytic system could be applied in the mono-reduction of 1,3-diketones (Scheme 1).
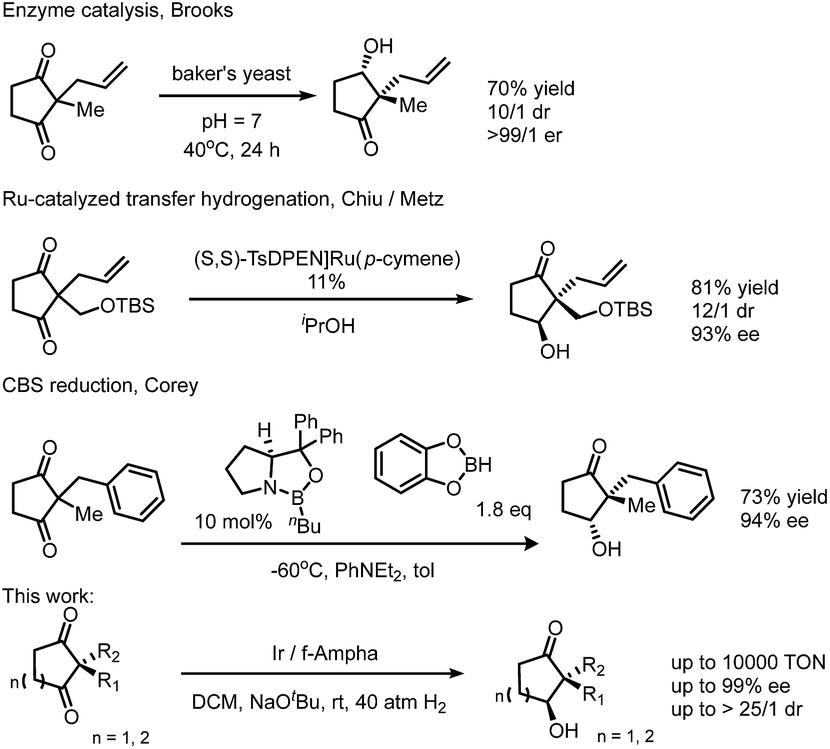 | ||
| Scheme 1 Methods of mono-reduction of a cyclic 1,3-diketone to generate a chiral quaternary carbon. | ||
Results and discussion
We initiated our investigation by screening a suitable ligand. Although simple ketones such as acetophenone could be reduced efficiently with all these ligands (f-amphox,16 f-amphol17 and f-ampha18), their performances in functionalized ketones were different. We selected 2-benzyl-2-methylcyclopentane-1,3-dione as the model substrate and conducted hydrogenation with iridium in isopropanol with potassium tert-butoxide. To prevent further reduction, less forcing conditions (20 atm H2) and a short reaction time (1 h) were applied. These ligands performed differently: f-amphox and f-ampha gave promising results in the preliminary assessment (Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Conversionb | drc | eec |
| a Reaction conditions: 1a (0.1 mmol, 0.1 M), 1a/[Ir(COD)Cl]2/ligand/base = 500/0.5/1.1/10, 20 atm H2, rt, 1 h. b Conversion was determined by 1H NMR analysis, no by-product was observed. c dr and ee were determined by HPLC on a chiral stationary phase. | ||||
| 1 | f-amphox | 24% | 7.3/1 | 93% |
| 2 | Indan-f-amphox | 11% | 5.4/1 | 28% |
| 3 | f-amphol | 50% | 4.1/1 | 37% |
| 4 | f-ampha | 84% | 10.1/1 | 95% |
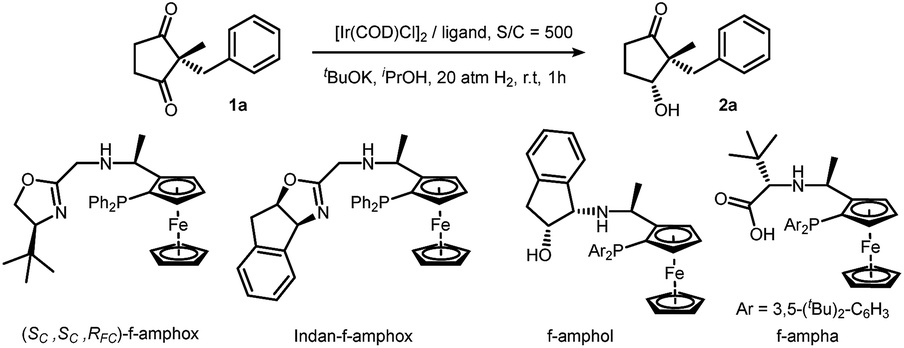
Extending the reaction time and increasing the hydrogen pressure drove this reaction to a full conversion. Interestingly, no over-reduction product (diol) was observed under harsher conditions. After careful optimization [for detailed condition screening, see ESI†], we finally obtained satisfactory conditions: catalyzed by an Ir/f-ampha complex (0.1% loading), the symmetric 1,3-diketone was reduced in dichloromethane in the presence of sodium tert-butoxide, giving the corresponding chiral hydroxy ketone with 99% ee and 21/1 dr. To our delight, no diol was observed in the crude reaction mixture. The turnover number of this reaction could reach 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 without obvious reduction of stereoselectivities (Scheme 2).19
000 without obvious reduction of stereoselectivities (Scheme 2).19
 | ||
| Scheme 2 Reaction scope of desymmetrization of cyclic 1,3-diketones via hydrogenation. Reaction conditions: 1 (0.2 mmol, 0.2 M), 1/[Ir(COD)Cl]2/f-amphox/tBuONa = 1000/0.5/1.1/10, 40 atm H2, rt, 14 h; isolated yields; dr and ee were determined by HPLC on a chiral stationary phase. aSubstrate/catalyst/base = 200/1/10, 15 min. bSubstrate/catalyst/base = 200/1/10, 14 h. cVolatile compound, >99% conversion. | ||
We applied the optimized conditions to explore the scope of this method with 0.1% catalyst loading. Various substitution groups on the benzene ring, no matter whether electron-withdrawing or electron-donating groups, did not bring significant changes in both the stereoselectivity and conversion (2a to 2j). 1,3-Diketones with an allyl group, instead of benzyl, were hydrogenated with high enantioselectivities as well as satisfactory diastereoselectivities (2k to 2m). In addition to alkenes, the alkynyl group also survives in this chemical transformation (2n). The preference of reducing polar CO bonds demonstrated its chemoselectivity. To our surprise, a dialkyl substrate also worked well in this reaction (2o and 2p). This excellent stereoselectivity indicated that this catalytic system could discriminate two different alkyl groups (methyl vs. ethyl and methyl vs. propyl). Discrimination between simple alkyl groups has always been a top challenge in asymmetric catalysis, while alkyl and aryl groups are easy to differentiate (2q). When we expanded the ring size of the substrate from five to six, the performance faded and moderate stereoselectivities were obtained (2r). α,α-Disubstituted 1,3-indandiones could also be hydrogenated, giving desired yields and stereoselectivities (2s and 2t).
This reaction could be scaled up smoothly (Scheme 3, top). In order to exploit the potential applications of this method in synthetic chemistry, we chose (+)-estrone as a target. This molecule plays a key role in steroidogenesis20 and chemical synthesis of steroids.7,21,22 We followed Corey's route,9 as well as List's route,23 to synthesize Torgov's 1,3-diketone24 in a sub-gram scale. Hydrogenation of this diketone under the optimized conditions quantitatively gives a hydroxy ketone with >99% ee and 8:1 dr. After Prins cyclization/dehydration and oxidation with IBX, Torgov's diene24 was obtained25 which could be easily converted to (+)-estrone by a two-step transformation.23
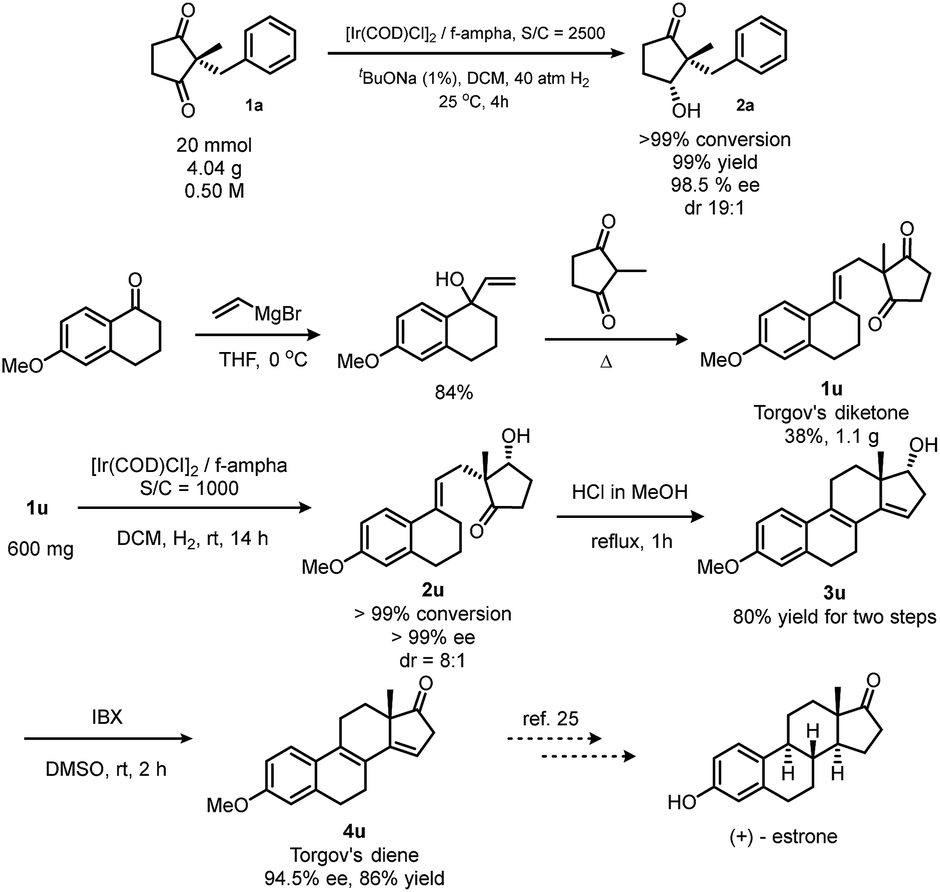 | ||
| Scheme 3 Scale-up reaction and application of desymmetrization via hydrogenation in the synthesis of (+)-estrone. | ||
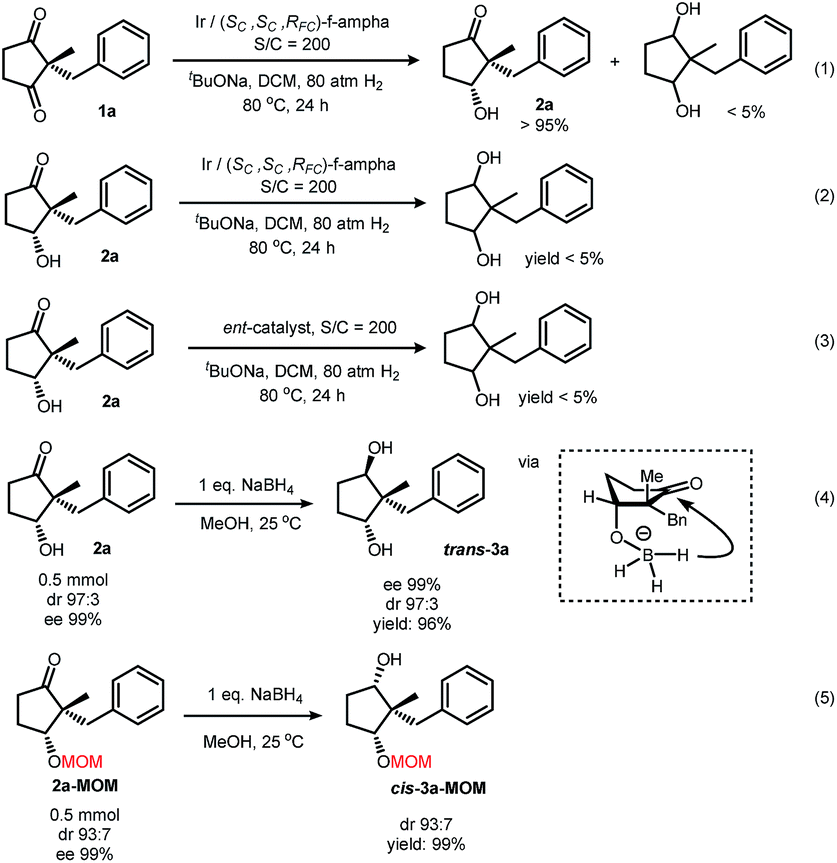 | ||
| Scheme 4 Attempts of further reduction of the hydroxy ketone. | ||
Our curiosity was drawn by the phenomenon of only one carbonyl group being reduced. When applying harsh conditions, it was also difficult to form diols (Scheme 4, eqn (1)). The purified product 2a could not yield a diol under these forcing conditions as well (eqn (2)). Hydrogenation under the same conditions with the other enantiomer of the ligand, however, also failed in this transformation (eqn (3)). The chiral pocket of both catalyst enantiomers seemed not to be compatible with the hydroxy ketone. Reduction of 2a with sodium borohydride exclusively gave a chiral trans-diol in a quantitative yield. After protecting the hydroxyl group, however, reduction of this ketone with sodium borohydride under the same conditions exclusively gave a cis-diol (Scheme 4, eqn (5)). Plausible explanations included an intramolecular hydride transfer after the formation of a boron alkoxide, which could be a result of transesterification of borate.26–28
While reduction of a five-membered-ring cyclic 1,3-diketone by both sodium borohydride and iridium catalyzed hydrogenation gave the same diastereoselectivity, the reduction of the six-membered-ring substrate was different. Hydrogenation under the optimized conditions gave an alcohol with the –OH cis to the larger benzyl group, but mono-reduction with sodium borohydride29 yielded the other diastereomer30 (Scheme 5). These results indicated the same facial preference of iridium catalyzed hydrogenation and sodium borohydride reduction in the five-membered ring but a different facial preference in the six-membered ring.
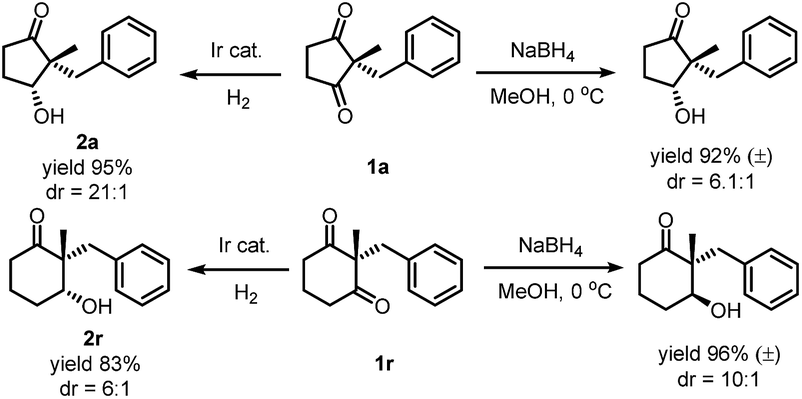 | ||
| Scheme 5 Comparison of Ir-catalyzed hydrogenation and sodium borohydride reduction in desymmetrization of cyclic diketones. | ||
Conclusions
We applied the strategy of transition metal catalyzed hydrogenation in the mono-reduction of cyclic 1,3-diketones. This desymmetrization reaction efficiently gave chiral hydroxy ketones with high stereoselectivities. Gratefully, further reduction that leads to diols was not observed in the hydrogenation step. This catalytic system was highly compatible with CC and CC bonds, therefore making it a practical method to prepare complicated molecules with a chiral quaternary carbon.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Yong-gui Zhou (Dalian Institute of Chemical Physics) and Prof. Pauline Chiu (the University of Hong Kong) for their constructive advice in the preparation of the manuscript. This work was supported by the SZDRC Discipline Construction Program, Shenzhen Nobel Prize Scientists Laboratory Project (C17783101) and Shenzhen Commission of Science, Technology and Innovation (JCYJ20170817104853520 and JSGG20160608140847864).Notes and references
- X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS
.
- Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS
- K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181 CrossRef CAS
- B. M. Trost and D. P. Curran, J. Am. Chem. Soc., 1981, 103, 7380–7381 CrossRef CAS
- D. W. Brooks, P. G. Grothaus and J. T. Palmer, Tetrahedron Lett., 1982, 23, 4187–4190 CrossRef CAS
- S. Breitler and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 11168–11171 CrossRef CAS PubMed
- R. J. Sharpe and J. S. Johnson, J. Am. Chem. Soc., 2015, 137, 4968–4971 CrossRef CAS PubMed
- R. J. Sharpe and J. S. Johnson, J. Org. Chem., 2015, 80, 9740–9766 CrossRef CAS PubMed
- Y.-Y. Yeung, R.-J. Chein and E. J. Corey, J. Am. Chem. Soc., 2007, 129, 10346–10347 CrossRef CAS
- L. L. Liu and P. Chiu, Chem. Commun., 2011, 47, 3416–3417 RSC
- Z. Lu, X. Zhang, Z. Guo, Y. Chen, T. Mu and A. Li, J. Am. Chem. Soc., 2018, 140, 9211–9218 CrossRef CAS
- D. W. Brooks, P. G. Grothaus and W. L. Irwin, J. Org. Chem., 1982, 47, 2820–2821 CrossRef CAS
- D. W. Brooks, H. Mazdiyasni and P. G. Grothaus, J. Org. Chem., 1987, 52, 3223–3232 CrossRef CAS
- B. Shi, S. Merten, D. K. Y. Wong, J. C. K. Chu, L. L. Liu, S. K. Lam, A. Jäger, W.-T. Wong, P. Chiu and P. Metz, Adv. Synth. Catal., 2009, 351, 3128–3132 CrossRef CAS
- L. Kuang, L. L. Liu and P. Chiu, Chem.–Eur. J., 2015, 21, 14287–14291 CrossRef CAS
- W. Wu, S. Liu, M. Duan, X. Tan, C. Chen, Y. Xie, Y. Lan, X.-Q. Dong and X. Zhang, Org. Lett., 2016, 18, 2938–2941 CrossRef CAS PubMed
- J. Yu, M. Duan, W. Wu, X. Qi, P. Xue, Y. Lan, X.-Q. Dong and X. Zhang, Chem.–Eur. J., 2017, 23, 970–975 CrossRef CAS
- J. Yu, J. Long, Y. Yang, W. Wu, P. Xue, L. W. Chung, X.-Q. Dong and X. Zhang, Org. Lett., 2017, 19, 690–693 CrossRef CAS
- Reaction conditions: 2.0 mmol of 1a, S/C = 10000, base 1%, 60 atm H2, rt, 48 h. Conversion 100%, yield 95%, 95% ee, 10:1 dr..
- G. M. Anstead, K. E. Carlson and J. A. Katzenellenbogen, Steroids, 1997, 62, 268–303 CrossRef CAS PubMed
- F. J. Zeelen, Nat. Prod. Rep., 1994, 11, 607–612 RSC
-
R. T. Blickenstaff, A. C. Ghosh and G. C. Wolf, Organic Chemistry: A Series of Monographs, Academic Press, 1974, vol. 30 Search PubMed
- S. Prévost, N. Dupré, M. Leutzsch, Q. Wang, V. Wakchaure and B. List, Angew. Chem., Int. Ed., 2014, 53, 8770–8773 CrossRef PubMed
- S. N. Ananchenko and I. V. Torgov, Tetrahedron Lett., 1963, 4, 1553–1558 CrossRef
- Slight erosion of ee was probably caused by the other pair of diastereomers which were hardly removed by flash chromatography. According to ref. 16, recrystallization could be a good alternative..
- For methanolysis of sodium borohydride, see: R. E. Davis and J. A. Gottbrath, J. Am. Chem. Soc., 1962, 84, 895–898 CrossRef CAS
- Exchange of ligands on substituted borane, see: D. A. Evans, K. T. Chapman and E. M. Carreira, J. Am. Chem. Soc., 1988, 110, 3560–3578 CrossRef CAS
- Substrate directed reaction, see review: A. H. Hoveyda, D. A. Evans and G. C. Fu, Chem. Rev., 1993, 93, 1307–1370 CrossRef CAS
- The results of sodium borohydride reduction of cyclic diketones are shown in the HPLC chromatograms of racemic samples in the ESI.†.
- R. V. Kolakowski, M. Manpadi, Y. Zhang, T. J. Emge and L. J. Williams, J. Am. Chem. Soc., 2009, 131, 12910–12911 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1906351, 1906353 and 1906354. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01769k |
| This journal is © The Royal Society of Chemistry 2019 |