
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA reactive coordinatively saturated Mo(III) complex: exploiting the hemi-lability of tris(tert-butoxy)silanolate ligands†
Margherita
Pucino
 ,
Florian
Allouche
,
Christopher P.
Gordon
,
Michael
Wӧrle
,
Victor
Mougel
* and
Christophe
Copéret
*
,
Florian
Allouche
,
Christopher P.
Gordon
,
Michael
Wӧrle
,
Victor
Mougel
* and
Christophe
Copéret
*
Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 2, 8093 Zurich, Switzerland. E-mail: ccoperet@ethz.ch; mougel@inorg.chem.ethz.ch
First published on 14th May 2019
Abstract
Coordinatively unsaturated Mo(III) complexes have been identified as highly reactive species able to activate dinitrogen without the need for a sacrificial reducing agent. Here, we report a coordinatively saturated octahedral Mo(III) complex stabilized by κ2-tris(tert-butoxy)silanolate ligands, which is yet highly reactive towards dinitrogen and small molecules. The combined high stability and activity are ascribed to the dual binding mode of the tris(tert-butoxy)silanolate ligands that allow unlocking a coordination site in the presence of reactive small molecules to promote their activation at low temperatures.
Introduction
The presence of a Mo center in the active site of the FeMo cofactor of nitrogenase enzymes has stimulated numerous studies on the synthesis of low valent Mo complexes and their reactivity towards small molecules.1 Tri-coordinated complexes with bulky amide ligands like Mo[N(R)Ar]3 (R = tBu, Ar = 3,5-Me2C6H3) have been shown to be particularly reactive and able to cleave dinitrogen.2 Such reactivity contrasts that observed for many mononuclear coordinatively saturated octahedral Mo(III) complexes, which are quite inert towards small molecules. To date, there are still a small number of examples3 of tri-coordinated unsaturated Mo(III) compounds due to their propensity to form rather unreactive triply bonded Mo![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Mo dimers.4 The use of bulky chelating ligands has thus been key to obtaining isolable, yet reactive coordinatively unsaturated Mo(III) centers.3c,5 This approach has enabled the synthesis of the first mononuclear Mo catalytic systems able to catalytically reduce N2 to ammonia.6
Mo dimers.4 The use of bulky chelating ligands has thus been key to obtaining isolable, yet reactive coordinatively unsaturated Mo(III) centers.3c,5 This approach has enabled the synthesis of the first mononuclear Mo catalytic systems able to catalytically reduce N2 to ammonia.6
The tris(tert-butoxy)silanolate (TBOS) ligand is not very sterically demanding but presents a variety of binding modes that allow for the formation of mono-nuclear complexes with a broad range of metals through coordination as a terminal κ1, terminal κ2, bridging κ1μ2 and bridging κ2μ2 ligand.7 Terminal κ1 binding modes are observed for small metal ions such as trivalent Cr or Ga, while terminal κ2 modes are found for larger metal ions such as trivalent Sc, Yb, Lu8 or Dy.9 Mo(III) possesses an intermediate ionic radius between that of these two groups; we thus reasoned that the TBOS ligands had the potential to stabilize Mo(III), while maintaining its reactivity towards small molecules, as such ligands can adopt different coordination modes and interconvert between them. Here, we present the synthesis and the reactivity of Mo(OSi(OtBu)3)3, 1, which displays a distorted octahedral geometry with three κ2-TBOS ligands. Coordination of the TBOS ligand in a bidentate κ2-fashion prevents dimerization and allows isolating a mononuclear Mo(III) complex, while the flexibility of its binding mode provides high reactivity as evidenced by the activation at low temperature of small molecules like N2, N2O and CO2, among others.
Results and discussion
Mo(OSi(OtBu)3)3, 1, is prepared by reaction of [Mo(N(R)Ar)3] with (tBuO)3SiO–H (TBOS–H) in a slurry of diethyl ether at −80 °C; it is isolated as dark brown crystals in 36% yield after evacuation of the solvent and aniline byproduct under high vacuum at low temperature, followed by crystallization from pentane. NMR studies indicate a quantitative reaction, but the tedious isolation of this compound, which is required to separate the highly soluble compound 1 from the residual amine, diminishes the overall isolated yield for the pure crystalline form significantly. Compound 1 is, however, stable for months when in the solid state under an argon atmosphere at −35 °C and is rather stable in apolar solvents such as benzene, allowing investigation of its reactivity with small molecules in solution (Scheme 1).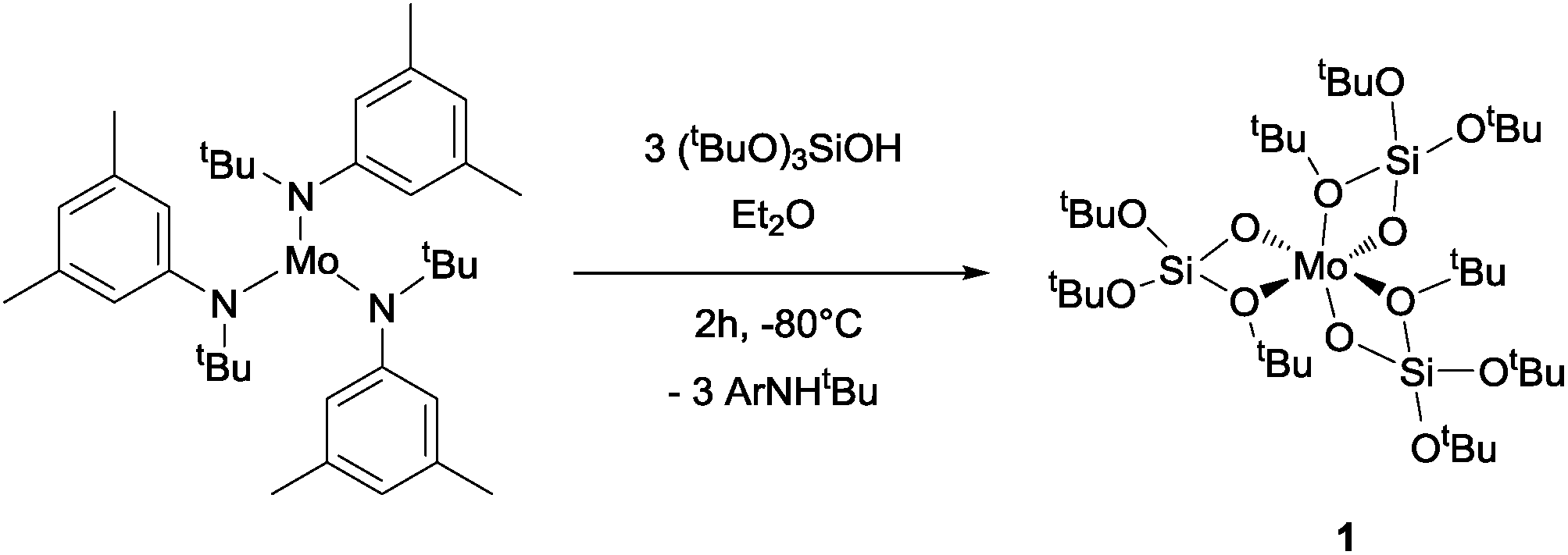 | ||
| Scheme 1 Synthesis of [Mo(TBOS)3] (1). | ||
Single-crystal X-ray diffraction10 shows that complex 1 adopts a distorted octahedral geometry with three κ2-TBOS ligands, where the Mo–O distance of the silanolate is ca. 0.3 Å shorter than that of the tert-butoxy moiety(2.020(4) Å vs. 2.302(4) Å) (Fig. 1). The 1H NMR spectrum of 1 shows a single very broad resonance at 3.82 ppm (ν½ = 164 Hz, RT), suggesting a fluxional coordination of the tert-butoxy moieties in solution. At −80 °C, the 1H NMR spectrum displays three peaks at 22.6, 4.9 and −1.36 ppm, in agreement with the three inequivalent tert-butoxy groups (see the ESI† for details). However, complex 1 still displays broad peaks at this temperature, suggesting fast dynamics of the methyl groups. The magnetic susceptibility (μeff = 3.74 μB) measured by the Evans method at 298 K is close to the expected value for a d3 complex with three unpaired electrons (3.75 μB).
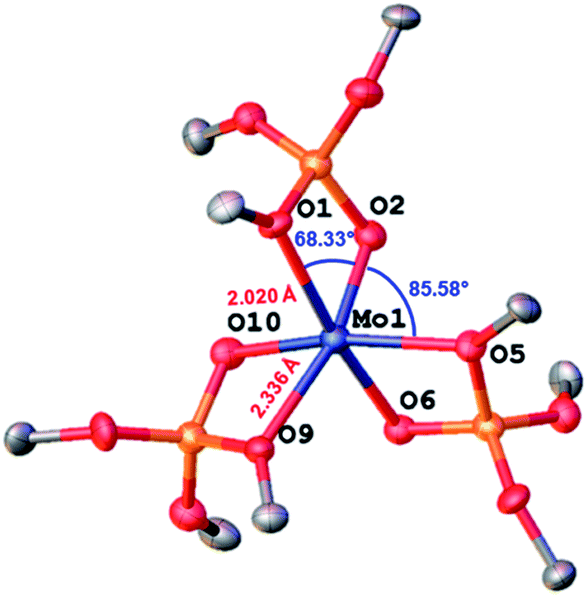 | ||
| Fig. 1 Thermal ellipsoid plot at 50% probability of [Mo(TBOS)3] (1). Hydrogen atoms and siloxide methyl groups have been omitted for clarity. | ||
We first explored the reaction of complex 1 with alkenes (ethylene and isobutene) as well as alkynes (2-butyne). By condensing the gas at a low temperature (−196 °C) in the solution and then warming to room temperature, the corresponding π-complexes were obtained in quantitative yields (Scheme 2, see the ESI† for details). The alkene and alkyne adducts [Mo(C2H4)(OSi(OtBu)3)3], 1-(ethylene), [Mo(C4H8)(OSi(OtBu)3)3], 1-(isobutene![[) with combining cedilla]](https://www.rsc.org/images/entities/char_0029_0327.gif) and [Mo(C4H6)(OSi(OtBu)3)3], crystallize in a tetrahedral geometry, while 1-(but-2-yne)11 presents a square-based pyramidal one. In the latter, the π-accepting ligand occupies the apical site and three monodentate silanolate ligands are in the basal plane (XRD structures are presented in Fig. S11–13†). The significant lengthening of the C–C bond upon coordination (1.406(7), 1.391(9) and 1.283(5) Å for 1-(ethylene), 1-(isobutene) and 1-(but-2-yne), respectively vs. 1.337(5) (ref. 12) 1.331(3) (ref. 13) and 1.202(5) (ref. 11) Å for the corresponding organic molecules) indicates a strong back-donation of the Mo(III) center to these π-acid ligands.14,15
and [Mo(C4H6)(OSi(OtBu)3)3], crystallize in a tetrahedral geometry, while 1-(but-2-yne)11 presents a square-based pyramidal one. In the latter, the π-accepting ligand occupies the apical site and three monodentate silanolate ligands are in the basal plane (XRD structures are presented in Fig. S11–13†). The significant lengthening of the C–C bond upon coordination (1.406(7), 1.391(9) and 1.283(5) Å for 1-(ethylene), 1-(isobutene) and 1-(but-2-yne), respectively vs. 1.337(5) (ref. 12) 1.331(3) (ref. 13) and 1.202(5) (ref. 11) Å for the corresponding organic molecules) indicates a strong back-donation of the Mo(III) center to these π-acid ligands.14,15
 | ||
| Scheme 2 Reaction of 1 with unsaturated hydrocarbons. | ||
The fluxionality of the ligand prompted us to further investigate the reactivity of 1 towards small molecules, namely N2, N2O and CO2. Exposing 1 to a N2 atmosphere at room temperature led to an immediate color change from amber to dark blue within seconds. This blue color fades away after 2 h at room temperature, leaving a colorless solution.
Removal of all volatiles followed by extraction in pentane and crystallization at −35 °C affords the Mo(VI) nitride complex [Mo(N)(TBOS)3], 2, in 40% yield. The relatively low isolated yield reflects the high solubility of the material, despite the quantitative reaction yield according to NMR. Single crystal X-ray analysis of 2 (Fig. 2a) shows a tetrahedral terminal nitride complex of Mo(VI) with three κ1-TBOS ligands (Scheme 3); the limited quality of the collected data however precludes an accurate discussion of bond distances. The presence of a nitride ligand was confirmed by FTIR, as evidenced by a band at 1011 cm−1 associated to ν(MoN).16 To unequivocally verify the signal, we carried out the reaction with 15N2. The infrared spectrum of 15NMo(TBOS)3 shows a band at 997 cm−1 that is red shifted compared to its 14N analogue, consistent with their attribution to MoN vibration. 1H NMR of 2 shows a single and sharp peak at 1.49 ppm, in agreement with a Mo(VI) diamagnetic species. The blue intermediate observed upon N2 addition can be isolated by rapid cooling of the reaction mixture (−196 °C) followed by fast solvent-exchange at 5 °C (from benzene-d6 to pentane) and crystallization at −35 °C. A dimeric Mo dinitrogen complex, (μ-N2)[Mo(TBOS)3]2, 3, is isolated as dark blue crystals. Single crystal X-ray analysis of 3 (Fig. 2b) displays two Mo ions in a distorted TBP (Trigonal BiPyramidal) geometry with two κ1 and one κ2 TBOS ligands along with the end-on bound dinitrogen molecule sandwiched between the two Mo centers (Mo1–N1–N2 and N1–N2–Mo2 angles of 172.5(3)° and 169.0(3)°, respectively). The N1–N2 distance is notably elongated with respect to the free N2 molecule (1.209(5) Å vs. 1.0976(2) (ref. 11) Å) indicating a significant degree of activation. When 3 is warmed to room temperature the subsequent step of the overall 6 e− reduction of N2 occurs, leading to the formation of two equivalents of 2 (Scheme 3). While this mechanism is similar to the one observed with coordinatively unsaturated tris-amido complexes,17 the complete process is much faster with 1 (2 h for the quantitative reaction at room temperature vs. less than 5% conversion after 12 h at 28 °C for Mo(NtBuAr)3).18 It is worth noting that the corresponding complex bearing three very large siloxy ligands – Mo(OSitBu3)3 – does not activate dinitrogen.5c This high reactivity towards N2 reduction is unusual for an octahedral Mo(III) complex, and highlights the particularity of the TBOS ligand, which can adopt mono- or bidentate binding modes while being relatively small compared to larger siloxy (e.g. tBu3SiO)19 or N(tBu) (3,5-Me2C6H3) ligands.7g
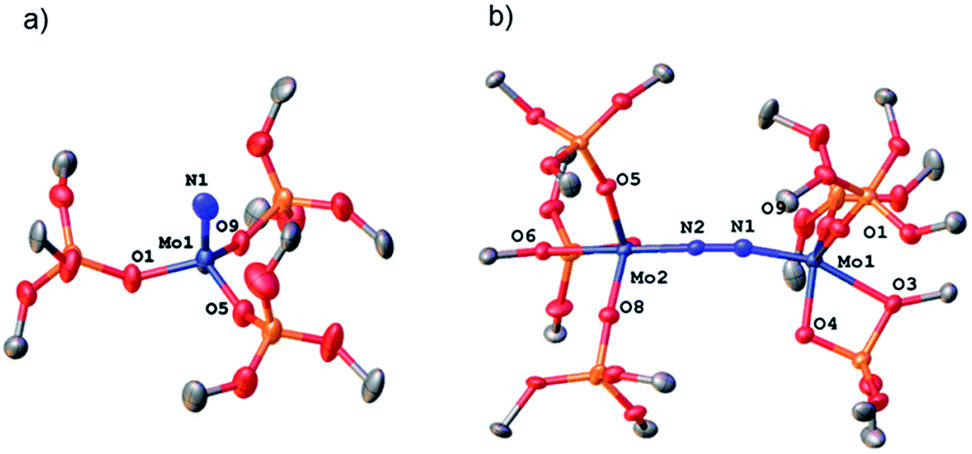 | ||
| Fig. 2 Thermal ellipsoid plot at 50% probability of (a) 2 and (b) 3. Hydrogen atoms, siloxide methyl groups and disorder have been omitted for clarity. | ||
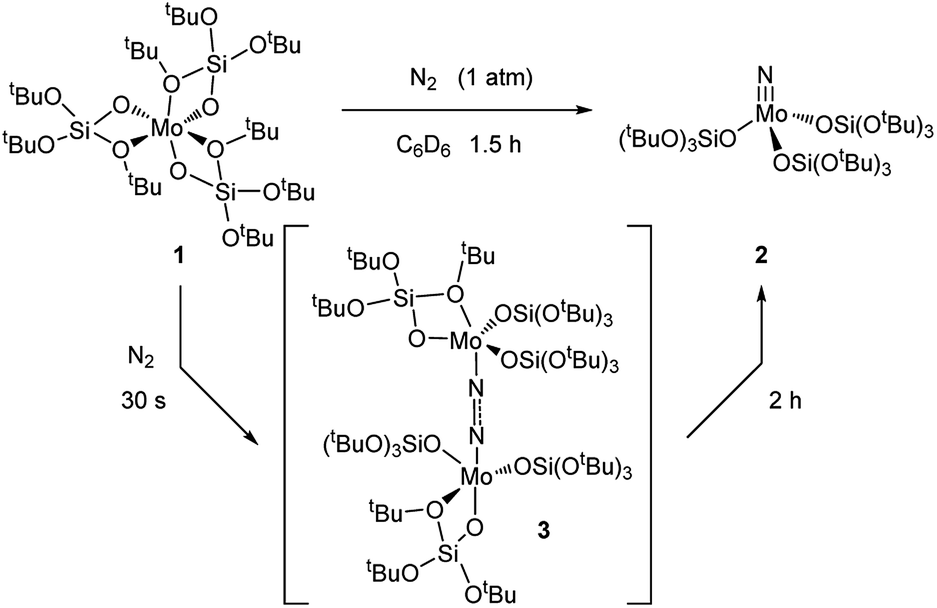 | ||
| Scheme 3 Reaction of 1 with N2 yields [Mo(N)(TBOS)3] (2) via a (μ-N2)[Mo(TBOS)3]2 intermediate (3). | ||
To corroborate our data, we performed DFT calculations in order to better understand the reactivity difference between these three systems. Due to the very large size of the investigated systems, we calculated the entire potential free energy surface with simplified models20 and calculated all reaction intermediates with the full ligand set (Scheme 4). The DFT calculations show that if electronic effects were the dominant factor (small models), one would predict the tris-amido system to be the most effective for N2 cleavage (TS energy of 17.2 kcal mol−1 above separated reactants), while the siloxy derivatives should have significantly lower reactivity (with TS energies of 27.4 and 30.9 kcal mol−1 for 1 and the model siloxy derivatives) in contrast with the observed trend, i.e.1 reacts much faster with N2 than the Cummins Mo(III), while the Wolczanski analog is unreactive towards N2 (Scheme 4).
 | ||
| Scheme 4 Top: calculated ΔG in kcal mol−1 for the three compared systems 1TL, 2TL and 3TL. Colored numbers refer to the small models and black numbers refer to the large ones. DFT models of complex 1 are in agreement with the XRD data, confirming the bond lengths and bond angles (calculated vs. experimental Mo–O1 2.410/2.302(4), Mo1–O6 2.043/2.046(4), Si–O1 1.707/1.677(5), O2–Mo–O1 67.2/68.33(16), O2–Mo1–O5 87.6/85.58(15)), so are the other values for intermediate c and for the final nitride e (see the ESI† for details). Bottom: calculated structure for the large model of complex 1 for the reaction intermediates. | ||
Once steric contributions are taken into consideration, it is notable how 1 yields more energetically accessible reaction intermediates and a significantly more stable nitrido complex: dinitrogen coordination costs only 6 kcal mol−1 for 1vs. 13–15 kcal mol−1 for the two others, and the formation of the dimeric species prior to N2 activation costs only 13 kcal mol−1 for 1vs. 27 and 33 kcal mol−1 for the Cummins and Wolczanski systems, respectively, in line with their relative difference of reactivity. Overall, the DFT calculations show that the TBOS ligand favors N2 activation because of its hemilability while protecting Mo(III) thanks to the possible κ2-configuration.
We then further explored the reactivity of 1 towards other small molecules. Upon exposure to N2O, a solution of 1 undergoes fast color change from amber to bright yellow upon warming up from liquid nitrogen temperature to room temperature (Scheme 4). Analysis of the reaction mixture by 1H NMR shows the presence of two peaks at 1.51 and 1.50 ppm. FTIR analysis of this reaction mixture after removal of the solvent in vacuo reveals the presence of an intense band at 1678 cm−1 which is attributed to Mo nitrosyl.16,21 Two species are obtained as crystals, and X-ray crystallography analyses revealed species 2 and [Mo(NO)(TBOS)3], 1-NO (Scheme 5). The nitrosyl compound 1-NO crystallizes in a distorted TBP geometry, with a linear NO ligand (Mo–N–O = 178.6(2)°) in the apical position and a κ2-TBOS ligand. The Mo–NO bond distance of 1.715(3) Å is consistent with the literature value for linear nitrosyl complexes of Cummins and Zubieta, two molybdenum thiolate species with metal–nitrogen distances of 1.769(13) and 1.766(6) Å, respectively.21a,22 It is slightly shorter, consistent with the weaker trans influence of the siloxy ligand. Despite a high kinetic barrier to reduction, the activation of N2O has been observed with numerous low valent metal complexes.23 However, the majority of metal-mediated activation of N2O generally involves the scission of the N–O bond, as the N–N bond is significantly stronger (∼481 kJ mol−1 and ∼167 kJ mol−1 for N–N and N–O, respectively) with a few examples of N–N bond activation,6a,21e,24 highlighting the high azophilicity of the Mo complex 1. The extremely challenging purification21a of the reaction mixture did not allow any further characterization of 1-NO and of the impurities which appear to be paramagnetic.
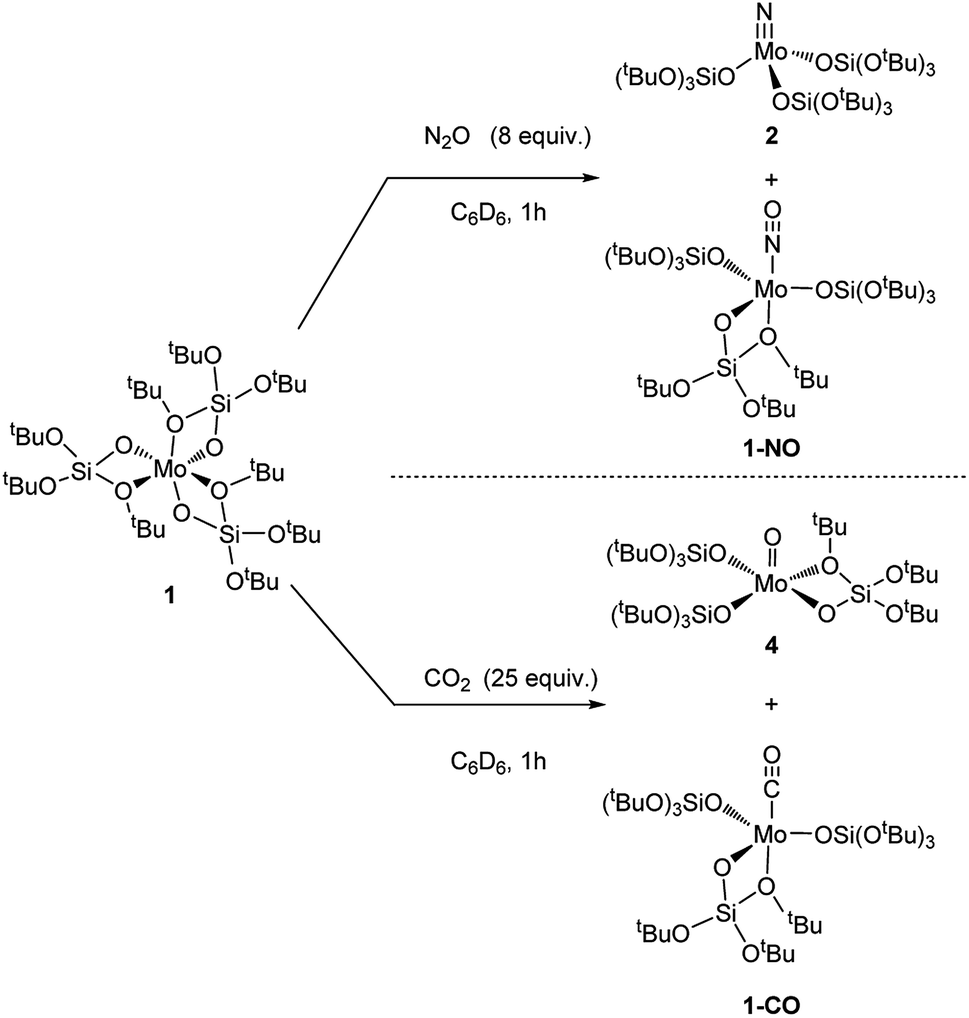 | ||
| Scheme 5 Reaction of 1 with N2O and CO2. | ||
Compound 1 when in contact with CO2 also results in a rapid color change from amber to red upon warming from the liquid nitrogen temperature to room temperature, to yield a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of two compounds according to NMR spectroscopy. Crystallization of the reaction mixture from pentane at −35 °C affords two sets of red crystals, assigned to [Mo(O)(TBOS)3], 4, and [Mo(CO)(TBOS)3], 1-CO (Fig. 3) according to X-ray single-crystal diffraction studies. The X-ray diffraction analysis of the mono-carbonyl complex 1-CO shows that the CO-adduct adopts a distorted TBP geometry with a κ2-TBOS ligand. The CO ligand occupies an axial position and is almost linear with a Mo–C–O angle of 175.2(3)°. The Mo–C and C–O distances of 1.931(3) Å and 1.156(3) Å, respectively, are in between those of Mo(CO)6 (Mo–C 2.059(3) Å and C–O 1.125(5) Å)25 and [Mo(CO)(Ph2PCH2CH2PPh2)2] (Mo–C 1.90(1) Å and C–O 1.19(1) Å)26 indicating a π-back-donation on the carbonyl, comparable to that of cis-(diethylenetriamine)molybdenum tricarbonyl.27 This is consistent with the red shift of the ν(C
:1 ratio of two compounds according to NMR spectroscopy. Crystallization of the reaction mixture from pentane at −35 °C affords two sets of red crystals, assigned to [Mo(O)(TBOS)3], 4, and [Mo(CO)(TBOS)3], 1-CO (Fig. 3) according to X-ray single-crystal diffraction studies. The X-ray diffraction analysis of the mono-carbonyl complex 1-CO shows that the CO-adduct adopts a distorted TBP geometry with a κ2-TBOS ligand. The CO ligand occupies an axial position and is almost linear with a Mo–C–O angle of 175.2(3)°. The Mo–C and C–O distances of 1.931(3) Å and 1.156(3) Å, respectively, are in between those of Mo(CO)6 (Mo–C 2.059(3) Å and C–O 1.125(5) Å)25 and [Mo(CO)(Ph2PCH2CH2PPh2)2] (Mo–C 1.90(1) Å and C–O 1.19(1) Å)26 indicating a π-back-donation on the carbonyl, comparable to that of cis-(diethylenetriamine)molybdenum tricarbonyl.27 This is consistent with the red shift of the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) band at 1869 cm−1 observed in the FTIR spectrum, which can be attributed to the weak σ-donor ability of the TBOS ligand.28 Complex 1-CO can also be synthesized upon exposing 1 to an excess of carbon monoxide, see the ESI† for details. The structure of 4 presents a Mo(V) complex in a distorted TBP geometry with an equatorial oxo moiety with no trans ligand. The MoO bond length is 1.665(2) Å, which lies in the usual range for terminal Mo(V) oxo complexes.29
O) band at 1869 cm−1 observed in the FTIR spectrum, which can be attributed to the weak σ-donor ability of the TBOS ligand.28 Complex 1-CO can also be synthesized upon exposing 1 to an excess of carbon monoxide, see the ESI† for details. The structure of 4 presents a Mo(V) complex in a distorted TBP geometry with an equatorial oxo moiety with no trans ligand. The MoO bond length is 1.665(2) Å, which lies in the usual range for terminal Mo(V) oxo complexes.29
 | ||
| Fig. 3 Thermal ellipsoid plot at 50% probability of (a) 1-NO, (b) 4 and (c) 1-CO. Hydrogen atoms, siloxide methyl groups and disorder have been omitted for clarity. | ||
Conclusions
In conclusion, we have shown that the TBOS ligand can bind Mo(III) in a bidentate κ2-fashion, providing an isolable – yet highly reactive – mononuclear octahedral Mo(III) complex. This complex thus activates a large variety of small – often rather inert – molecules, cleaving N–N and C–O bonds at low temperatures. An example is the 6-electron reduction of N2, which occurs faster than with other tri-coordinated Mo(III) complexes. The high reactivity of this coordinatively saturated complex illustrates the high adaptability of the TBOS ligand, which is able to protect highly reactive Mo(III) site from dimerization, without quenching its reactivity. The use of the TBOS ligand thus constitutes an alternative strategy to employ highly coordinatively unsaturated complexes towards small molecule activation at Mo centers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Dr Pavel Zhizhko for fruitful discussions. MP is thankful to the SNF (Swiss National Science Foundation) (200021_169134). VM was supported by an ETH Zürich-Marie Curie action for people (FEL-08 12-2). CPG acknowledges the Scholarship Fund of the Swiss Chemical Industry. We thank Corinna Braghieri for help with the TOC graphic.Notes and references
- (a) D. C. K. V. Rajagopalan, E. Stiefel and W. E. Newton, ACS Symp. Ser., 1993, 535, 50–68 CrossRef; (b) M. Hidai and Y. Mizobe, Chem. Rev., 1995, 95, 1115–1133 CrossRef CAS; (c) S. C. Lee and R. H. Holm, Chem. Rev., 2004, 104, 1135–1158 CrossRef CAS PubMed.
- C. E. Laplaza and C. C. Cummins, Science, 1995, 268, 861–863 CrossRef CAS PubMed.
- (a) B. S. Buyuktas, M. O. Marilyn and P. Power, Chem. Commun., 1998, 1689–1690 RSC; (b) C. C. Cummins, Prog. Inorg. Chem., 1998, 47, 685–836 CAS; (c) D. V. Yandulov, R. R. Schrock, A. L. Rheingold, C. Ceccarelli and W. M. Davis, Inorg. Chem., 2003, 42, 796–813 CrossRef CAS; (d) D. V. Yandulov, R. R. Schrock, A. L. Rheingold, C. Ceccarelli and W. M. Davis, Inorg. Chem., 2003, 42, 796–813 CrossRef CAS PubMed; (e) D. Kuiper, P. Wolczanski, E. B. Lobokovsky and T. Cundari, J. Am. Chem. Soc., 2008, 130, 12931–12943 CrossRef CAS PubMed.
- F. A. Cotton, Gen. Rev., 2005, 69–182 CAS.
- (a) K. Arashiba, Y. Miyake and Y. Nishibayashi, Nature, 2011, 3, 120–125 CAS; (b) M. J. Bezdek, S. Guo and P. J. Chirik, Inorg. Chem., 2016, 55, 3117–3127 CrossRef CAS PubMed; (c) D. S. Kuiper, P. T. Wolczanski, E. B. Lobkovsky and T. R. Cundari, J. Am. Chem. Soc., 2008, 130, 12931–12943 CrossRef CAS PubMed.
- (a) D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS; (b) L. A. Wickramasinghe, T. Ogawa, R. R. Schrock and P. Müller, J. Am. Chem. Soc., 2017, 139, 9132–9135 CrossRef CAS.
- (a) K. L. Fujdala and T. D. Tilley, Chem. Mater., 2001, 13, 1817–1827 CrossRef CAS; (b) K. L. Fujdala, I. J. Drake, A. T. Bell and T. D. Tilley, J. Am. Chem. Soc., 2004, 126, 10864–10866 CrossRef CAS PubMed; (c) J. Jarupatrakorn and T. D. Tilley, Dalton Trans., 2004, 2808–2813 RSC; (d) A. Fischbach, G. Eickerling, W. Scherer, E. Herdtweck and R. Anwander, Synthesis and derivatization of homoleptic dinuclear lanthanide siloxide complexes, 2004, vol. 59 Search PubMed; (e) R. L. Brutchey, I. J. Drake, A. T. Bel and T. D. Tilley, Chem. Commun., 2005, 29, 3736–3738 RSC; (f) R. L. Brutchey, C. G. Lugmair, L. O. Schebaum and T. D. Tilley, J. Catal., 2005, 229, 72–84 CrossRef CAS; (g) K. L. Fujdala, R. L. Brutchey and T. D. Tilley, in Surface and Interfacial Organometallic Chemistry and Catalysis, ed., C. Copéret and B. Chaudret, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 69–115 Search PubMed; (h) D. A. Ruddy, N. L. Ohler, A. T. Bell and T. D. Tilley, J. Catal., 2006, 238, 277–282 CrossRef CAS; (i) M. Zimmermann, N. Å. Frøystein, A. Fischbach, P. Sirsch, H. M. Dietrich, K. W. Törnroos, E. Herdtweck and R. Anwander, Chem.–Eur. J., 2007, 13, 8784–8800 CrossRef CAS PubMed; (j) V. Mougel, C. Camp, J. Pécaut, C. Copéret, L. Maron, C. E. Kefalidis and M. Mazzanti, Angew. Chem., Int. Ed., 2012, 51, 12280–12284 CrossRef CAS PubMed; (k) M. P. Conley, M. F. Delley, G. Siddiqi, G. Lapadula, S. Norsic, V. Monteil, O. V. Safonova and C. Copéret, Angew. Chem., Int. Ed., 2014, 53, 1872–1876 CrossRef PubMed; (l) D. P. Estes, G. Siddiqi, F. Allouche, K. V. Kovtunov, O. V. Safonova, A. L. Trigub, I. V. Koptyug and C. Copéret, J. Am. Chem. Soc., 2016, 138, 14987–14997 CrossRef CAS PubMed; (m) F. Allouche, D. Klose, C. P. Gordon, A. Ashuiev, M. Wörle, V. Kalendra, V. Mougel, C. Copéret and G. Jeschke, Angew. Chem., Int. Ed., 2018, 57, 14533–14537 CrossRef CAS PubMed.
- G. Lapadula, M. P. Conley, C. Copéret and R. A. Andersen, Organometallics, 2015, 34, 2271–2277 CrossRef CAS.
- F. Allouche, G. Lapadula, G. Siddiqi, W. W. Lukens, O. Maury, B. Le Guennic, F. Pointillart, J. Dreiser, V. Mougel, O. Cador and C. Copéret, ACS Cent. Sci., 2017, 3, 244–249 CrossRef CAS PubMed.
- Experimental details for X-ray data crystal structure analysis collections of all complexes are given in ESI.† CCDC 1826972 (1), CCDC 1826973 (2), CCDC 1826967 (3), CCDC 1826964 (1-(ethylene)), CCDC 1826971 (1-(isobutene)), CCDC 1826963 (1-(but-2-yne)), CCDC 1826968 (1-NO), CCDC 1826969 (4), CCDC 1826965 (1-CO).
- (a) P. M. Boorman, M. Wang and M. Parvez, Dalton Trans., 1996, 4533–4542 RSC; (b) E. Le Grognec, R. Poli and P. Richard, Dalton Trans., 2000, 1499–1506 RSC.
- H. J. Bowen, Tables of interatomic distances and configuration in molecules and ions The Chemical Society, Burlington House, London, 1958 Search PubMed.
- L. Pauling and L. O. Brockway, J. Am. Chem. Soc., 1937, 59, 1223–1236 CrossRef CAS.
- M. J. Byrnes, X. Dai, R. R. Schrock, A. S. Hock and P. Müller, Organometallics, 2005, 24, 4437–4450 CrossRef CAS.
- Reacting 1 with a large excess of 2-butyne (1000 equiv.) affords a gel, likely resulting from butyne polymerization.
- L. McAfee, J. Chem. Educ., 2000, 77, 1122 CrossRef CAS.
- J. J. Curley, T. R. Cook, S. Y. Reece, P. Müller and C. C. Cummins, J. Am. Chem. Soc., 2008, 130, 9394–9405 CrossRef CAS PubMed.
- C. E. Laplaza, M. J. A. Johnson, J. C. Peters, A. L. Odom, E. Kim, C. C. Cummins, G. N. George and I. J. Pickering, J. Am. Chem. Soc., 1996, 118, 8623–8638 CrossRef CAS.
- D. S. Kuiper, P. T. Wolczanski, E. B. Lobkovsky and T. R. Cundari, Inorg. Chem., 2008, 47, 10542–10553 CrossRef CAS PubMed.
- Q. Cui, D. G. Musaev, M. Svensson, S. Sieber and K. Morokuma, J. Am. Chem. Soc., 1995, 117, 12366–12367 CrossRef CAS.
- (a) T. Agapie, A. L. Odom and C. C. Cummins, Inorg. Chem., 2000, 39, 174–179 CrossRef CAS PubMed; (b) J.-P. F. Cherry, A. R. Johnson, L. M. Baraldo, Y.-C. Tsai, C. C. Cummins, S. V. Kryatov, E. V. Rybak-Akimova, K. B. Capps, C. D. Hoff, C. M. Haar and S. P. Nolan, J. Am. Chem. Soc., 2001, 123, 7271–7286 CrossRef CAS PubMed; (c) I. J. Blackmore, X. Jin and P. Legzdins, Organometallics, 2005, 24, 4088–4098 CrossRef CAS; (d) T. W. Hayton, P. Legzdins and W. B. Sharp, Chem. Rev., 2002, 102, 935–992 CrossRef CAS PubMed; (e) C. E. Laplaza, A. L. Odom, W. M. Davis, C. C. Cummins and J. D. Protasiewicz, J. Am. Chem. Soc., 1995, 117, 4999–5000 CrossRef CAS.
- (a) A. Proust, P. Gouzerh and F. Robert, Inorg. Chem., 1993, 32, 5291–5298 CrossRef CAS; (b) P. T. Bishop, J. R. Dilworth, J. Hutchinson and J. Zubieta, Dalton Trans., 1986, 967–973 RSC.
- W. B. Tolman, Angew. Chem., Int. Ed., 2010, 49, 1018–1024 CrossRef CAS.
- (a) M. E. Vol'pin and V. B. Shur, Organomet. React., 1970, 1, 55–117 Search PubMed; (b) Y. Akio, G. Shigeo, O. Masaharu, T. Mitsuaki, I. Sakuji and K. Tominaga, Bull. Chem. Soc. Jpn., 1972, 45, 3110–3117 CrossRef; (c) W. H. Bernskoetter, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2005, 127, 14051–14061 CrossRef CAS PubMed; (d) M. Falcone, L. Chatelain, R. Scopelliti, I. Živković and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS PubMed.
- C. W. Mak Thomas, Z. Kristallogr. Cryst. Mater., 1984, 166, 277 Search PubMed.
- M. Sato, T. Tatsumi, T. Kodama, M. Hidai, T. Uchida and Y. Uchida, J. Am. Chem. Soc., 1978, 100, 4447–4452 CrossRef CAS.
- F. A. Cotton and R. M. Wing, Inorg. Chem., 1965, 4, 314–317 CrossRef CAS.
- (a) V. Mougel and C. Coperet, Chem. Sci., 2014, 5, 2475–2481 RSC; (b) D. P. Estes, C. P. Gordon, A. Fedorov, W.-C. Liao, H. Ehrhorn, C. Bittner, M. L. Zier, D. Bockfeld, K. W. Chan, O. Eisenstein, C. Raynaud, M. Tamm and C. Copéret, J. Am. Chem. Soc., 2017, 139, 17597–17607 CrossRef CAS PubMed.
- (a) M. G. B. Drew and I. B. Tomkins, J. Chem. Soc. A, 1970, 22–25 RSC; (b) F. E. Inscore, H. K. Joshi, A. E. McElhaney and J. H. Enemark, Inorg. Chim. Acta, 2002, 331, 246–256 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1826963–1826965, 1826967–1826969, 1826971–1826973. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01955c |
| This journal is © The Royal Society of Chemistry 2019 |