
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective single C–F bond arylation of trifluoromethylalkene derivatives†
Luning
Tang
,
Ze-Yao
Liu
,
Wenzhi
She
and
Chao
Feng
 *
*
Institute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing 211816, P. R. China. E-mail: iamcfeng@njtech.edu.cn
First published on 5th August 2019
Abstract
A strategically novel single C–F bond functionalization of CF3-derived molecules, which shows a prominent advantage for the expedient construction of difluoromethylene-bridged organic scaffolds, is disclosed. The reported protocol consists of SN2′ amination, N-alkylation and palladium-catalyzed allylic substitution reactions, which enables straightforward arylation and alkenylation of vinyltrifluoromethane derivatives. Furthermore, this strategy is characterized by its broad substrate scope with respect to both CF3-alkene and arylboronic acid derivatives.
Introduction
Trifluoromethyl (CF3) ranks among the most popular fluorinated alkyl groups and finds wide applications in pharmaceutical chemistry, materials science, and agrochemistry owing to its unique physicochemical properties.1 As such, the development of effective synthetic methods for the incorporation of CF3 into organic molecules has witnessed a remarkable advancement during the past few decades.2 In addition, trifluoromethylated compounds could also virtually serve as promising progenitors for the direct assembly of gem-difluoroalkene bridged organic frameworks, which widely occur in drugs and bioactive compounds, provided that selective single C–F bond transformation of the CF3 group could be readily accomplished (Fig. 1).3,4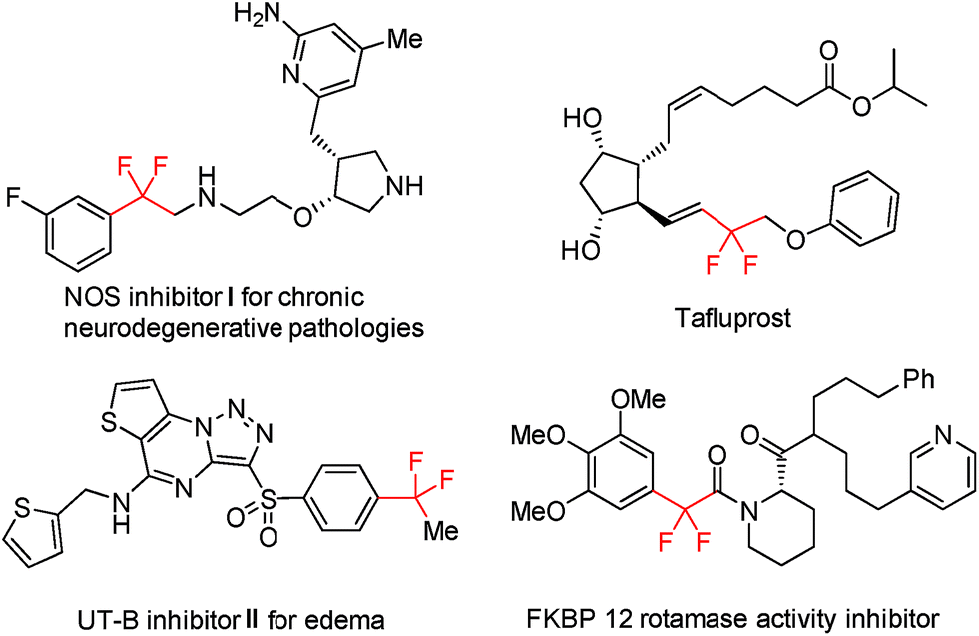 | ||
| Fig. 1 Representative bioactive CF2-containing molecules. | ||
Compared with conventional synthetic methods that resort to halodifluoromethylated precursors, the direct utilization of CF3-congeners tends to be more beneficial considering both step-economy and starting material accessibility.5 While synthetically more appealing, the development of efficacious scenarios, which are amenable to the selective functionalization of the single C(sp3)–F bond of the CF3 group, progresses very slowly and still remains a daunting challenge. The underlying reason for this situation comes in part from the extremely high bond dissociation energy (BDE) of C–F bonds6 (e.g., 128 kcal mol−1 for HCF3) but more from the difficulty to well control the chemoselectivity by suppressing post-defluorination. The intrinsic fact that the strength of C–F bonds becomes attenuated progressively with the shrinking of F-substitutions on the same carbon atom also adds to such a difficulty.7 Therefore, it is not surprising that in most reported examples of CF3-derivative transformations, non-fluorinated products were always produced by unselective triple C–F bond cleavage.8
Notwithstanding the prominent challenges encountered in the selective C–F bond functionalization of CF3-containing molecules, prominent studies by making use of different strategies were disclosed during the past several years.4,9 For example, by taking advantage of intramolecular F-abstraction by an in situ generated neighboring silylium group, Hosoya and co-workers reported an elegant example of C–F bond allylation of ortho-silylated ArCF3 derivatives.4a Very recently, photoredox-catalysis was also revealed to be a promising choice for this aim. In this respect, the groups of König4b and Jui4c have successfully utilized this manifold for the smooth generation of an α,α-difluorobenzylic radical intermediate, which further engaged in subsequent intramolecular cyclization or intermolecular alkylation reactions (Scheme 1a). In spite of these groundbreaking studies of the selective functionalization of CF3-derivatives, the analogous transformation with respect to trifluoromethylalkenes, however, still remains elusive. The discrepancy of reactivity between electronically activated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds with C–F bonds in these molecules, towards either radical or anionic species, is believed to be the fundamental root for such a dilemma. This competitively dominant reaction profile is visually illustrated by a considerable amount of precedents of the SN2′-type defluorofunctionalization of CF3-alkene derivatives over the past several years, which, however, only enables the introduction of functionalities into the distal olefinic carbon atom, thus affording gem-difluoroalkene products (Scheme 1b).9b–k Thus far, the general approach for ipso-C–F functionalization of trifluoromethylalkenes has yet to be achieved. Inspired by the well-established SN2′ reaction manifold of CF3-alkenes, we envision the feasibility of using amines as a traceless mediator for ipso-C–F bond functionalization of these molecules. We assume that with a sequence of SN2′ amination and N-alkylation, gem-difluoroallyl ammonium 3 could be obtained directly from CF3-alkene derivatives. In view of the precedents of using ammonium functionalities as the leaving groups in transition-metal-catalyzed cross-coupling reactions,10 we anticipate that an elegant combination of SN2′ amination, N-alkylation and palladium-catalyzed allylic substitution could potentially provide an expedient strategy for the construction of α,α-difluoroallylarenes,11 provided that nucleophilic substitution of gem-difluoroallylpalladium species could be steered to occur selectively on the fluorine-containing carbon atom (Scheme 1c).
C double bonds with C–F bonds in these molecules, towards either radical or anionic species, is believed to be the fundamental root for such a dilemma. This competitively dominant reaction profile is visually illustrated by a considerable amount of precedents of the SN2′-type defluorofunctionalization of CF3-alkene derivatives over the past several years, which, however, only enables the introduction of functionalities into the distal olefinic carbon atom, thus affording gem-difluoroalkene products (Scheme 1b).9b–k Thus far, the general approach for ipso-C–F functionalization of trifluoromethylalkenes has yet to be achieved. Inspired by the well-established SN2′ reaction manifold of CF3-alkenes, we envision the feasibility of using amines as a traceless mediator for ipso-C–F bond functionalization of these molecules. We assume that with a sequence of SN2′ amination and N-alkylation, gem-difluoroallyl ammonium 3 could be obtained directly from CF3-alkene derivatives. In view of the precedents of using ammonium functionalities as the leaving groups in transition-metal-catalyzed cross-coupling reactions,10 we anticipate that an elegant combination of SN2′ amination, N-alkylation and palladium-catalyzed allylic substitution could potentially provide an expedient strategy for the construction of α,α-difluoroallylarenes,11 provided that nucleophilic substitution of gem-difluoroallylpalladium species could be steered to occur selectively on the fluorine-containing carbon atom (Scheme 1c).
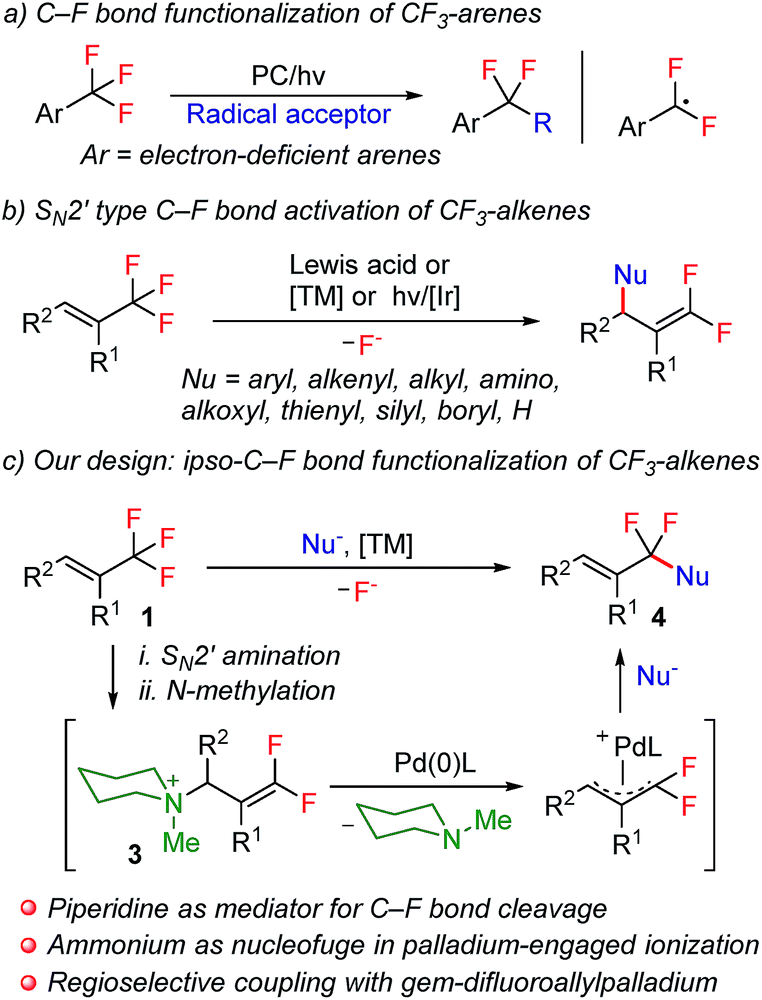 | ||
| Scheme 1 Selective single C–F bond functionalization of CF3-derivatives. (a) C–F bond functionalization of CF3-arenes. (b) SN2′ type C–F bond activation of CF3-alkenes. (c) Our ipso-C–F bond functionalization of CF3-alkenes. | ||
Results and discussion
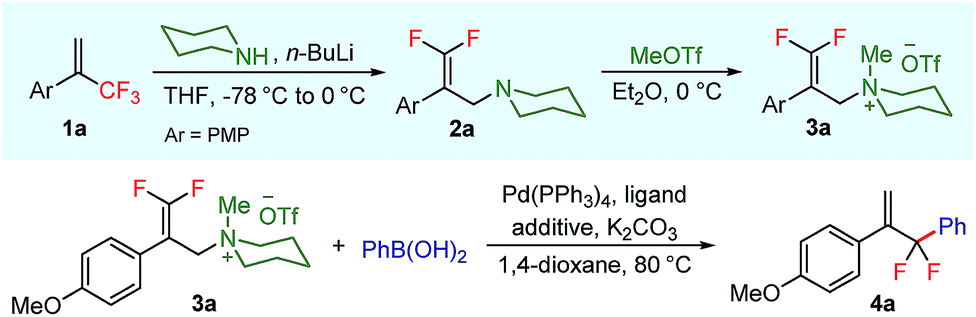
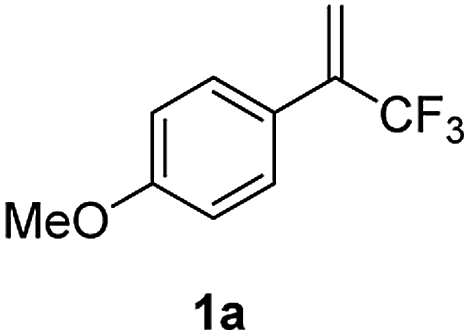
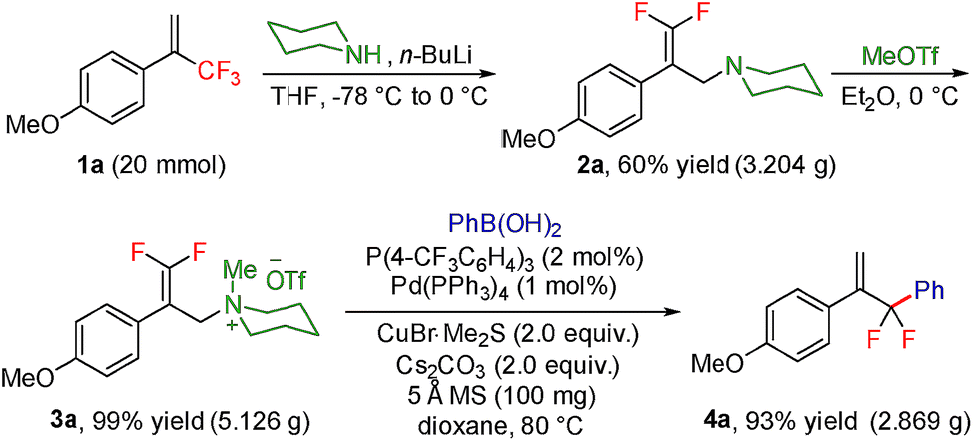
To test our hypothesis, we started our investigation by using 1-methoxy-4-(3,3,3-trifluoroprop-1-en-2-yl)benzene 1a as the model substrate (Table 1). The SN2′ amination reaction of 1a proceeded smoothly with the treatment of piperidine in the presence of n-BuLi, yielding 65% of 3,3-difluoroallylamine 2a (Table 1).12 The following methylation with MeOTf readily afforded the quaternary ammonium salt 3a in a nearly quantitative yield. Encouraged by these productive results, we moved on to evaluate the feasibility of aryl substitution with regard to quaternary ammonium salt 3a by using phenylboronic acid with different ligands and additives. Delightfully, under the reaction conditions of 10 mol% Pd(PPh3)4, 20 mol% X-Phos, 30 mol% CuI, and 2.0 equiv. K2CO3 in the presence of dioxane at 80 °C for 20 hours, 3a underwent regioselective allylic substitution with phenylboronic acid to afford the desired gem-difluoroallylation product 4a in 41% yield (entry 1). For improving the reaction efficiency, various ancillary ligands were then examined (entries 2–4), among which P(4-CF3C6H5)3 turned out to be the appropriate choice (entry 2). It is worth pointing out that the identity of the copper salt additive and its stoichiometry are of vital importance to the occurrence of allylic substitution, and the employment of 2.0 equiv. of CuBr·Me2S was ultimately revealed to be optimal (entries 5–12). Further fine tuning the reaction conditions, by using Cs2CO3 as a base, led to a slight improvement in the reaction yield (entry 13). While the exact role of the copper salt in the arylation step was unclear, we believed that it could promote this transformation in two aspects by acting either as the transmetalation agent to promote the transfer of the aryl group from boron to palladium or as the tertiary amine scavenger to prevent the active palladium catalyst from being deactivated by the amine byproduct. Pleasingly, the yield of 4a increased dramatically upon the addition of 5 Å molecular sieves (entry 14).13 Furthermore, the catalyst loading of palladium could be lowered to 1.0 mol%, with only marginal influence on the product yield being observed (entry 15).14|
|
|||
|---|---|---|---|
| Entry | Ligand | Additive (mol%) | Yieldb [%] |
| a Reaction conditions (unless otherwise specified): 3a (0.1 mmol, 1.0 equiv.), PhB(OH)2 (2.0 equiv.), Pd(PPh3)4 (10 mol%), ligand (20 mol%), additive (30 mol%), K2CO3 (2.0 equiv.), 1,4-dioxane (1 mL), 80 °C, and 20 h. b Yields determined by 19F-NMR spectroscopy using trifluoromethylbenzene as an internal standard. c Cs2CO3 instead of K2CO3. d 5 Å molecular sieve (100 mg). e Pd(PPh3)4 (1 mol%) and P(4-CF3C6H4)3 (2 mol%). f Isolated yield. | |||
| 1 | X-Phos | CuI (30%) | 41 |
| 2 | P(4-CF3C6H5)3 | CuI (30%) | 50 |
| 3 | P(C6F5)3 | CuI (30%) | 39 |
| 4 | C-Phos | CuI (30%) | 32 |
| 5 | P(4-CF3C6H4)3 | CuI (10%) | 5 |
| 6 | P(4-CF3C6H4)3 | CuI (50%) | 56 |
| 7 | P(4-CF3C6H4)3 | CuI (100%) | 72 |
| 8 | P(4-CF3C6H4)3 | CuI (200%) | 80 |
| 9 | P(4-CF3C6H4)3 | CuI (250%) | 80 |
| 10 | P(4-CF3C6H4)3 | CuBr·Me2S (200%) | 83 |
| 11 | P(4-CF3C6H4)3 | CuTc (200%) | 31 |
| 12 | P(4-CF3C6H4)3 | CuOAc (200%) | 29 |
| 13c | P(4-CF3C6H4)3 | CuBr·Me2S (200%) | 85 |
| 14c,d | P(4-CF3C6H4)3 | CuBr·Me2S (200%) | 99 |
| 15c,e | P(4-CF3C6H4)3 | CuBr·Me2S (200%) | 95 (92)f |
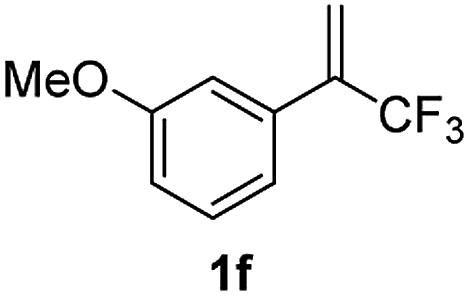
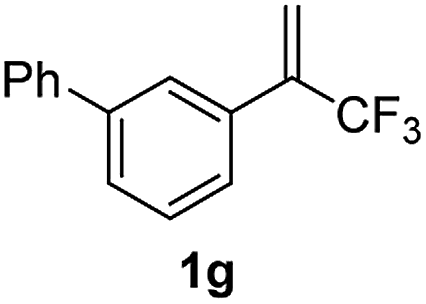
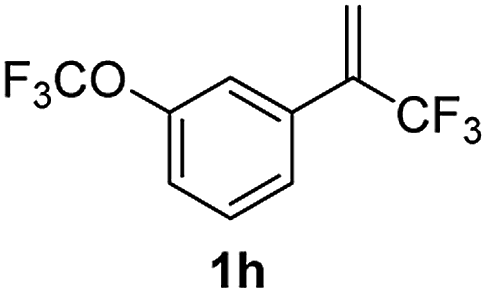
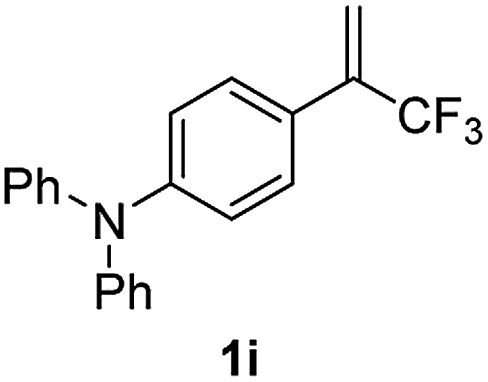
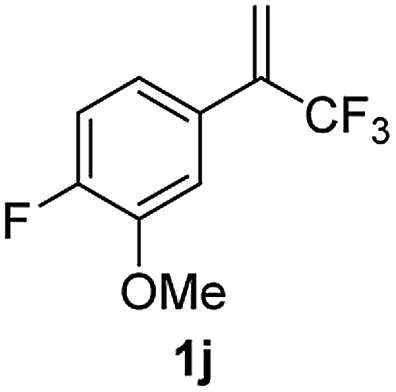
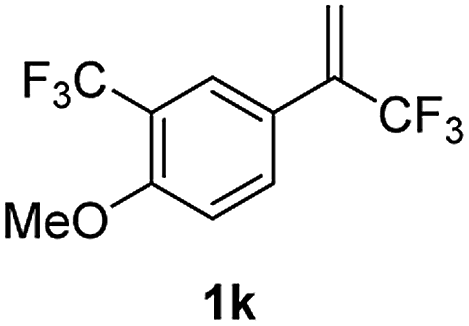
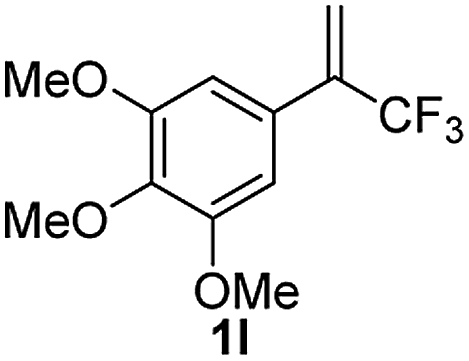
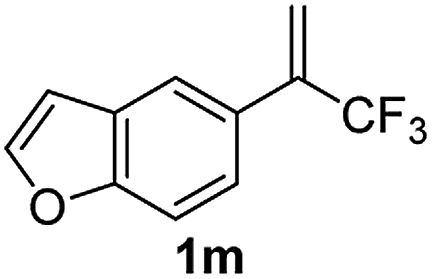
With the optimized reaction conditions in hand, the reaction generality with respect to trifluoromethylalkene 1 was first examined. As tabulated in Table 2 a variety of α-CF3-styrenes were shown to be compatible with the sequential SN2′ amination/N-methylation/allylic substitution. Vinyltrifluoromethane derivatives bearing alkyl, methoxyl, phenyl, trifluoromethoxy and chloride groups on the α-phenyl substituent readily participated in the amination and afforded products 2a–2h in 46–80% yields. N-methylation of these 3,3-difluoroallylamines 2, generally proceeded with ease to furnish the quaternary ammonium salts 3a–3h in excellent yields (93–99%), and the subsequent arylation with the as-obtained ammonium intermediates also proceeded smoothly to deliver the desired products 4a–4h in 30–92% yields. Intriguingly, the diphenylamine derived substrate 1i was also compatible with this tandem reaction, with no obvious interference in N-methylation being observed. Moreover, in the case of substrate 1j, which is potentially sensitive towards the amination process because of the presence of C(Ar)–F bonds, the reaction showed excellent chemoselectivity for allylic substitution. Furthermore, poly-substituted α-CF3-styrene derivatives were also competent substrates in this transformation as represented by the examples of 1j–1l. In addition, heterocycle-derived substrates such as benzofuran 1m and benzo[b][1,4]dioxine 1n were also well accommodated and afforded the desired products 4m and 4n in overall moderate yields.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Substrate | Products | Substrate | Products | ||||
| 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 |
| a See the ESI for experimental details. | |||||||
|
65% | 97% | 92% |
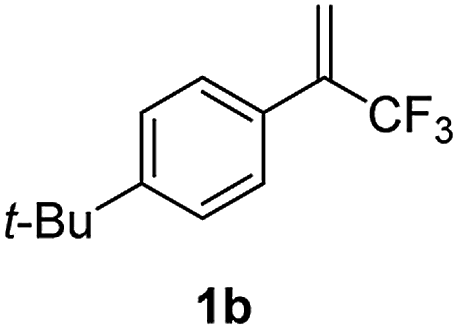
|
64% | 98% | 73% |
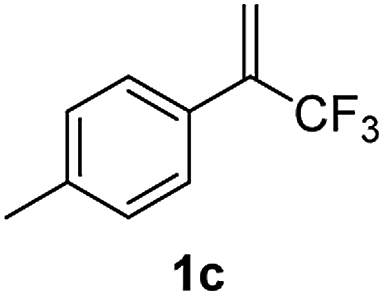
|
59% | 95% | 62% |
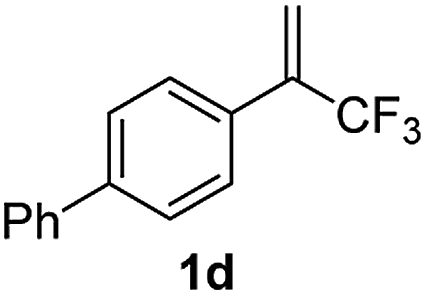
|
80% | 93% | 72% |
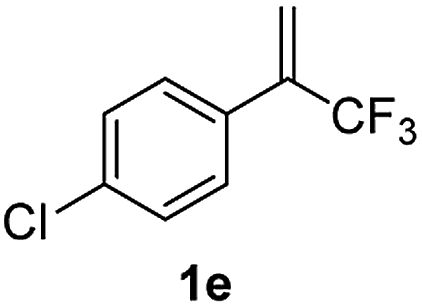
|
70% | 97% | 61% |
|
46% | 99% | 68% |
|
53% | 98% | 90% |
|
48% | 94% | 30% |
|
64% | 85% | 66% |
|
68% | 93% | 73% |
|
56% | 95% | 87% |
|
48% | 95% | 75% |
|
79% | 99% | 73% |
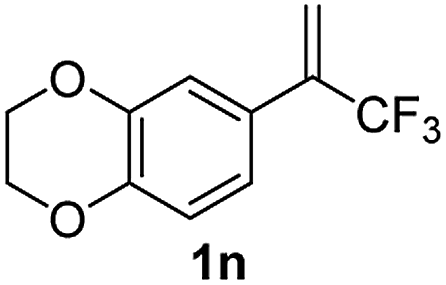
|
63% | 99% | 58% |
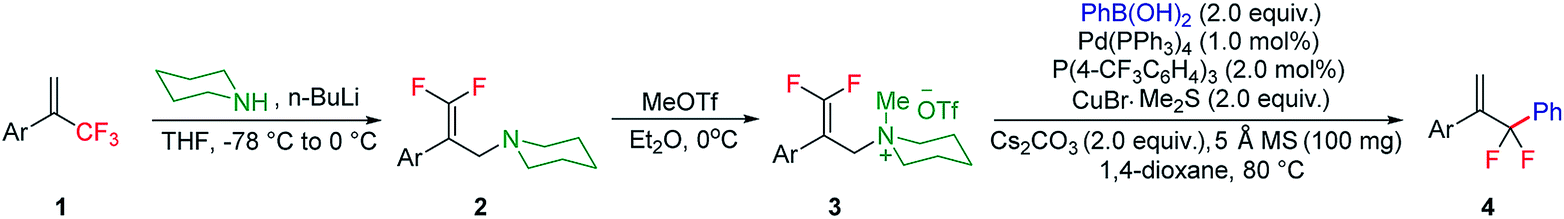
To further evaluate the generality of this transformation, the reaction scope of arylboronic acid was subsequently investigated (Scheme 2). By and large, arylboronic acids bearing either electron-donating or electron-withdrawing groups proved to be well amenable to this gem-difluoroallylation process, and the desired products 4aa–4al could be obtained in 39–99% yields. However, arylboronic acids with ortho-substituents took part in this reaction sluggishly as showcased by 4am and 4an, thus indicating that the gem-difluoroallylation reaction is quite sensitive to steric hindrance. The presence of the amino functionality in the molecule of arylboronic acid was well tolerated without notable impediment to palladium catalysis (4ao). Furthermore, naphthylboronic acids, either α or β isomers, were revealed to be appropriate coupling partners, albeit giving rise to the desired products in yields with a contrasting difference (4ap and 4aq). In addition, arylboronic acids with poly-substituents or those derived from benzoheterocycles also participated in this allylic substitution uneventfully, affording the desired products 4ar–4au in moderate to high yields. Notably, (2-fluoropyridin-4-yl)boronic acid engaged well in this reaction and delivered the product 4av in 50% yield. Pleasingly, alkenylboronic acids also proved to be competent reaction partners, which provide a straightforward synthetic avenue for the assembly of 3,3-difluoropenta-1,4-diene derivatives, albeit with somewhat decreased reaction efficiency. To further demonstrate the synthetic potential of the reaction protocol, elaboration of complex molecules such as estrone was carried out, which delivered the desired product 4az in 57% yield.
 | ||
| Scheme 2 Reaction scope of boronic acid. | ||
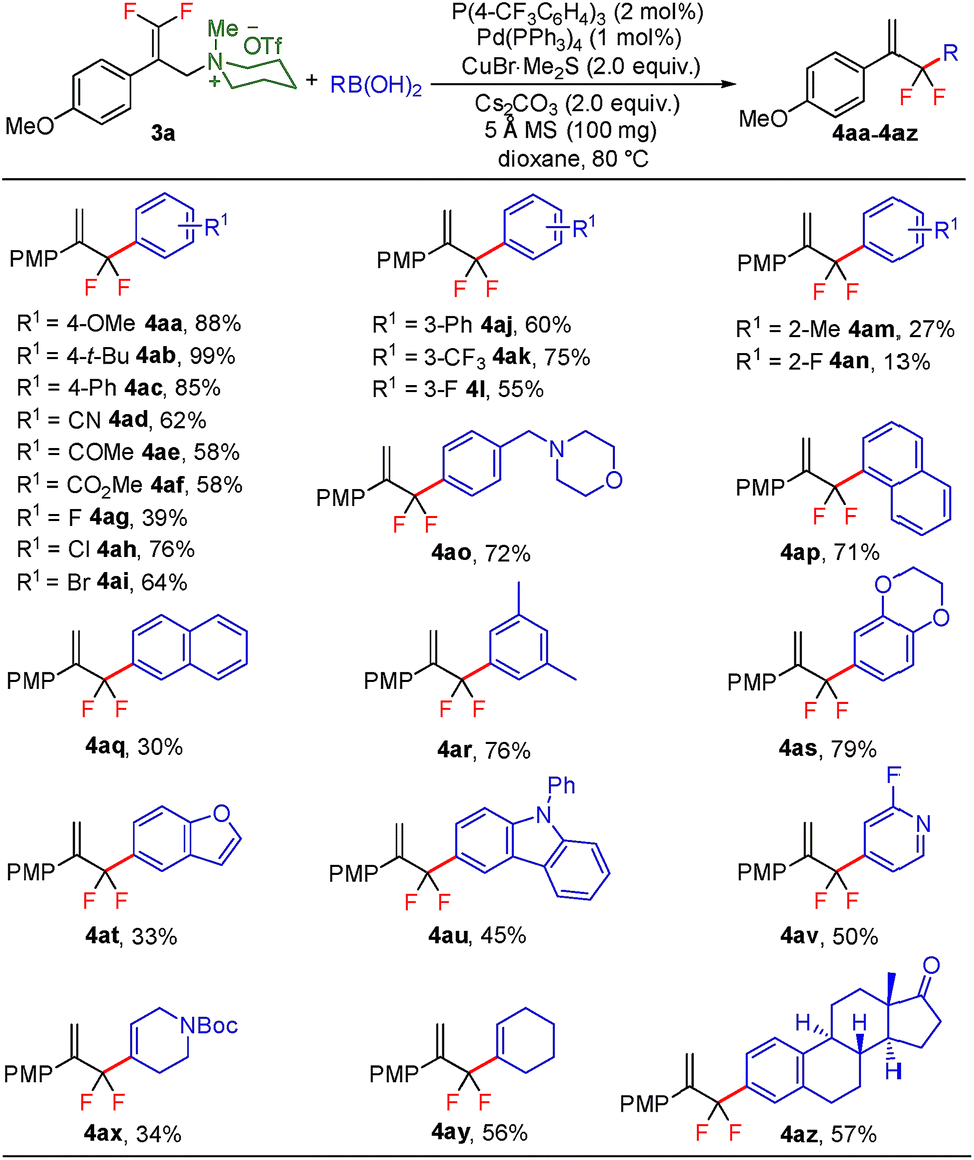
The practicality of this method was showcased by the gram-scale transformation of 1a, which provided the desired product 4a in 92% yield (Scheme 3). Moreover, a telescoped procedure that avoided intermediate purification worked well, delivering 4a in 42% overall yield (three steps, see the ESI† for details).
 | ||
| Scheme 3 Gram-scale reaction. | ||
As the obtained gem-difluoroallylarenes contain both the alkenyl and difluoromethylene functionalities, further synthetic elaborations were carried out. Osmium-catalyzed dihydroxylation of compound 4a readily occurred to provide the product 5b in moderate yield, without affecting the difluoromethylene moiety, while in the presence of a rhodium catalyst, 4a underwent SN2′ type C–F bond functionalization with arylboronic esters, affording the tetra-substituted fluoroalkene 5a in 48% yield (Scheme 4).
 | ||
| Scheme 4 Synthetic transformation of gem-difluoroallylarene 4a. | ||
Conclusions
In summary, we have developed a novel method, which enables the selective single C–F bond arylation/alkenylation of trifluoromethylalkene derivatives. By directly making use of CF3-alkenes as the reaction substrates and strategically merging SN2′ amination, N-alkylation and palladium-catalyzed allylic substitution, an unprecedented synthetic protocol for easy access to α,α-difluoroallylarene/alkene structures was formulated. Furthermore, the high regioselectivities of both amination and allylic substitution processes are deemed responsible for the success of this transformation. Because of its wide substrate scope and practicality, this method is expected to find more potential applications in the discovery of new lead compounds for pharmaceutical innovations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support of the “Thousand Talents Plan” Youth Program, the “Jiangsu Specially-Appointed Professor Plan”, the National Natural Science Foundation of China (21871138), and the Natural Science Foundation of Jiangsu Province (BK20170984).Notes and references
- (a) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed; (b) D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071–1081 CrossRef; (c) W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Aceña, K. Izawa, H. Liu and V. A. Soloshonok, J. Fluorine Chem., 2014, 167, 37–54 CrossRef CAS; (d) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- (a) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683–730 CrossRef CAS PubMed; (b) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294–8308 CrossRef CAS PubMed; (c) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC; (d) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 CrossRef CAS PubMed; (e) S. Barata-Vallejo and A. Postigo, Coord. Chem. Rev., 2013, 257, 3051–3069 CrossRef CAS.
- (a) F. Xue, H. Li, S. L. Delker, J. Fang, P. Martasek, L. J. Roman, T. L. Poulos and R. B. Silverman, J. Am. Chem. Soc., 2010, 132, 14229–14238 CrossRef CAS PubMed; (b) N. Tadashi, M. Takeshi, G. Wakana, K. Masaaki, M. Nobuaki, M. Yasushi and H. Hideaki, Biol. Pharm. Bull., 2003, 26, 1691–1695 CrossRef PubMed; (c) C. L. Lynch, C. A. Willoughby, J. J. Hale, E. J. Holson, R. J. Budhu, A. L. Gentry, K. G. Rosauer, C. G. Caldwell, P. Chen, S. G. Mills, M. MacCoss, S. Berk, L. Chen, K. T. Chapman, L. Malkowitz, M. S. Springer, S. L. Gould, J. A. DeMartino, S. J. Siciliano, M. A. Cascieri, A. Carella, G. Carver, K. Holmes, W. A. Schleif, R. Danzeisen, D. Hazuda, J. Kessler, J. Lineberger, M. Millerc and E. A. Emini, Bioorg. Med. Chem. Lett., 2003, 13, 119–123 CrossRef CAS PubMed; (d) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- (a) S. Yoshida, K. Shimomori, Y. Kim and T. Hosoya, Angew. Chem., Int. Ed., 2016, 55, 10406–10409 CrossRef CAS PubMed; (b) K. Chen, N. Berg, R. Gschwind and B. König, J. Am. Chem. Soc., 2017, 139, 18444–18447 CrossRef CAS PubMed; (c) H.-B. Wang and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 163–166 CrossRef CAS PubMed; (d) H. Dang, A. M. Whittaker and G. Lalic, Chem. Sci., 2016, 7, 505–509 RSC.
- (a) Y.-B. Yu, G.-Z. He and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 10457–10461 CrossRef CAS PubMed; (b) Q.-Q. Min, Z. Yin, Z. Feng, W.-H. Guo and X. Zhang, J. Am. Chem. Soc., 2014, 136, 1230–1233 CrossRef CAS PubMed; (c) Y.-L. Xiao, Q.-Q. Min, C. Xu, R.-W. Wang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5837–5841 CrossRef CAS PubMed; (d) J.-W. Gu, W.-H. Guo and X. Zhang, Org. Chem. Front., 2015, 2, 38–41 RSC; (e) Y.-L. Xiao, W.-H. Guo, G.-Z. He, Q. Pan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9909–9913 CrossRef CAS PubMed; (f) T. L. Andersen, M. W. Frederiksen, K. Domino and T. Skrydstrup, Angew. Chem., Int. Ed., 2016, 55, 10396–10400 CrossRef CAS PubMed.
- J. Burdeniuc, B. Jedlicka and R. H. Crabtree, Chem. Ber., 1997, 130, 145–154 CrossRef CAS.
- (a) K. B. Wiberg and P. R. Rablen, J. Am. Chem. Soc., 1993, 115, 614–625 CrossRef CAS; (b) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (c) C. P. Andrieux, C. Combellas, F. Kanoufi, J.-M. Savéant and A. Thiébault, J. Am. Chem. Soc., 1997, 119, 9527–9540 CrossRef CAS.
- (a) V. J. Scott, R. Çelenligil-Çetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852–2853 CrossRef CAS PubMed; (b) K. Fuchibe and T. Akiyama, J. Am. Chem. Soc., 2006, 128, 1434–1435 CrossRef CAS PubMed; (c) C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed; (d) J. Terao, M. Nakamura and N. Kambe, Chem. Commun., 2009, 6011–6013 RSC; (e) T. Stahl, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 1248–1251 CrossRef CAS PubMed; (f) J. Zhu, M. Perez, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2016, 55, 1417–1421 CrossRef CAS PubMed.
- (a) R. Doi, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2016, 55, 341–344 CrossRef CAS PubMed; (b) K. Fuchibe, H. Hatta, K. Oh, R. Oki and J. Ichikawa, Angew. Chem., Int. Ed., 2017, 56, 5890–5893 CrossRef CAS PubMed; (c) S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073–15077 CrossRef CAS PubMed; (d) T. Ichitsuka, T. Fujita and J. Ichikawa, ACS Catal., 2015, 5, 5947–5950 CrossRef CAS; (e) W. Dai, Y. Lin, Y. Wan and S. Cao, Org. Chem. Front., 2018, 5, 55–58 RSC; (f) Y. Lan, F. Yang and C. Wang, ACS Catal., 2018, 8, 9245–9251 CrossRef CAS; (g) T. Fujita, M. Takazawa, K. Sugiyama, N. Suzuki and J. Ichikawa, Org. Lett., 2017, 19, 588–591 CrossRef CAS PubMed; (h) T. Ichitsuka, T. Fujita, T. Arita and J. Ichikawa, Angew. Chem., Int. Ed., 2014, 53, 7564–7568 CrossRef CAS PubMed; (i) Y. Liu, Y. Zhou, Y. Zhao and J. Qu, Org. Lett., 2017, 19, 946–949 CrossRef CAS PubMed; (j) M. Wang, X. Pu, Y. i. Zhao, P. Wang, Z. Li, C. Zhu and Z. Shi, J. Am. Chem. Soc., 2018, 140, 9061–9065 CrossRef CAS PubMed; (k) P. Gao, C. Yuan, Y. Zhao and Z. Shi, Chem, 2018, 4, 1–11 CrossRef.
- (a) L.-G. Xie and Z.-X. Wang, Angew. Chem., Int. Ed., 2011, 50, 4901–4904 CrossRef CAS PubMed; (b) P. Maity, D. M. Shacklady-McAtee, G. P. Yap, E. R. Sirianni and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 280–285 CrossRef CAS PubMed; (c) T. Moragas, M. Gaydou and R. Martin, Angew. Chem., Int. Ed., 2016, 55, 5053–5057 CrossRef CAS PubMed; (d) D. Y. Wang, Z. K. Yang, C. Wang, A. Zhang and M. Uchiyama, Angew. Chem., Int. Ed., 2018, 57, 3641–3645 CrossRef CAS PubMed; (e) Y.-Q.-Q. Yi, W.-C. Yang, D.-D. Zhai, X.-Y. Zhang, S.-Q. Li and B.-T. Guan, Chem. Commun., 2016, 52, 10894–10897 RSC.
- (a) J. Tsuji, I. Shimizu, I. Minami, Y. Ohashi, T. Sugiura and K. Takahashi, J. Org. Chem., 1985, 50, 1523–1529 CrossRef CAS; (b) K. J. Schwarz, C. M. Pearson, G. A. Cintron-Rosado, P. Liu and T. N. Snaddon, Angew. Chem., Int. Ed., 2018, 57, 7800–7803 CrossRef CAS PubMed; (c) Y. Chen, J. P. Romaire and T. R. Newhouse, J. Am. Chem. Soc., 2015, 137, 5875–5878 CrossRef CAS PubMed; (d) B. Zhang and X. Zhang, Chem. Commun., 2016, 52, 1238–1241 RSC; (e) G. P. Boldrini, M. Mengoli, E. Tagliavini, C. Trombini and A. Umani-Ronchi, Tetrahedron Lett., 1986, 27, 4223–4226 CrossRef CAS.
- (a) T. A. Hamlin, C. B. Kelly, R. M. Cywar and N. E. Leadbeater, J. Org. Chem., 2014, 79, 1145–1155 CrossRef CAS PubMed; (b) J.-P. Bégué, D. Bonnet-Delpon and M. H. Rock, Synlett, 1995, 6, 659–660 CrossRef.
- The addition of external H2O was found to be detrimental to the arylation reaction.
- For the proposed reaction mechanism of arylation step, see the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01966a |
| This journal is © The Royal Society of Chemistry 2019 |