
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstituent-controlled, mild oxidative fluorination of iodoarenes: synthesis and structural study of aryl I(III)- and I(V)-fluorides†
Joel
Häfliger‡
,
Cody Ross
Pitts‡
 ,
Dustin
Bornemann
,
Roland
Käser
,
Nico
Santschi
,
Julie
Charpentier
,
Elisabeth
Otth
,
Nils
Trapp
,
René
Verel
,
Hans Peter
Lüthi
and
Antonio
Togni
*
,
Dustin
Bornemann
,
Roland
Käser
,
Nico
Santschi
,
Julie
Charpentier
,
Elisabeth
Otth
,
Nils
Trapp
,
René
Verel
,
Hans Peter
Lüthi
and
Antonio
Togni
*
Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 2, 8093 Zürich, Switzerland. E-mail: atogni@ethz.ch
First published on 5th June 2019
Abstract
We report a mild approach to the synthesis of difluoro(aryl)-λ3-iodanes (aryl-IF2 compounds) and tetrafluoro(aryl)-λ5-iodanes (aryl-IF4 compounds) using trichloroisocyanuric acid (TCICA) and potassium fluoride (KF). Under these reaction conditions, selective access to either the I(III)- or I(V)-derivatives is predictable based solely on the substitution pattern of the iodoarene starting material. Moreover, the discovery of this TCICA/KF approach prompted detailed dynamic NMR, kinetic, computational, and crystallographic studies on the relationship between the IF2 group and the ortho-substituents on carefully designed probe molecules. It was during these experiments that the role of the ortho-substituent in inhibiting further oxidative fluorination of I(III)-compounds to I(V)-compounds during the reaction with TCICA and KF was revealed. Additionally, a notable exception to this empirical trend is discussed herein.
Oxidative fluorination chemistry has often stipulated the use of harsh reagents. Regarding the fluorination of aryl iodides, synthetic limitations arguably have had significant downstream effects on the ability to study structure and reactivity of difluoro(aryl)-λ3-iodanes (aryl-IF2 compounds) and, to an even greater extent, tetrafluoro(aryl)-λ5-iodanes (aryl-IF4 compounds) in the last several decades.1 Aside from two important exceptions from the Shreeve2 and Gilmour3 laboratories (employing Selectfluor in the synthesis of electron-rich aryl-IF2 compounds), the vast majority of methods for aryl-IF2 synthesis rely on F2, a source of HF, or a number of other notoriously hazardous reagents (e.g. SF4, XeF2, etc.).1,4,5 Aryl-IF4 synthesis is equally if not more challenging.1,6
As one possible solution to this accessibility problem, we present a mild, safe, and inexpensive oxidative fluorination of aryl iodides that requires only trichloroisocyanuric acid (TCICA) and potassium fluoride. Somewhat serendipitously, we found that the selectivity between aryl-IF2 and aryl-IF4 formation can be controlled reliably by the substitution pattern of the arene (Fig. 1). This discovery was made during an in-depth study on the relationship between the IF2 group and ortho-substituents, whereby we conducted several variable-temperature NMR, computational, and crystallographic analyses, reported herein.
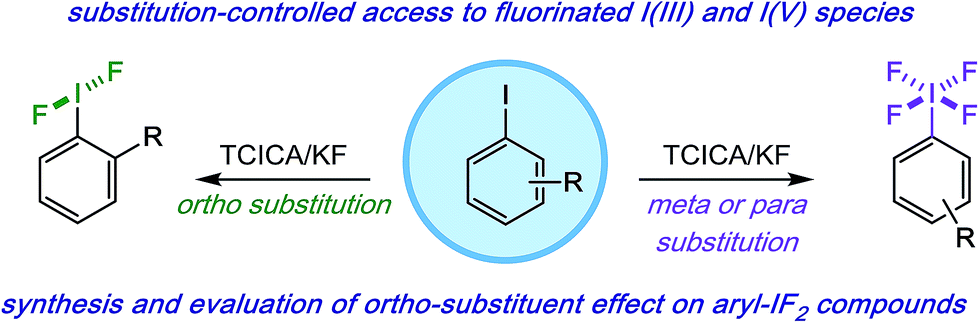 | ||
| Fig. 1 Mild and predictable access to aryl-IF2 and aryl-IF4 compounds based on iodoarene substitution pattern. | ||
Reaction design and optimization
Originally, the reaction design was inspired by our recent foray into aryl-SF4Cl compound synthesis, whereby we accomplished a mild, gas reagent-free oxidative fluorination of diaryl disulfides using TCICA, KF, and catalytic TFA.7 Using a similar approach, we began screening with iodobenzotrifluoride isomers: this would potentially allow analysis of the material balance of the reaction mixtures by 19F NMR. After stirring the reaction mixtures at rt in MeCN for 24 h using TCICA (1.0 equiv.), KF (3.0 equiv.), and TFA (10 mol%), we made two important observations: (1) when making the NMR samples in the fume hood (i.e. not under rigorously air- and moisture-free conditions), we noticed rapid formation of a precipitate in the filtered samples that employed the meta- and para-iodobenzotrifluoride isomers as substrates, but not in the ortho-isomer sample (specifically upon addition of an internal standard solution with slightly wet CD3CN) and (2) although 19F NMR analysis indeed showed formation of the corresponding difluoro(aryl)-λ3-iodanes (i.e. aryl-IF2 compounds) in all cases, the products were observed in only trace yields for the meta- and para-isomers and in about 20% for the ortho-isomer, the majority of the observable material balance being unreacted starting material.At the time, we assumed the identity of the precipitate was the corresponding iodosoarene for each the meta- and para-iodobenzotrifluoride isomers. In this light, our observations suggested that the ortho-substituted aryl-IF2 compound may be less susceptible to degradation, and thus easier to handle during the screening process. Accordingly, we continued to screen for optimized conditions using ortho-iodobenzotrifluoride 1 (Table 1) (notably, it was not until much later in the study that we determined the precipitate was not primarily a byproduct of aryl-IF2 compound hydrolysis, but rather a byproduct of aryl-IF4 compound hydrolysis, as discussed in more detail below. While our initial assumption was erroneous, the following series of experiments led us to this discovery of the effect of substitution on oxidative fluorination).
|
|
|||||
|---|---|---|---|---|---|
| Entry | TCICA (equiv.) | KF (equiv.) | Additive | Temp. (°C) | Yield (%) |
| a Yields determined by 19F NMR using fluorobenzene as an internal standard. b MeCN not dry. | |||||
| 1 | 2.0 | — | — | rt | — |
| 2 | 2.0 | 5.0 | — | rt | 29 |
| 3 | 2.0 | 5.0 | 0.1 TFA | rt | 27 |
| 4 | 4.0 | 5.0 | — | rt | 49 |
| 5 | 6.0 | 5.0 | — | rt | 79 |
| 6 | 6.0 | 7.0 | — | rt | 81 |
| 7 | 6.0 | 3.0 | — | rt | 75 |
| 8 | 2.0 | 3.0 | — | 50 | 61 |
| 9 | 4.0 | 5.0 | — | 50 | 62 |
| 10 | 4.0 | 6.0 | — | 40 | 94 |
| 11 | 6.0 | 5.0 | — | 40 | 26b |
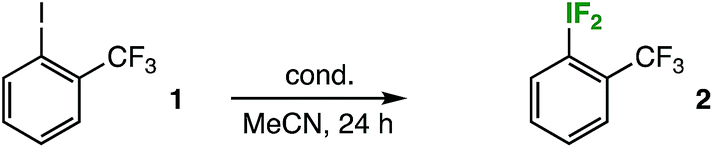
Immediately, we discovered that although an acid catalyst was beneficial for aryl-SF4Cl formation in our previous work, it had no positive impact on aryl-IF2 (2) product formation. Yet, we found that using TCICA and KF in slight excess had a considerable impact, bringing the yield up to 79% at room temperature in the absence of a catalyst. Next, we attempted the reaction at slightly elevated temperatures. To our surprise, the aryl-IF2 product 2 not only survived heating the reaction mixture to 40 °C in a borosilicate vial for 24 h, but it formed in 94% yield by 19F NMR (using 4.0 equiv. TCICA and 6.0 equiv. KF).
We proceeded to screen with these optimized conditions. Additionally, we found that: (1) the reaction does not proceed in solvents such as DMF, DCM, EtOAc, and toluene, and the product yield is poor when the MeCN is not dry; (2) N-chlorosuccinimide and N-chlorophthalimide are not suitable replacements for TCICA under optimized conditions (i.e. no aryl-IF2 product formation was observed in either case); (3) reaction times of less than 24 h (e.g. 16 h) may result in inferior product yields; and (4) the aryl-IF2 product, over 2–3 days, decomposes more quickly when exposed to light in a filtered solution of the crude reaction mixture in CD3CN at room temperature.
Substrate scope of difluoro(aryl)-λ3-iodanes and the importance of ortho-substitution
The most remarkable aspect of the optimization process was the consistent absence of the expected decomposition product – 2-iodosobenzotrifluoride – as determined by 19F NMR and lack of precipitate formation in the NMR tube. Suspecting that the substitution pattern plays an important role, we investigated the substrate scope by first holding the ortho-trifluoromethyl substituent constant (Table 2).
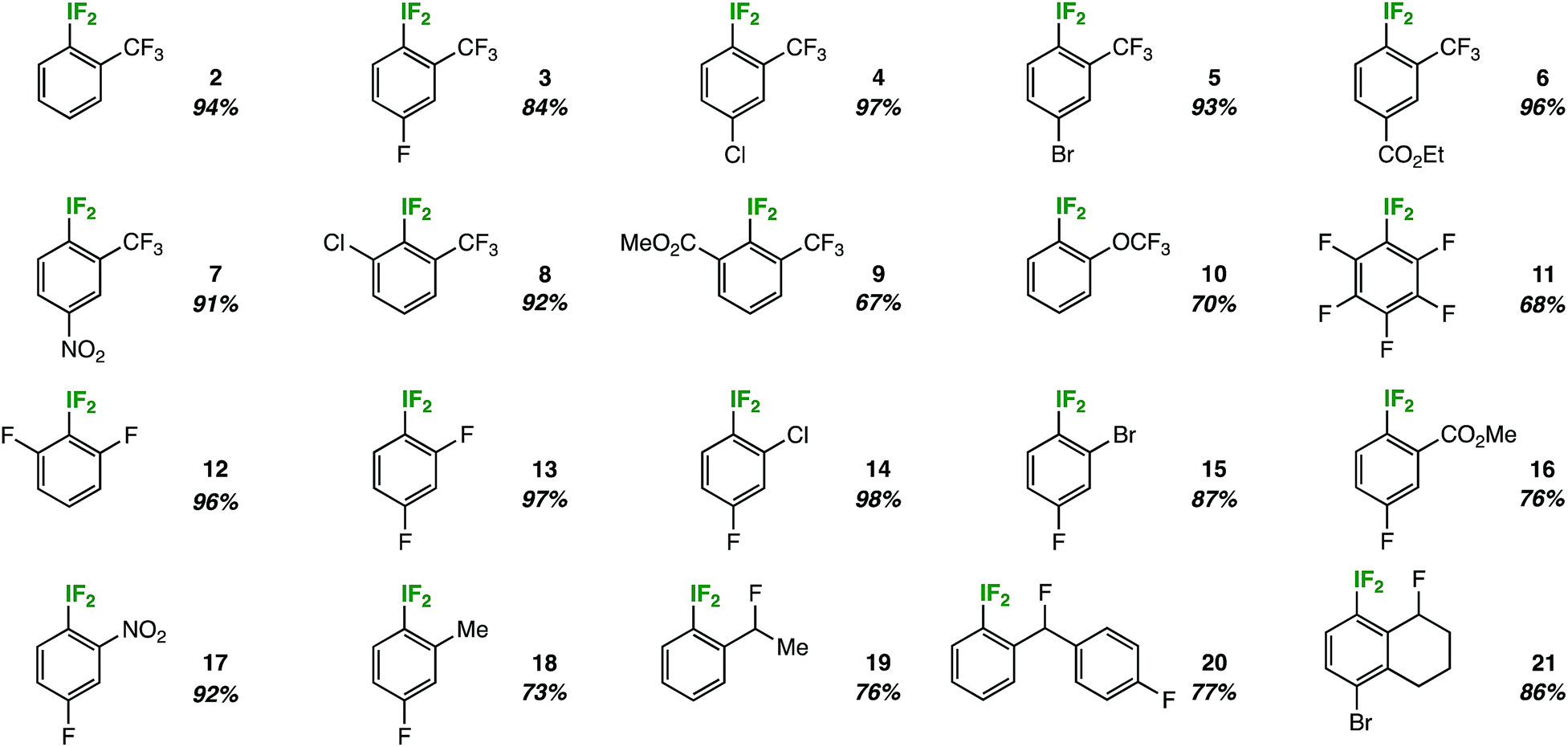
|
In doing so, we noted that the reaction tolerates substrates adorned with additional electron-withdrawing substituents, such as fluorine, chlorine, bromine, esters, and nitro groups (compounds 3–7). Also, ortho-disubstitution does not inhibit aryl-IF2 formation, as demonstrated by products 8 and 9. This substrate class has been popularized recently as a privileged platform to access chiral aryl-IF2 reagents.8 On the other hand, the reaction does not tolerate strong electron-donating groups, as these substrates are prone to competitive ring and/or benzylic chlorination in the presence of TCICA.9 Also, aryl-IF2 product formation is not observed (or observed in very poor yield) in the presence of substituents with acidic protons, such as carboxylic acids or secondary amides.
Beyond ortho-trifluoromethyl substitution, we also discovered that aryl-IF2 compounds containing other fluorinated substituents, such as ortho-trifluoromethoxy substituents (10) or ortho-difluoro substituents (11 and 12), may be synthesized in good yields under our conditions (Table 2).
Subsequently, we continued to vary systematically the ortho-substituent to electron-withdrawing groups of various sizes, such as halogens (compounds 13–15), an ester (compound 16), and a nitro group (compound 17). The reaction performed well in all cases. Additionally, we found that mild donating groups (e.g. a methyl group) in the ortho-position are compatible under reaction conditions. That is, no notable background benzylic or ring chlorination was observed in the synthesis of compound 18.
In nearly all cases, no decomposition or precipitation was observed upon analyzing the aryl-IF2 products in Table 2, which is in stark contrast to the aforementioned meta- and para-substituted substrates. This prompted an important question: is ortho-substitution, in general, somehow important in relation to the hydrolytic stability of aryl-IF2 compounds?
Probing an IF2 interaction
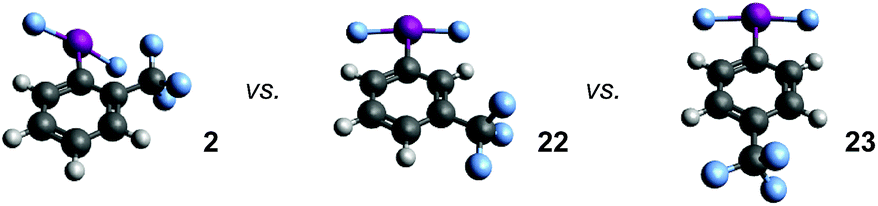
Preliminary evidence for an interaction between the IF2 group and the ortho-substituent stems from the observed J-coupling of 3.2 Hz between the IF2 fluorine atoms and CF3 fluorine atoms in the 19F{1H} NMR spectrum of compound 2. Note that we also observed a J-coupling of 2.3 Hz between the IF2 fluorine atoms and the ortho-fluorine atoms in compound 12. Although initial attempts to clarify the results through 19F–19F NOESY experiments were unsuccessful, these J-values are likely attributed to a lone-pair interaction.We examined the relationship between the IF2 and CF3 group in compound 2 further using DFT calculations. Geometry optimizations were performed using Gaussian10 at the wB97xD/cc-pvdz level of theory,11 using a cc-pvdz-PP basis set for the iodine atom,12 on the ortho-, meta-, and para-trifluoromethyl aryl-IF2 isomers 2, 22, and 23. Interestingly, compound 2 is predicted to be the least thermodynamically stable by 9.8 kcal mol−1 (for 22 and 23, ΔE ≈ 0), which rules out the possibility of a stabilizing interaction between the fluorine atoms on the CF3 group and the iodine atom. A closer look at the structures reveals similar bond distances, angles, and natural charges predicted for all isomers, but one property is drastically different – the Cortho–Cipso–I–F dihedral angle (Table 3). In compounds 22 and 23, the lowest energy conformations both have dihedral angles of virtually 0°, while the dihedral angle of compound 2 is 40° – forced out of plane with the ring. In this particular instance, the preferred angle of 0° in 22 and 23 calculated in the gas phase is likely attributed to a favorable interaction between the iodine lone pairs and the π-system, which the ortho-substituent in 2 seems to hinder. The calculated angles notably differ from the preferred orientation of the IF2 group (and many related IX2 groups) in a number of X-ray crystal structures.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Subs. | d(C–I) | d(I–F) | q C | q I | q F | θ C–I–F | θ F–I–F | ϕ C–C–I–F |
| a Calculations peformed at wB97xD/cc-pvdz, with a cc-pvdz-PP basis set used for the iodine atom. C refers to ipso carbon atom and F refers to fluorine atom bound to iodine, unless otherwise specified. b Dihedral angle of Cortho–Cipso–I–F. c Average between 2.00 and 2.02 (not equal bond lengths). d Average between −0.621 and −0.633. e Average between 85° and 87°. f F atom proximal to substituent (dihedral angle is 38° for distal F). g Structure is nearly symmetric about F–I–F (mirror plane). h Structure is symmetric about F–I–F (mirror plane). | ||||||||
| o-CF3 | 2.13 Å | 2.01 Åc | −0.248 | 1.419 | −0.627d | 86°e | 172° | 40°f |
| m-CF3 | 2.13 Å | 2.02 Åg | −0.251 | 1.409 | −0.628 | 87° | 174° | 0° |
| p-CF3 | 2.13 Å | 2.03 Åh | −0.300 | 1.502 | −0.649 | 87° | 175° | 0° |
Overall, this provides little indication of what is going on in solution, but suggests that the CF3 group may, in fact, have an influence on the preferred orientation of the IF2 group. Specifically, in solution, this could affect the rotation about the C–I bond. One postulate could be that if the preferred IF2 orientation is out of the plane of the ring, this would leave the iodine atom less susceptible to nucleophilic attack, thus offering one explanation for our observations thus far. An analogy may be drawn to the stabilizing effects of the ortho-methyl groups on the SF3 moiety in Fluolead, for instance.13
Dynamic NMR probe
In order to examine the influence of the ortho-substituent on the conformation of the IF2 group in solution, we designed probe molecules suitable for variable-temperature NMR (VT-NMR) studies. Initially, we synthesized compounds 1914 and 2014a,15 (Table 2), each with a stereogenic center and fluorine atom in the benzylic position, as possible probes. Theoretically, if the C–I bond in these molecules is not freely rotating, the fluorine atoms on the IF2 moiety would become diastereotopic, thus leading to two spectroscopically distinct 19F NMR signals.In MeCN, no dynamic behavior was observed in the IF2 signal by 19F NMR down to approximately 233 K for compound 19. However, this result may be complicated by free rotation about the Cortho–Cbenzylic bond. In an attempt to limit this rotation, we replaced the methyl group in the benzylic position with a larger, aryl substituent (compound 20); still, no dynamic behavior was observed down to 233 K.
Assuming rotation about the Cortho–Cbenzylic bond was, indeed, still complicating the picture, we synthesized a less-trivial tetrahydronaphthalene derivative 21 (Table 2) in order to eliminate this variable by locking the benzylic position into place with a more rigid ring structure (Fig. 2, top panel). This probe molecule 21 was made in 8 steps in 9% overall yield from commercially available 5,6,7,8-tetrahydro-1-naphthylamine 24 (see ESI† for details).14,16 Not only was it designed to inhibit Cortho–Cbenzylic bond rotation, but 21 was also adorned with an electron withdrawing group (i.e. a bromine atom) in the 4-position to deactivate the aromatic ring and benzylic position distal to the IF2 moiety toward background chlorination from TCICA (Fig. 2, bottom panel).
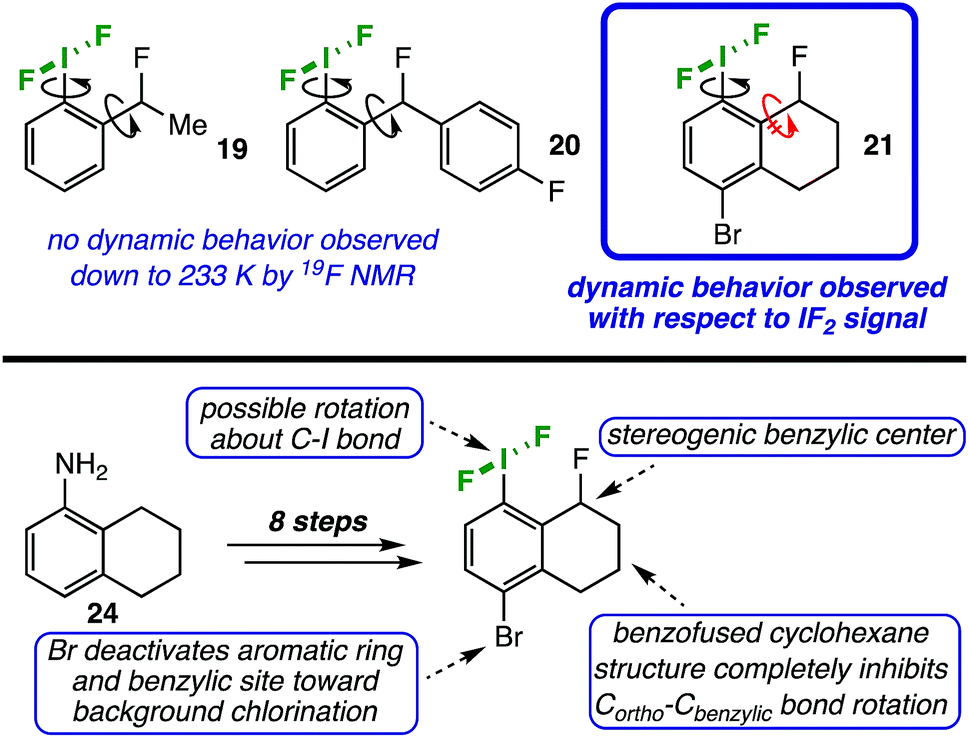 | ||
| Fig. 2 (Top panel) Probe molecules used to investigate rotation of IF2 moiety about C–I bond via VT-NMR. (Bottom panel) Highlighting design features of probe molecule 21. | ||
To our satisfaction, we observed significant line broadening of the IF2 signal in 21 upon cooling the sample and approached the coalescence temperature, which was likely just below 233 K. As this is near the freezing point of the solvent, we concentrated the reaction mixture and redissolved the residue in deuterated dichloromethane to access lower temperatures.
Upon cooling a solution of 21 in CD2Cl2, we determined a coalescence temperature (TC) of 228 K and observed complete separation of the “IF2” 19F NMR signal into two distinct doublets with a clear roofing effect at temperatures below 202 K (Fig. 3).17 Here, the fluorine atoms bound to iodine are seen as diastereotopic on the NMR time-scale with 2JFF = 98.3 Hz, and the distance between the two signals, i.e. |(νA − νB)|, is 1152 Hz. Note that no coupling constant between the benzylic fluorine atom and either of the iodine-bound fluorine atoms is observable at any temperature within our data set; however, as this coupling constant is expected to be small (ca. 2–3 Hz), it would likely be subsumed in the broader parent signals.
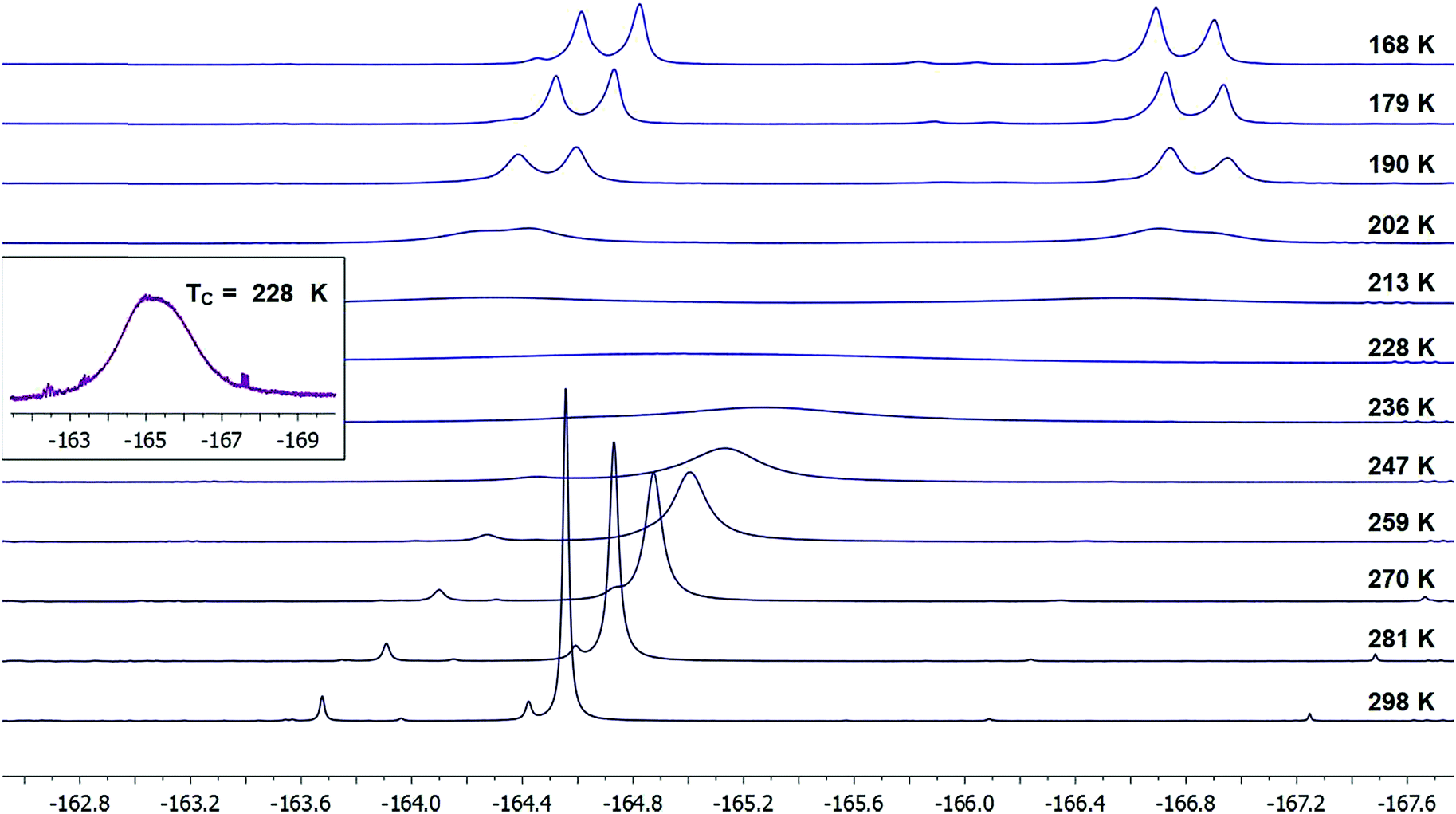 | ||
| Fig. 3 Stacked 19F NMR spectra of the IF2 signal in probe molecule 21 over a range of temperatures from 298 K to 168 K. A zoomed-in spectrum is displayed for the coalescence temperature (TC) at 228 K. | ||
Additionally, it is important to state that we did not observe dynamic activity of either the benzylic fluorine or hydrogen atoms by either 19F or 1H NMR at any temperature down to 168 K, indicating that the benzo-fused cyclohexane ring-flipping dynamics have a negligible impact on the IF2 dynamics.
At temperatures below 202 K, we also noted an apparent convergence of the two IF219F signals down to 168 K, with |(νA − νB)| = 989 Hz. This is likely due to a more dramatic temperature-dependent shift of one of the fluorine signals over the other.18 Although this phenomenon is not surprising, it complicates the accurate determination of rate constants (k), and thus activation energy (Ea), as the equations for the T = TC and T > TC temperature regimes both depend on the maximum chemical shift difference |(νA − νB)| (see ESI† for details).19 Accordingly, we have analyzed our data using two different assumptions: (1) |(νA − νB)| = signal separation we observed at lowest measurable temperature (989 Hz) and (2) |(νA − νB)| = largest signal separation we observed (1152 Hz) in our data set. Under these assumptions, we have determined activation energies from Arrhenius analyses, respectively, of 7.0 ± 0.9 and 7.4 ± 0.9 kcal mol−1. We also note that respective enthalpies of activation (ΔH‡) of 6.5 ± 0.9 and 6.9 ± 0.9 kcal mol−1 and entropies of activation (ΔS‡) of −13 ± 4 and −11 ± 4 cal mol−1 K−1 were calculated for each assumption via Eyring plots. Considering there is a convergence of the Δ|(νA − νB)| that is exacerbated at lower temperatures, we believe that assumption (2) may be more appropriate, though both assumptions lead to values that are very close in agreement.
Although these thermodynamic parameters presumably describe rotation about the C–I bond, one must consider alternative mechanisms. For one, we ruled out the possibility of an intermolecular exchange mechanism by performing the VT-NMR studies at different concentrations (see ESI† for details).20 A more challenging issue to address, however, is pseudorotation. Very few detailed dynamic NMR studies on I(III) compounds (and, notably, none on aryl-IF2 compounds) have surfaced in the literature, but one can attribute the observed dynamic activity of diacetoxyaryl- and triaryl-substituted iodanes to pseudorotation in the works of Reich and Cooperman21 and Ochiai and coworkers,20 respectively. Their probe molecules are incomparable to compound 21 in terms of ligand substitution patterns, core skeletons, etc.; thus, there can be no straightforward comparison of our experimentally-determined thermodynamic values to the existing literature. In order to probe the likelihood of C–I rotation being the relevant mechanism over pseudorotation for our aryl-IF2 model compound 21, we turned to DFT calculations.
DFT analysis of rotational barrier
Compound 21 was optimized at the wB97xD/cc-pvdz level of theory (cc-pvdz-PP for the iodine atom), and then the IF2 group was rotated 180° about the C–I bond in 12° increments. The structure was relaxed at each step of the rotation scan to determine the lowest energy conformations. From the plotted potential curve, we calculated an activation energy of 5.9 kcal mol−1 that comports well with our experimental value.For the sake of comparison, we were able to find an alternative iodane structure on the potential energy surface (with a Cipso–I–F three-centered bond) that represents a plausible pseudorotation intermediate (21-iso);22 it is 25 kcal mol−1 higher in energy than the optimized F–I–F three-centered bond structure (Fig. 4). This number serves as a lower-bound estimate of the barrier to pseudorotation, which would be prohibitively high in energy. Therefore, we can conclude that the observed dynamic activity more likely arises from C–I bond rotation.
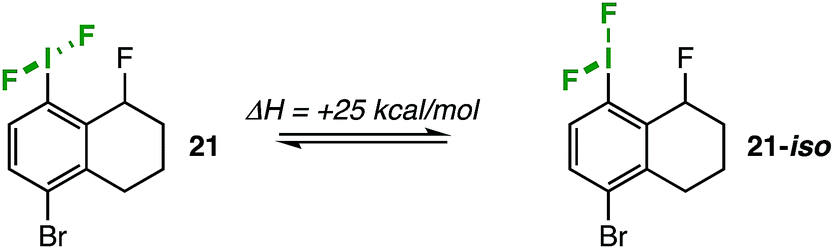 | ||
| Fig. 4 Isodesmic relationship at wB97xD/cc-pvdz (cc-pvdz-PP for the iodine atom). | ||
Solid-state structure of 21
In addition to analyzing probe molecule 21 in solution, we obtained single crystals suitable for X-ray structure determination (Fig. 5). The typical, slightly distorted T-shape arrangement about the iodine atom was observed, with θF–I–F = 171.9°, an average θC–I–F = 86.0°, d(C–I) = 2.097 Å, and d(I–F) = 1.998 Å. The Cortho–Cipso–I–F torsion angle (ϕC–C–I–F) is 82.9°, placing the IF2 group out of plane with the arene, but not quite perpendicular to it.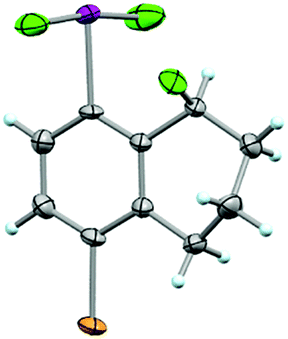 | ||
| Fig. 5 Crystal structure of compound 21 determined from single-crystal X-ray diffraction (displacement ellipsoids depicted at 50% probability level). | ||
Although it is plausible the ortho-substituent influences the orientation of the IF2 group here, it is difficult to separate this phenomenon from packing effects. For instance, only a small number of aryl-IF2 molecular structures have been reported in the CCDC to date, and there are no clear trends yet, for instance, in Cortho–Cipso–I–F torsion angles based on substitution patterns (i.e. presence vs. absence of ortho-substituents, considering ortho-substituents of various sizes, etc.).23 Thus, in the solid state, the orientation of the IF2 group is more likely governed by various interactions between the fluorine and iodine atoms of neighboring molecules in the extended lattice than arene substitution pattern (e.g. intermolecular C–I⋯F–I contacts of 2.888 Å and I–F⋯F–I contacts of 2.891 Å were observed in the packing motif of our probe molecule – note ΣrV[I,F] = 3.45 Å and ΣrV[F,F] = 2.94 Å – and others have reported similar short contacts in aryl-IF2 structures). It is also worth mentioning that no short intermolecular contacts involving the Br substituent in 21 were observed.
An unexpected role of the ortho-substituent
Although it is clear that the ortho-substituent has an influence on the IF2 group in solution (and potentially in the solid state) from the experiments outlined above, it is not completely understood whether the ortho-substituent actually influences the hydrolytic stability of aryl-IF2 compounds. Thus, we performed a series of controlled hydrolysis experiments, whereby the NMR samples were prepared first under Ar and rigorously dry conditions, then flooded with water immediately prior to the measurements in an attempt to simulate pseudo-first order decays of the aryl-IF2 compounds.24Initially, we varied systematically the size and electronegativity of the ortho-substituents, performing the hydrolysis experiments on compounds 3, 13–15, and 17 and 18. However, the precipitation of the byproducts seriously complicates the picture (even at lower concentrations), so it was not possible to obtain reliable, quantitative kinetic information or to establish definitive trends. Yet, qualitatively, we can at least note that the ortho-methyl-substituted compound 18 appears to decay at a similar rate to either the ortho-chloro- or ortho-bromo-substituted compounds 14 and 15 (see ESI† for details). This is an indication that, with respect to hydrolytic stability, the size of the ortho-substituent could play a more important role than electronegativity.
In addition, we varied the position of the trifluoromethyl substituent in the CF3-substituted aryl-IF2 compounds, i.e. investigating compounds 2, 22, and 23. If our hypothesis that the presence of an ortho-substituent has a strong positive effect on hydrolytic stability is correct, then one would expect that compound 2 would decay at the slowest rate. Surprisingly, this is not the case. It appears the relative rates of decay based on substitution are: meta-CF3 > ortho-CF3 > para-CF3 (with the para-CF3 isomer being the most stable of the three). Based on a thermodynamic argument alone, one would expect the ortho-CF3 isomer to decay the fastest, but this is also not the case. Thus, the ortho-substituent may, in fact, have a positive kinetic influence on hydrolytic stability, but it is not nearly as substantial as we anticipated at the outset of this study.
However, by analyzing the reaction mixtures under rigorously air- and moisture-free conditions during the hydrolysis experiments (prior to water addition), we unveiled an interesting and unexpected result when investigating the meta- and para-substituted substrates: formation of tetrafluoro(aryl)-λ5-iodanes (aryl-IF4 compounds). Aryl-IF4 compounds are remarkably more sensitive toward hydrolysis than aryl-IF2 compounds, so extremely careful preparation of the NMR samples was necessary in order to observe them (admittedly, that is how we had completely overlooked this finding earlier in this study). Thus, our initial assumption that the identity of the precipitate forming in the NMR samples prepared in air was strictly the iodosoarene (from hydrolysis of aryl-IF2) was incorrect; the precipitate is actually made up of a more significant amount of iodylarene formation for samples employing meta- and para-substituted substrates.
This key observation puts the role of the ortho-substituent into a new perspective. The hydrolysis experiments suggest that the ortho-substituent only has a minor positive influence on hydrolytic stability of aryl-IF2 products once they are formed, but it plays a more noteworthy role during the reaction with TCICA and KF in inhibition of further oxidation of iodine(III) to iodine(V).
Substrate scope of tetrafluoro(aryl)-λ5-iodanes
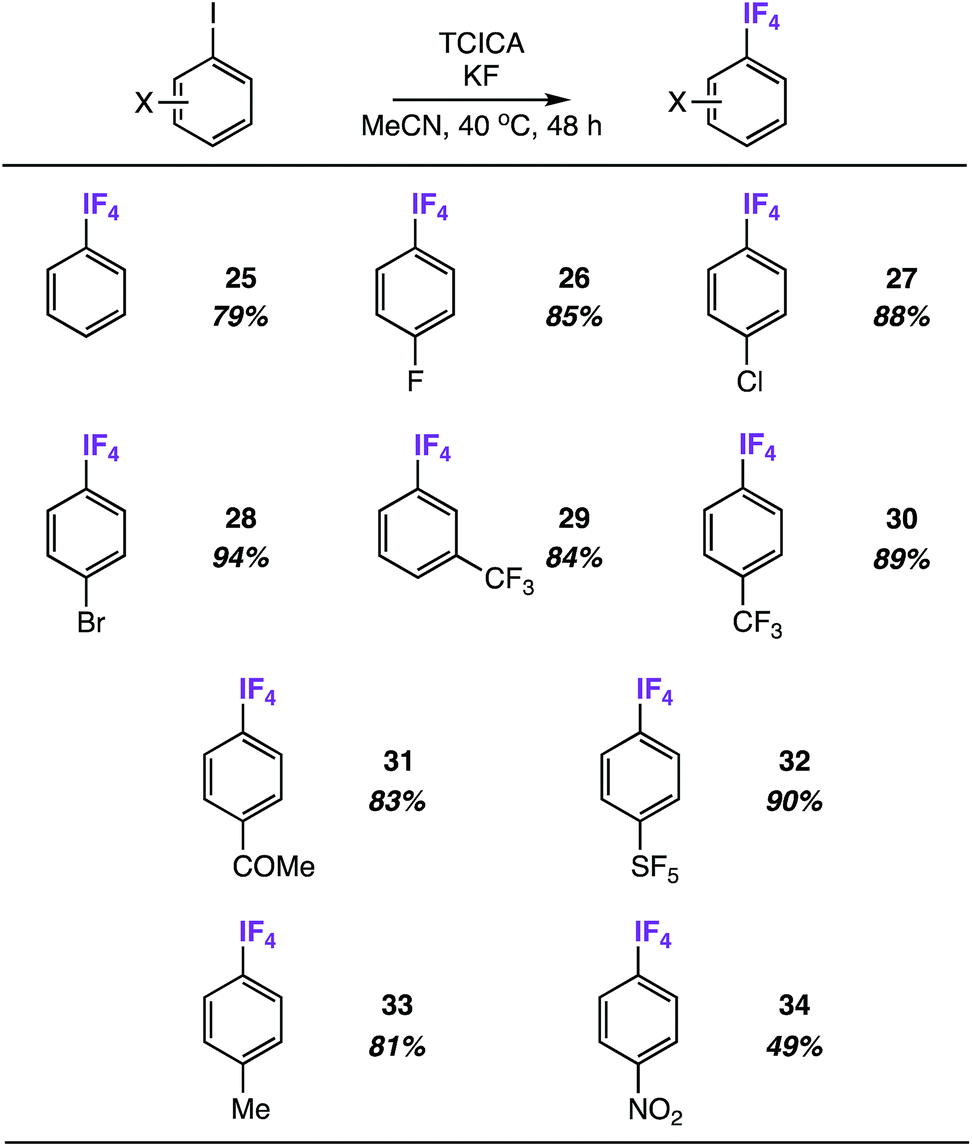
We discovered quickly that satisfactory yields of meta- and para-substituted aryl-IF4 compounds can be obtained under nearly identical reaction conditions to those in Table 2 (i.e. 4.0 equiv. TCICA and 6.0 equiv. KF in MeCN at 40 °C) by simply letting the reactions stir for 48 h instead of 24 h.Under these conditions, we synthesized a variety of aryl-IF4 compounds in good yields (79–94%, Table 4). As anticipated, the functional group compatibility is similar to what we noted in Table 2: aryl-IF4 formation from the unsubstituted iodobenzene (compound 25) and iodoarenes substituted with either electron-withdrawing groups (e.g. halogens, ketones, trifluoromethyl, and pentafluorosulfanyl groups in compounds 26–32) or mild electron-donating groups (e.g. a methyl group in compound 33) proceeds smoothly. On the other hand, the substrate with the most extreme electron-withdrawing group – a para-nitro group – only provided compound 34 in 49%.
| a Yields determined by 19F NMR using fluorobenzene or benzotrifluoride as an internal standard. |
|---|
|
Remarkably, only one ortho-fluorine substituent is enough to stifle aryl-IF4 formation. For instance, letting 2,4-difluoro-iodobenzene stir for 48 h under reaction conditions results in less than 10% of the corresponding aryl-IF4 product by 19F NMR (note the IF4 signal was identified at −21.21 ppm in the 19F{1H} spectrum as a doublet with a J-coupling of 19.2 Hz to the ortho-fluorine atom). In nearly all other instances when the ortho-substituent is larger than a fluorine atom, the aryl-IF2 product is formed exclusively.
We also discovered that these aryl-IF4 compounds, although less hydrolytically stable than aryl-IF2 compounds, may be more easily extracted from the reaction mixture by comparison (see ESI† for details). For instance, we extracted compound 26 from the reaction mixture under N2 atmosphere in a glovebox using n-hexane and obtained a white solid in 77% isolated yield.
From this, single crystals were grown that proved suitable for X-ray structure determination (Fig. 6). As anticipated, a slightly distorted square pyramidal geometry was observed about the iodine atom with the arene in the apical position. In all, the structure displays similar features to the aryl-IF4 compounds originally reported by Frohn25 and Seppelt26 (e.g. d(C–I) = 2.078 Å, average d(I–F) = 1.939 Å, average trans θF–I–F = 170.1°, average cis θF–I–F = 89.6°, average θC–I–F = 85.1°, ϕC–C–I–F = 44.7°, and short intermolecular I⋯F and F⋯F contacts can be observed in the packing motif).
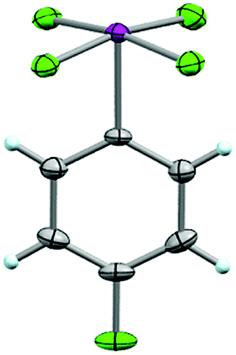 | ||
| Fig. 6 Crystal structure of compound 26 determined from single-crystal X-ray diffraction (displacement ellipsoids depicted at 50% probability level). | ||
Synthesis of another aryl-λ5-iodane: an ortho-substituted exception
Inspired by the known reactivity of TCICA and KF in the synthesis of fluoroiodane 36 from alcohol 35 (Fig. 7),27 we also wondered if 36 could be further oxidized to I(V) compound 37 under our reaction conditions. Compound 37 was originally synthesized by Amey and Martin from 35 using excess CF3OF, and it was briefly explored as an oxidant.28 More recently, it was synthesized in our laboratory using either XeF2 or Cl2/KF.29 The TCICA/KF approach would make 37 significantly more accessible for future studies.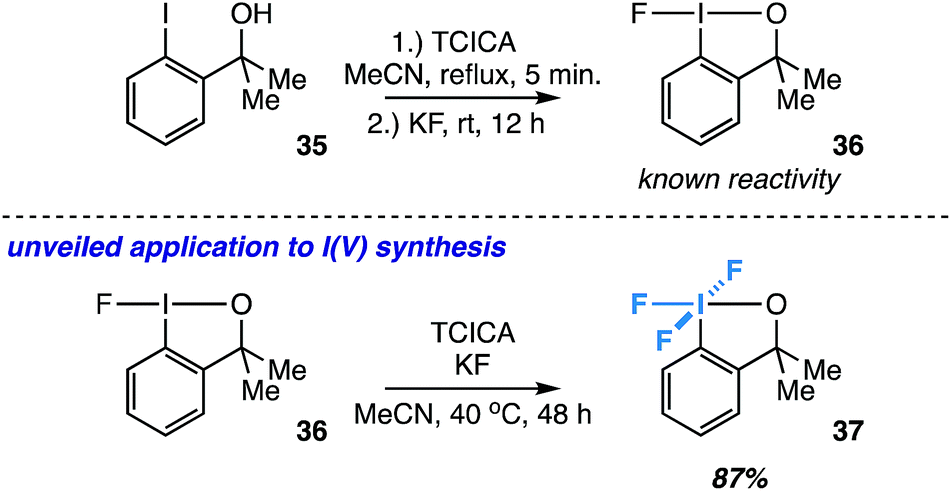 | ||
| Fig. 7 Extended application to the synthesis of an ortho-substituted I(V) compound (37) as an exception to the rule. Yield determined by 19F NMR. | ||
To our satisfaction, fluoroiodane 36 undergoes further oxidative fluorination under TCICA/KF conditions to provide 37 in 87% yield by 19F NMR (Fig. 7). Moreover, under identical reaction conditions, alcohol 35 can also be converted into 37 directly in 69% yield. In all, our synthesis of 37 indicates that the TCICA/KF approach can be useful in the synthesis of fluorinated I(V)-compounds beyond aryl-IF4, and it presents a notable “exception to the rule” that ortho-substituted iodoarenes show higher selectivity for the I(III) products over the I(V) products under these reaction conditions.
Solid-state structure of 37
Additionally, single crystals of 37 suitable for X-ray structure determination were analyzed that display some interesting features (Fig. 8) (note that there were two symmetry-independent moieties in the asymmetric unit). Due to the participation of the oxygen atom on the ortho-substituent in a three-centered bonding interaction, the average Cortho–Cipso–I–Ftrans torsion angle (ϕC–C–I–Ft) is 171.9° and the average Cortho–Cipso–I–Fcis torsion angle (ϕC–C–I–Fc) is 87.4°. This is a noteworthy deviation from the preferred orientation of the IF4 moiety in aryl-IF4 compounds such as 26, whereby ϕC–C–I–F ≈ 45°.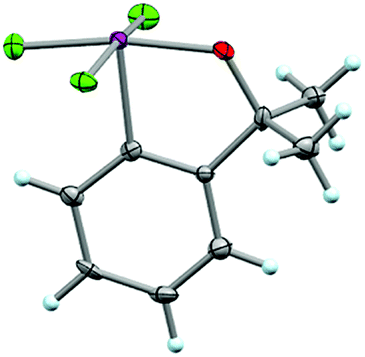 | ||
| Fig. 8 Crystal structure of compound 37 determined from single-crystal X-ray diffraction (displacement ellipsoids depicted at 50% probability level, only one of two asymmetric moieties from asymmetric unit shown). | ||
Also, the average C–I bond distance (2.072 Å) is in close accord with aryl-IF4 compound 26, but the I–F bond distances in 37 are slightly longer, i.e. average d(I–Fcis) = 1.963 Å and average d(I–Ftrans) = 1.979 Å (the latter bond conceivably elongated due to the greater trans effect of the oxygen atom; note that the average d(O–I) = 1.924 Å).30 However, for another comparison, both the I–Ftrans and O–I bonds in 37 are significantly shorter than those in the fluoroiodane precursor 36 (reported as 2.073 Å and 2.033 Å, respectively).29
We also noted that the bond angles across the three-centered bonds, on average (trans θF–I–F = 168.5° and trans θO–I–F = 167.9°), deviate from linearity to a greater extent than aryl-IF4 compound 26 (trans θF–I–F = 170.1°), but slightly less than that of fluoroiodane 36 (trans θO–I–F = 167.9°) (additional details about bond angle comparisons can be found in the ESI†). Lastly, it is important to mention that the crystal structure of 37 displayed short intermolecular I⋯F and F⋯F contacts in the packing motif and the tendency to form similar “chain” structures as Frohn,25 Seppelt,26 and we have observed (see ESI†).
Conclusions
Remarkably, this TCICA/KF approach to oxidative fluorination of iodoarenes can be employed to synthesize both aryl-IF2 and aryl-IF4 compounds. Under our reaction conditions, access to either the I(III)- or I(V)-derivatives is controlled primarily by the substitution pattern on the iodoarene substrate, i.e. an ortho-substituent inhibits further conversion of aryl-IF2 to the corresponding aryl-IF4 compound. An exception to this trend was also presented in the form of compound 37, whereby the ortho-substituent participates in a three-centered bonding interaction with the iodine atom, thus making the I(V)-derivative accessible. Moreover, this method arguably presents the mildest synthesis of aryl-IF4 compounds reported to date, as well as the mildest approach to electron-deficient aryl-IF2 compounds, as a complement to the methods of Shreeve and Gilmour that can be used to access more electron-rich aryl-IF2 compounds using Selectfluor.Beyond the development of the TCICA/KF approach as an oxidative fluorination method, this study also raised several questions about the relationship between the IF2 group and ortho-substituents on an arene. This resulted in a series of controlled hydrolysis experiments, computational studies, X-ray crystallographic analyses, and the synthesis of a probe molecule that allotted the first detailed dynamic NMR study on rotation about the C–I bond on substituted aryl-IF2 compounds, from which, rotational barriers and thermodynamic parameters were reported herein.
In all, we hope that our easy access to and increased structural understanding of aryl-IF2 and aryl-IF4 compounds in both solution and the solid state will stimulate more research in this area. Future studies will be focused on studying the oxidative fluorination mechanism, as well as exploring applications of aryl-IF2 and aryl-IF4 compounds as possible reagents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the ETH transfer office for support in filing a patent application on this work, in which C. R. P., N. S., and A. T. are listed as inventors. MoBiAS (ETH) is acknowledged for assistance with HRMS analyses. Financial support was provided by ETH Zürich and the ETH Postdoctoral Fellowship Program (C. R. P.).Notes and references
- V. V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure and Synthetic Applications of Polyvalent Iodine Compounds, John Wiley & Sons, West Sussex, UK, 2014 Search PubMed.
- C. Ye, B. Twamley and J. M. Shreeve, Org. Lett., 2005, 7, 3961 CrossRef CAS PubMed.
- J. C. Sarie, C. Thiehoff, R. J. Mudd, C. G. Daniliuc, G. Kehr and R. Gilmour, J. Org. Chem., 2017, 82, 11792 CrossRef CAS PubMed.
- For reviews, see: (a) P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123 CrossRef CAS; (b) V. V. Zhdankin, ARKIVOC, 2009, 1 Search PubMed; (c) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS PubMed.
- For some examples, see: (a) M. Zupan and A. Pollak, J. Fluorine Chem., 1976, 7, 445 CrossRef CAS; (b) I. Ruppert, J. Fluorine Chem., 1980, 15, 173 CrossRef CAS; (c) D. Naumann and G. Rüther, J. Fluorine Chem., 1980, 15, 213 CrossRef CAS; (d) K. Alam and A. F. Janzen, J. Fluorine Chem., 1987, 36, 179 CrossRef CAS; (e) S. Hara, M. Yoshida, T. Fukuhara and N. Yoneda, Chem. Commun., 1998, 965 RSC; (f) V. Padelidakis, W. Tyrra and D. Naumann, J. Fluorine Chem., 1999, 99, 9 CrossRef CAS; (g) H. J. Frohn and V. V. Bardin, J. Fluorine Chem., 2005, 126, 1036 CrossRef CAS; (h) C. Ye, B. Twamley and J. M. Shreeve, Org. Lett., 2005, 7, 3961 CrossRef CAS PubMed; (i) S. Suzuki, T. Kamo, K. Fukushi, T. Hiramatsu, E. Tokunaga, T. Dohi, Y. Kita and N. Shibata, Chem. Sci., 2014, 5, 2754 RSC; (j) S. M. Banik, J. W. Medley and E. N. Jacobsen, J. Am. Chem. Soc., 2016, 138, 5000 CrossRef CAS PubMed; (k) J. D. Haupt, M. Berger and S. R. Waldvogel, Org. Lett., 2019, 21, 242 CrossRef CAS PubMed.
- (a) L. M. Yagupol'skii, V. V. Lyalin, V. V. Orda and L. A. Alekseeva, Zh. Obshch. Khim., 1968, 38, 2813 Search PubMed; (b) I. I. Maletina, V. V. Orda, N. N. Aleinikov, B. L. Korsunskii and L. M. Yagupol'skii, Zh. Org. Khim., 1976, 12, 1371 CAS; (c) H. J. Frohn, Chem.-Ztg., 1984, 108, 146 CAS.
- C. R. Pitts, D. Bornemann, P. Liebing, N. Santschi and A. Togni, Angew. Chem., Int. Ed., 2019, 58, 1950 CrossRef CAS PubMed.
- S. Haubenreisser, T. H. Wöste, C. Martínez, K. Ishihara and K. Muñiz, Angew. Chem., Int. Ed., 2016, 55, 413 CrossRef CAS PubMed.
- U. Tilstam and H. Weinmann, Org. Process Res. Dev., 2002, 6, 384 CrossRef CAS.
- Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007 CrossRef.
- T. Umemoto, R. P. Singh, Y. Xu and N. Saito, J. Am. Chem. Soc., 2010, 132, 18199 CrossRef CAS PubMed.
- (a) A. L. Johnsen, J. Org. Chem., 1982, 47, 5220 CrossRef; (b) J. R. Wolstenhulme, J. Rosenqvist, O. Lozano, J. Ilupeju, N. Wurz, K. M. Engle, G. W. Pidgeon, P. R. Moore, G. Sandford and V. Gouverneur, Angew. Chem., Int. Ed., 2013, 52, 9796 CrossRef CAS PubMed.
- M. Wang, Q. Fan and X. Jiang, Org. Lett., 2018, 20, 216 CrossRef CAS PubMed.
- The synthesis of one of the tetralone intermediates required to make 21 is loosely based on a synthesis reported by Kutateladze and co-workers: W. C. Cronk, O. A. Mukhina and A. G. Kutateladze, J. Org. Chem., 2014, 79, 1235 CrossRef CAS PubMed . Additional modifications were adapted from procedures in ref. 14, as well as: P. Nguyen, E. Corpuz, T. M. Heidelbaugh, K. Chow and M. E. Garst, J. Org. Chem., 2003, 68, 10195 CrossRef PubMed.
- Note that a temperature calibration was performed prior to the following experiments using 4% MeOH in MeOH-d4 and a sample of pure MeOH-d4, see: A. L. Van Geet, Anal. Chem., 1970, 42, 679 CrossRef CAS.
- For an example of temperature-dependent shifts in 19F NMR spectra, see: A. Dimitrov, U. Groß, S. Rüdiger, W. Storek and J. Burdon, J. Fluorine Chem., 1996, 78, 1 CrossRef CAS.
- (a) T. Drakenberg, K. Dahlqvist and S. Forsén, Acta Chem. Scand., 1970, 24, 694 CrossRef CAS; (b) F. P. Gasparro and N. H. Kolodny, J. Chem. Educ., 1977, 54, 259 CrossRef; (c) F. A. Bovey, Nuclear Magnetic Resonance Spectroscopy, Academic Press, New York, 1988 Search PubMed.
- M. Ochiai, T. Yoshikazu and Y. Masaki, J. Am. Chem. Soc., 1990, 112, 5677 CrossRef CAS.
- H. J. Reich and C. S. Cooperman, J. Am. Chem. Soc., 1973, 95, 5077 CrossRef CAS.
- For a review on pseudorotation, see: H. L. Strauss, Annu. Rev. Phys. Chem., 1983, 34, 301 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed . The CCDC search was conducted on May 3rd, 2019 using the online version, whereby a similarity search was performed on the core Ph-IF2 structure. A total of 17 results were found (excluding our contributions), of which, only 8 represented aryl-IF2 compounds (CCDC deposition numbers: 273254, 273255, 1555065, 139385, 938893, 760658, 760659, and 760660) and only 3 represented aryl-IF4 compounds (CCDC deposition numbers: 216480, 1409048, and 1308449).
- J. F. Corbett, J. Chem. Educ., 1972, 49, 663 CrossRef CAS , and references cited therein.
- H. J. Frohn, S. Görg, G. Henkel and M. Läge, Z. Anorg. Allg. Chem., 1995, 621, 1251 CrossRef CAS.
- S. Hoyer and K. Seppelt, J. Fluorine Chem., 2004, 125, 989 CrossRef CAS.
- (a) V. Matoušek, E. Pietrasiak, R. Schwenk and A. Togni, J. Org. Chem., 2013, 78, 6763 CrossRef; (b) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS.
- R. L. Amey and J. C. Martin, J. Am. Chem. Soc., 1979, 101, 5294 CrossRef CAS.
- J. Charpentier, PhD thesis no. 23352, ETH Zürich, 2016.
- (a) J. V. Quagliano and L. Schubert, Chem. Rev., 1952, 50, 201 CrossRef CAS; (b) M. Ochiai, T. Sueda, K. Miyamoto, P. Kiprof and V. V. Zhdankin, Angew. Chem., Int. Ed., 2006, 118, 8383 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1913138–1913140. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02162k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |