
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical oxidation induced selective tyrosine bioconjugation for the modification of biomolecules†
Chunlan
Song
a,
Kun
Liu
a,
Zhongjie
Wang
a,
Bo
Ding
a,
Shengchun
Wang
a,
Yue
Weng
*ab,
Chien-Wei
Chiang
*a and
Aiwen
Lei
 *ac
*ac
aCollege of Chemistry and Molecular Sciences, Institute for Advanced Studies (IAS), Wuhan University, Wuhan 430072, P. R. China. E-mail: aiwenlei@whu.edu.cn
bNational Synchrotron Radiation Research Center (NSRRC), Hsinchu Science Park, Hsinchu, Taiwan
cNational Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang 330022, P. R. China
First published on 8th July 2019
Abstract
Directly introducing a beneficial functional group into biomolecules under mild, clean and easy-to-handle conditions is of great importance in the field of chemical biology and pharmacology. Herein, we described an electrochemical strategy to perform the bioconjugation of tyrosine residues with phenothiazine derivatives in a rapid and simple manner. In this electrochemical system, various polypeptides and proteins were successfully labelled with excellent site- and chemo-selectivity, and metals, oxidants or additives were also avoided.
Introduction
Developing a site-selective, mild and biocompatible reaction for biomolecule modification is an important pursuit in the field of chemical biology, medical chemistry and clinical pharmacology.1 Through the attachment of synthetic molecules to a specific position on proteins, bioconjugation can be applied to DNA labeling, antibody-drug modification and protein visualization studies.2 The advantage of naturally low abundance of aromatic amino acid residues on protein surfaces would lead to a higher degree of bioconjugation, without changing the overall charge state or redox sensitivity.3 However, due to the similarity of their redox potentials and the difficulty of C(sp2)–H functionalization,4 techniques for labeling these aromatic amino acid residues were fewer than those of cysteine and lysine.4b,c,5 Thus, aromatic amino acid residues still lack efficiency as well as chemo- and site-selective methods to achieve bioconjugation reactions.As one of the important amino acids, tyrosine can be found in many polypeptides and proteins, such as tyrosine protein kinases, kisspeptin and myoglobin. Because of its low natural abundance in native proteins, tyrosine is also considered as an attractive target for labeling biomolecules.4d,6 Previously, some approaches have been developed for tyrosine modification, such as Mannich-type reactions,7 Pd catalysts,6a,8 Ru photocatalysts,9 click-like reactions,10etc.11 Recently, several strategies for site-selective protein modification based on oxidative coupling have been reported, which could significantly enrich the methodologies of bioconjugation chemistry.6c,12 Accompanied by a good momentum of organic electrosynthesis in the field of oxidative coupling reactions, electrochemical anodic oxidation provides a green option to prevent the usage of hazardous oxidants and harsh conditions, and sensitive functional groups could also be well tolerated.13 Merging electrochemical organic synthesis and bioconjugation chemistry is very promising, and therefore developing multifarious protocols for peptide and protein modification is highly desirable, especially when excellent selectivity and efficiency could be obtained.
On the other hand, as valuable labeling targets, phenothiazine derivatives have been recognized as highly bioactive drugs and chromophores.14 The incorporation of phenothiazine into biomolecules would not only demonstrate the advantage of phenothiazines-containing protein drugs, but also potentially lead to chemical probes. Recently, the group of Gouin has demonstrated the electrochemically promoted tyrosine-click-chemistry for protein labeling with a urazole reagent, which significantly improved both the yield and selectivity compared with the original conditions by using a pre-activated reagent.10e Herein, we described an electrochemically promoted labeling strategy of tyrosine-containing biomolecules with phenothiazine derivatives under simple, mild and clean conditions (Fig. 1a). Initially, in order to gain insight into the relative redox activity of phenothiazine and aromatic amino acids, cyclic voltammetry experiments have been performed (Fig. 1b). The results showed that the oxidation potential of phenothiazine was much lower than that of Tyr, Trp, Phe and His, which indicated that the reaction proceeded through the single-electron oxidation of phenothiazine (PTZ) to generate the nitrogen radical. Following radical addition to the ortho-position of phenol would achieve the modification of tyrosine. Due to the less electron-rich position of other aromatic amino acid residues, the radical addition would not be favored. Thus, through choosing phenothiazine as a valuable ‘tag’ to generate an active species, excellent site- and chemo-selectivity on the protein modification could be obtained.
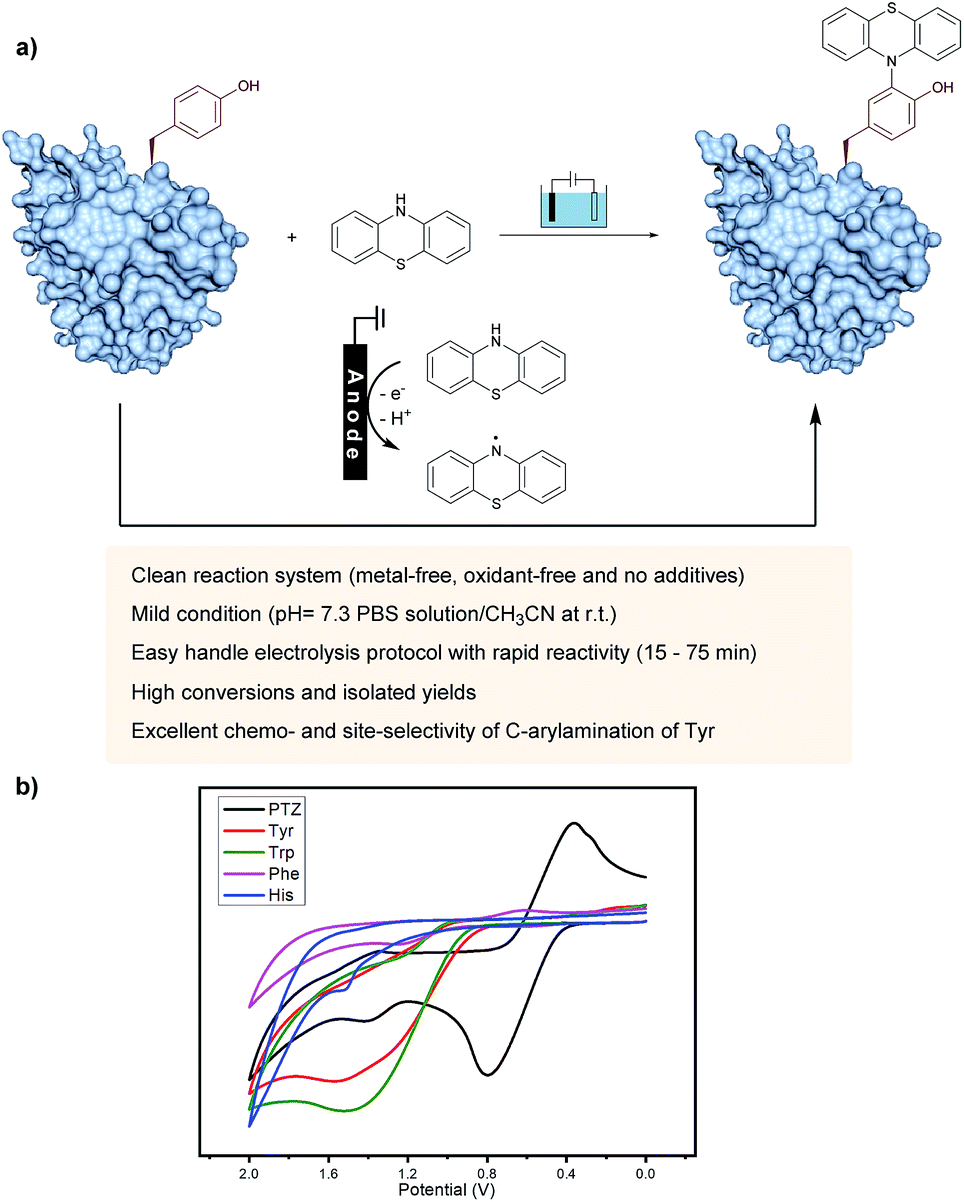 | ||
| Fig. 1 Discovery of an electrochemical oxidative tyrosine bioconjugation. (a) Proposed pathway: phenothiazine underwent single-electron oxidization by the anode to generate a radical species, and then coupled with the tyrosine residue of biomolecules to obtain the labeling product. (b) Cyclic voltammograms of 0.005 M PTZ (black), Tyr (red), Trp (green), Phe (purple) and His (blue) at a glassy carbon electrode, in 0.05 M nBu4NBF4 in CH3CN/H2O. Scan rate = 100 mV s−1. Reference electrode: Ag/AgCl. | ||
Results and discussion
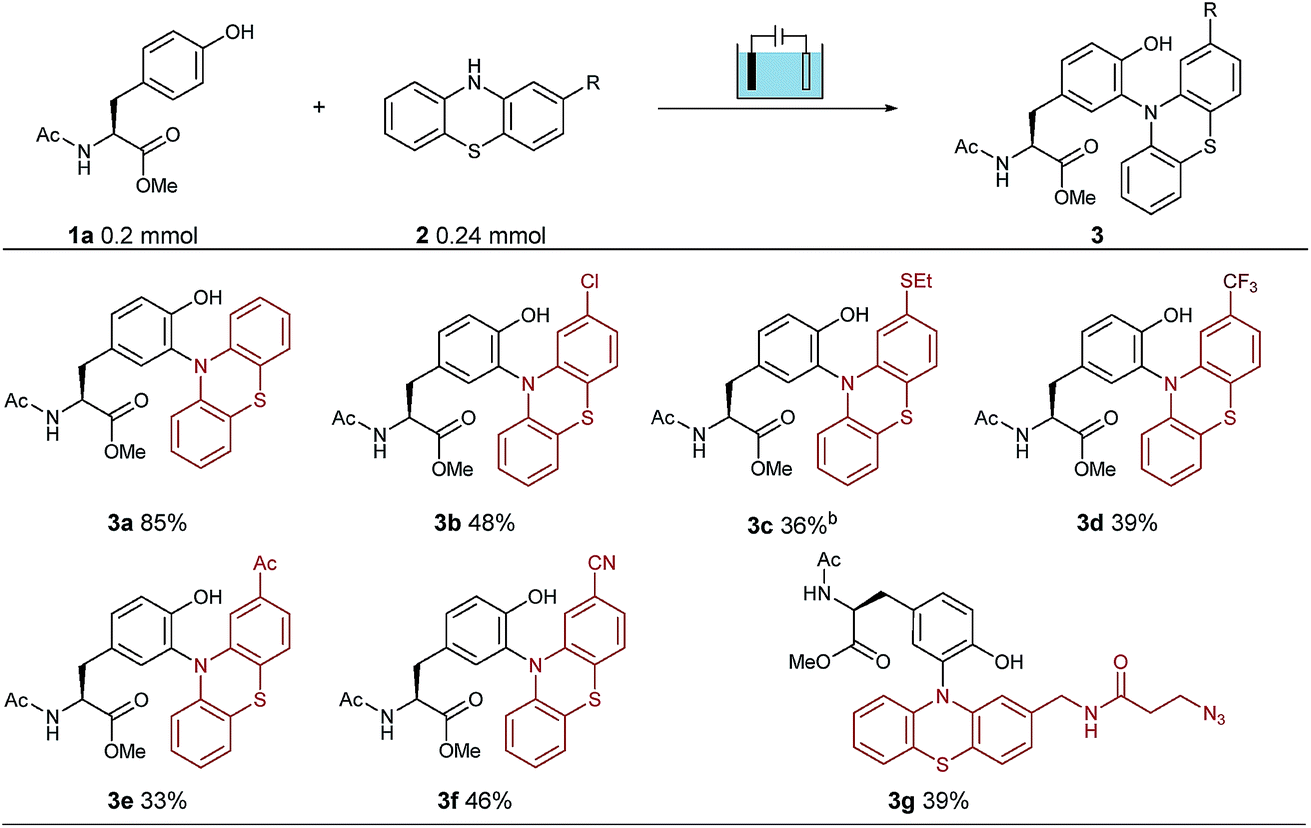
To establish suitable conditions for this electro-oxidative bioconjugation reaction, a protected tyrosine 1a and phenothiazine 2a were chosen as the model reaction substrates for optimization. Under the electrolysis conditions, the reaction was conducted in a three-necked undivided cell with a graphite rod anode and a nickel plate cathode at 10 mA constant current for 75 minutes. Product 3a could be obtained in 85% isolated yield with Na2SO4 as the electrolyte in CH3CN/H2O at room temperature (Table 1). To extend the scope of phenothiazine derivatives, a variety of substituted phenothiazines were then applied to couple with tyrosine 1a under the optimized reaction conditions, which bear halogen (3b), sulfide (3c), trifluoromethyl (3d), acetyl (3e), cyano (3f) and azide (3g) groups. Under these reaction conditions, different electron-donating or electron-withdrawing substituted phenothiazines were all found to be suitable substrates in moderate isolated yields. Remarkably, this method could be extended to azide group substituted phenothiazine which could be applied in further bio-orthogonal reactions.| a Reaction conditions: graphite rod anode, nickel plate cathode, constant current = 10 mA, 1a (1.0 equiv., 0.20 mmol), 2 (1.2 equiv., 0.24 mmol), Na2SO4 (2 equiv., 0.40 mmol), CH3CN/H2O (6.0 mL/4.0 mL), room temperature, N2, and 75 min (Q = 45 C, 2.3 F). Yields of isolated products are shown. b 10 mL CH3CN. |
|---|
|
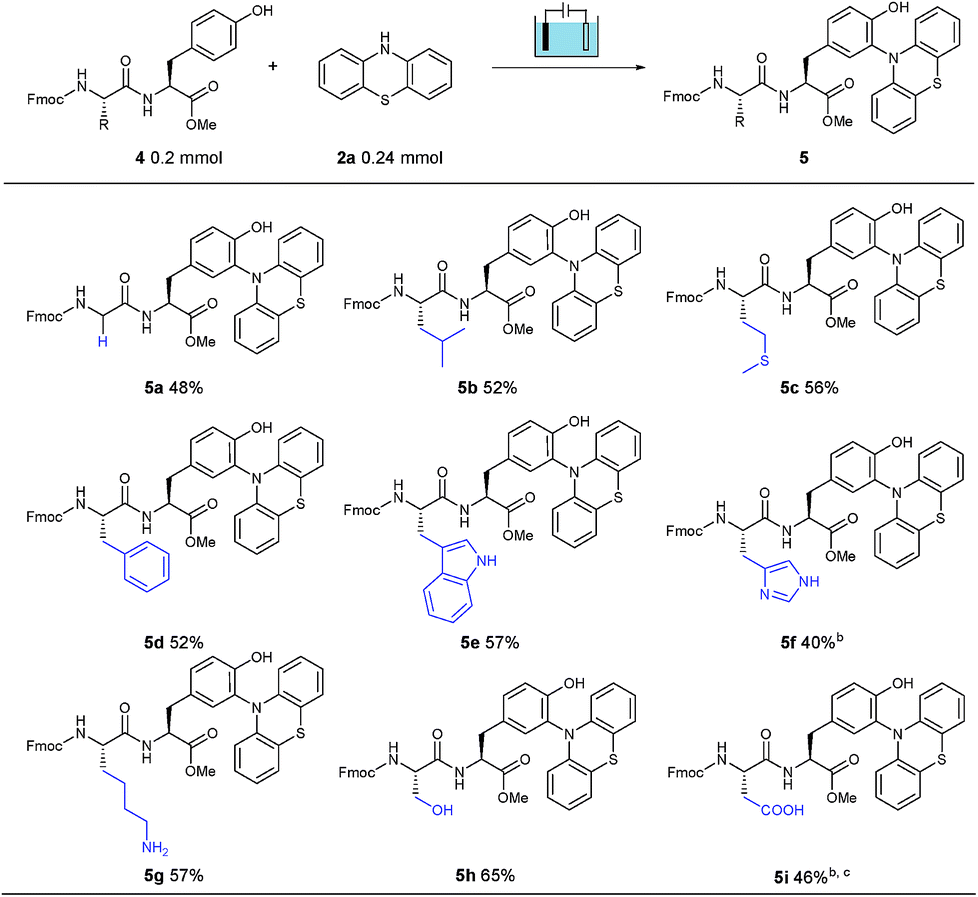
Furthermore, to explore the polypeptide selectivity and tolerance of this tyrosine labeling reaction, various dipeptides containing tyrosine were introduced into this system. With phenothiazine as the coupling partner, these dipeptides made a contribution to the desired bioconjugation reaction in CH3CN/PBS (pH = 7.3) solution without addition of Na2SO4 (Table 2). Besides the relatively inert glycine (4a) and leucine (4b), this established electrosynthesis protocol was also compatible with the thioether of methionine (4c), phenyl group of phenylalanine (4d), indole NH of tryptophan (4e), imidazole NH of histidine (4f), amino group of lysine (4g), hydroxyl group of serine (4h) and carboxylic acid of aspartic acid (4i), indicating a broad functional group tolerance. Meanwhile, the labeling was observed neither on the other aromatic amino acids Trp, His and Phe, nor on the amino, hydroxyl and carboxylic groups of Lys, Ser and Asp. Unfortunately, the Cys-containing dipeptide could only obtain trace amounts of the corresponding product, possibly due to the easily oxidizable property of cysteine under oxidation conditions, leading to the decomposition of the dipeptide. In most cases studied, this electrochemical bioconjugation reaction proceeded perfectly, selectively and cleanly with good isolated yields.
| a Reaction conditions: graphite rod anode, nickel plate cathode, constant current = 10 mA, 4 (1.0 equiv., 0.20 mmol), 2a (1.2 equiv., 0.24 mmol), CH3CN/PBS (pH = 7.3) (6.0 mL/4.0 mL), room temperature, N2, and 75 min (Q = 45 C, 2.3 F). Yields of isolated products are shown. b 105 min. c CH3CN/PBS (pH = 8.0). |
|---|
|
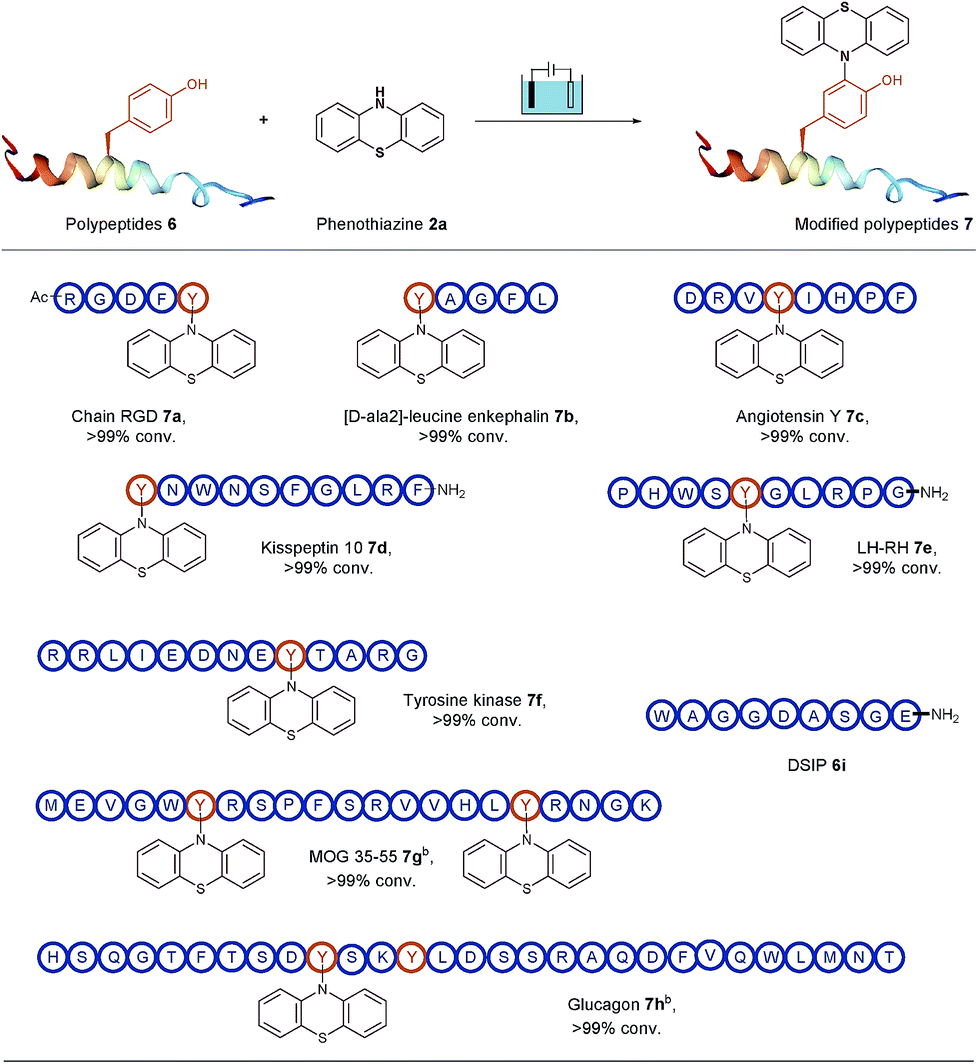
To open an electrosynthetic pathway towards biomolecules such as larger peptides and proteins, we next evaluated the applicability of this electro-oxidative bioconjugation procedure in the synthesis of phenothiazine-modified polypeptides. As a proof-of-concept, the modification of a variety of unprotected peptides 6 from 5-mers to 29-mers have been achieved effectively with excellent conversions (Table 3). We chose the Tyr-containing RGD peptide as the substrate originally, which was considered as an attractive target for tumor treatment by inducing apoptosis and caspase-3 activation. This extracellular RGD peptide was used in CH3CN/PBS (pH = 7.3) to furnish phenothiazine-conjugate 7a in 99% conversion. Having established the electrochemical transformation of phenothiazine 2a with RGD, we then turned our attention to other Tyr-containing polypeptides. For example, acyclic pentapeptide YAGFL was reacted with phenothiazine 2a under electrolysis conditions, and the bioconjugated product 7b was obtained in full conversion. We next examined the applicability of this electrolysis methodology for endogenous peptides with biological activity, such as angiotensin Y, kisspeptin 10, LH-RH, tyrosine protein kinases, MOG 35–55 and glucagon. The above peptides contained at least one tyrosine at the positions of the N-terminus, the C-terminus, or a loop. To our delight, all these polypeptides (6c–6h) were successfully tagged with phenothiazine within 30 min at room temperature. It was worth noting that if one equiv. of phenothiazine was added to the reaction solution of 6g and 6h, respectively, two different results could be observed. Since the two tyrosine residues of 6g are located in different areas of the peptide, the modification could occur on the both tyrosine residues. For 6h, two tyrosine residues are just located on the i and i + 3 positions, and the steric influence would make phenothiazine specifically labelled on a less steric hindrance position of Y10. Eventually, the modification of 29-mer peptide glucagon 6h was conducted the single modified peptide 7h. Meanwhile, a peptide DSIP (WAGGDASGE) 6i which lacks tyrosine residue was used as a control experiment substrate to confirm the selectivity of the reaction. No desired bioconjugated product could be observed, demonstrating that this protocol had the ability to specifically label tyrosine residues on biomolecules with good site- and chemo-selectivity.
| a Reaction conditions: graphite rod anode, nickel plate cathode, constant current = 10 mA, polypeptides 6 (5 mg), phenothiazine 2a (10 mg), CH3CN/PBS (pH = 7.3) (0.75 mL/0.75 mL), room temperature, N2, and 30 min. Conversion of products are shown, as determined by HPLC. b One equiv. of phenothiazine was used. |
|---|
|
Therefore, all these examples revealed the exceptional site- and chemo-selectivity of the reaction and its application to the peptide-labelled chemistry. In comparison with reported bioconjugation methods, this electrolysis method provided fast kinetics and high productivity in a metal- and additive-free manner. It is particularly noteworthy that this protocol provides a direct access to late-stage derivatization of valuable drugs (Fig. 2). For instance, when the biotin-containing phenothiazine 2h and probenecid-containing (uricosuric drugs) phenothiazine 2i were introduced to react with pentapeptide YAGFL 6b, respectively, the desired products 7i and 7j could be obtained. Noticeably, these reactions were permitted by the direct electro-oxidative bioconjugation reaction, and exhibit a remarkable substituent tolerance. In addition, to demonstrate that the phenothiazine-labelled peptide might be utilized as a fluorophore, molecule 7k from the reaction of 2-acetylphenothiazine 2e and RGDFY 6a was prepared. The photograph showed that phenothiazine-modified peptide 7k apparently presented the ability of fluorescence emission under irradiation of UV light, clearly indicating that phenothiazine labelled biomolecules had the potential to be a possible biological fluorophore.
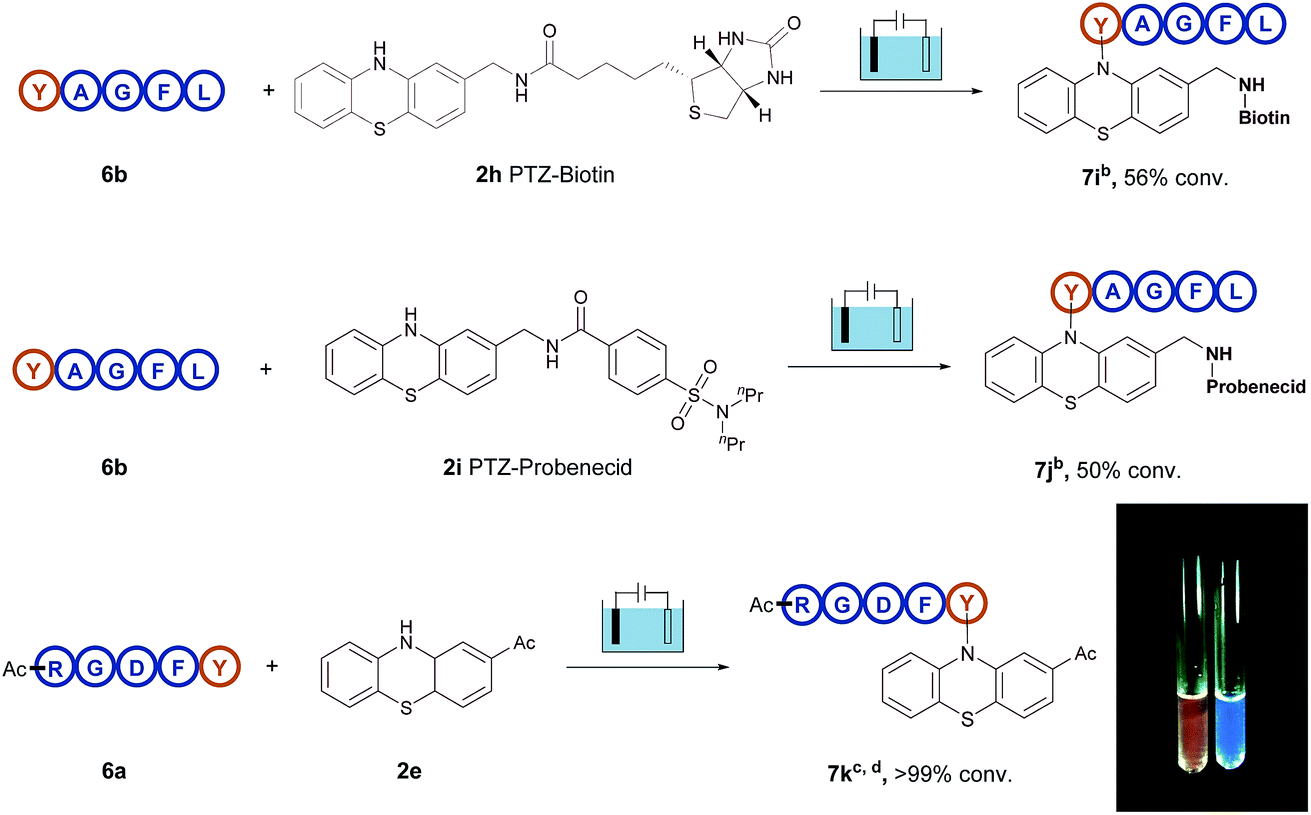 | ||
| Fig. 2 Additional application of electrochemical oxidative bioconjugation. aReaction conditions: graphite rod anode, nickel plate cathode, constant current = 10 mA, polypeptides (5 mg), phenothiazine (10 mg), CH3CN/PBS (pH = 7.3) (0.75 mL/0.75 mL), room temperature, N2, and 30 min. bConversion yields of products are shown, as determined by HPLC using diphenylamine as the standard. cConversion yields of products are shown, as determined by HPLC using anisic acid as the standard. dPhotographs of the solution of 6a (right) and modified product 7k (left) with UV lamp excitation. | ||
Afterward, we turned to discover the modification of proteins. Two proteins, insulin and myoglobin, containing at least one exposed tyrosine on the protein surface were electrolyzed with phenothiazine. When an excess amount of phenothiazine was added to the solution of insulin under electrolysis, four potentially reactive tyrosine residues on insulin were all tagged (Fig. 3a). Next, we tested the feasibility of our developed method for a larger protein, myoglobin. The electro-oxidative bioconjugation could be well performed within 15 min and generate single phenothiazine tagged myoglobin at −20 °C (Fig. 3b). Importantly, CD spectroscopy revealed that the electrochemical methodology had negligible influence on the structure of both insulin and myoglobin (Fig. 3c and d). These satisfactory results revealed that the exposed tyrosine could be well-tagged by phenothiazine under our electro-oxidative conditions without decomposition.
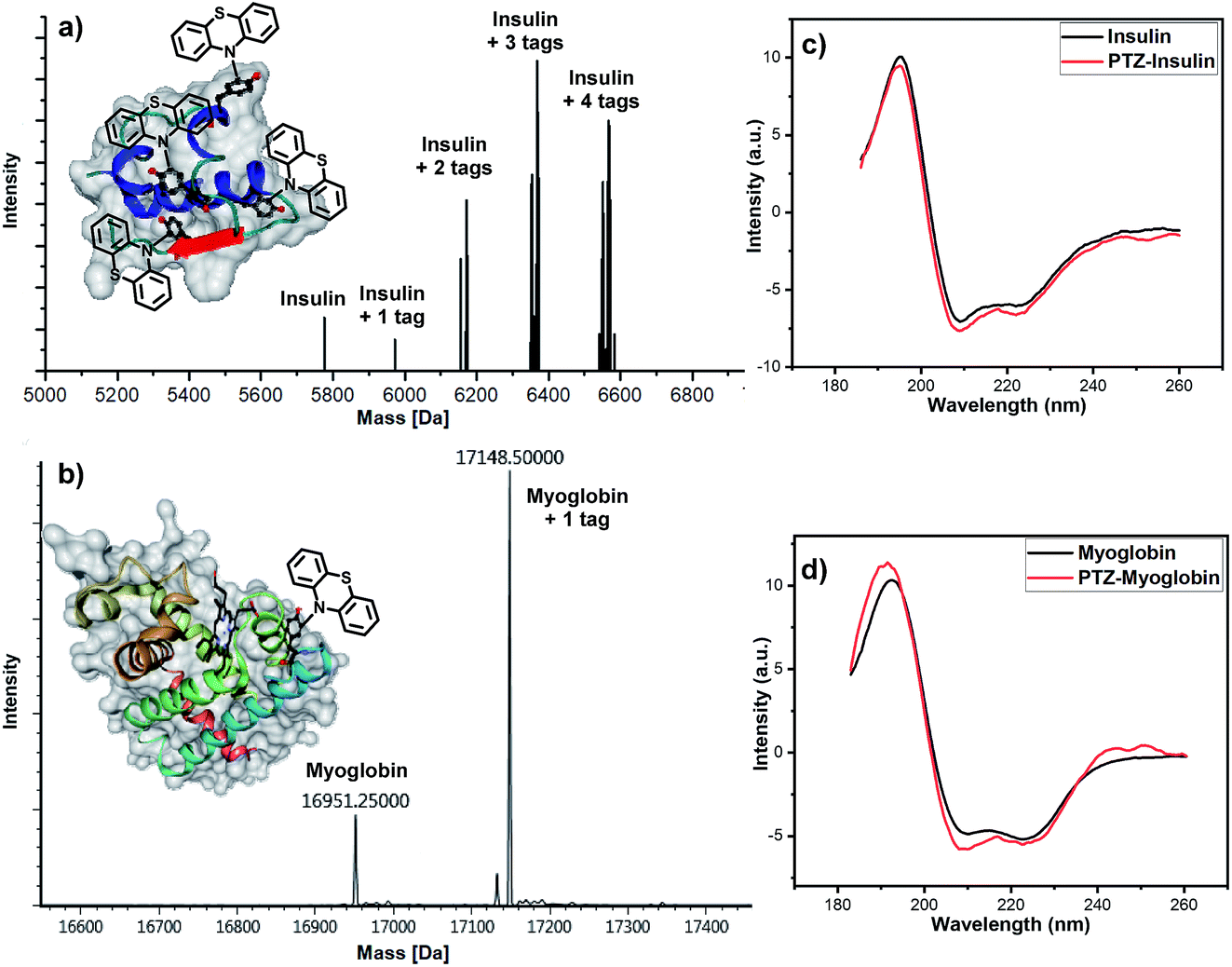 | ||
| Fig. 3 Electrochemical modification of proteins. Reaction conditions for the bioconjugation of insulin: graphite rod anode, nickel plate cathode, constant current = 10 mA, insulin (5 mg), phenothiazine (10 mg), CH3CN/PBS (pH = 7.3) (0.75 mL/0.75 mL), room temperature, N2, and 30 min. Reaction conditions for the bioconjugation of myoglobin: graphite rod anode, nickel plate cathode, constant current = 0.5 mA, myoglobin (5 mg), phenothiazine (20 mg), CH3CN/PBS (pH = 7.3) (0.75 mL/0.75 mL), −20 °C, N2, and 15 min. (a) Maldi-Tof MS analysis of modified insulin. (b) LC-MS analysis of modified myoglobin. (c) Effect of electrolytic arylamination on the structure of insulin. (d) Effect of electrolytic arylamination on the structure of myoglobin. | ||
Conclusions
By introducing organic electrochemistry, tyrosine residues can be well-labelled with phenothiazine derivatives with high chemo- and site-selectivity as well as excellent conversion under mild conditions. This strategy has been successfully applied to the modification of valuable biomolecules such as polypeptides, insulin and myoglobin. Moreover, this technique could promote the development of safer and more biological sustainable bioconjugation reactions that are operated under metal-, oxidant- and additive free conditions. We anticipate that advances in electrochemically induced bioconjugation will lead to an expanding library of interdisciplinary methodologies.Experimental
A general procedure for bioconjugation of tyrosine and phenothiazine derivatives: in an oven-dried undivided three-necked bottle (25 mL) equipped with a stir bar, protected tyrosine (0.20 mmol), phenothiazine (0.24 mmol), Na2SO4 (0.40 mmol) and CH3CN/H2O (6 mL/4 mL) were combined and added. The bottle was equipped with a graphite rod (ϕ 6 mm, about 15 mm immersion depth in solution) as the anode and a nickel plate (15 mm × 15 mm × 1.0 mm) as the cathode and then charged with nitrogen. The reaction mixture was stirred and electrolyzed at a constant current of 10 mA under room temperature for 75 min (2.3 F). When the reaction was finished, the reaction mixture was extracted with ethyl acetate (10 mL × 3). The organic layers were combined, dried over Na2SO4, and concentrated. The pure product was obtained by flash column chromatography on silica gel (dichloromethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol = 100:1).
:methanol = 100:1).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21701127, 21520102003, and 21390402), the Hubei Province Natural Science Foundation of China (2017CFB152 and 2017CFA010), the Fundamental Research Funds for the Central Universities (213413000050), and the Scientific Research Foundation of Wuhan University (413100043 and 413100021). The Program of Introducing Talents of Discipline to Universities of China (111 Program) is also appreciated.Notes and references
- (a) C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed; (b) O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174 CrossRef CAS; (c) O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495 RSC; (d) Q.-Y. Hu, F. Berti and R. Adamo, Chem. Soc. Rev., 2016, 45, 1691 RSC; (e) K. Kubota, P. Dai, B. L. Pentelute and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 3128 CrossRef CAS; (f) D. T. Cohen, C. Zhang, C. M. Fadzen, A. J. Mijalis, L. Hie, K. D. Johnson, Z. Shriver, O. Plante, S. J. Miller, S. L. Buchwald and B. L. Pentelute, Nat. Chem., 2019, 11, 78 CrossRef CAS.
- (a) P. Akkapeddi, S.-A. Azizi, A. M. Freedy, P. M. S. D. Cal, P. M. P. Gois and G. J. L. Bernardes, Chem. Sci., 2016, 7, 2954 RSC; (b) X. Chen and Y.-W. Wu, Org. Biomol. Chem., 2016, 14, 5417 RSC.
- N. S. Joshi, L. R. Whitaker and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 15942 CrossRef CAS.
- (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS; (b) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775 CrossRef CAS; (c) Y. Seki, T. Ishiyama, D. Sasaki, J. Abe, Y. Sohma, K. Oisaki and M. Kanai, J. Am. Chem. Soc., 2016, 138, 10798 CrossRef CAS; (d) J. Wang, S. Zheng, Y. Liu, Z. Zhang, Z. Lin, J. Li, G. Zhang, X. Wang, J. Li and P. R. Chen, J. Am. Chem. Soc., 2016, 138, 15118 CrossRef CAS; (e) J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863 CrossRef CAS; (f) W. Zhao, H. G. Lee, S. L. Buchwald and J. M. Hooker, J. Am. Chem. Soc., 2017, 139, 7152 Search PubMed.
- (a) G. Lautrette, F. Touti, H. G. Lee, P. Dai and B. L. Pentelute, J. Am. Chem. Soc., 2016, 138, 8340 CrossRef CAS; (b) J. Willwacher, R. Raj, S. Mohammed and B. G. Davis, J. Am. Chem. Soc., 2016, 138, 8678 CrossRef CAS; (c) H. G. Lee, G. Lautrette, B. L. Pentelute and S. L. Buchwald, Angew. Chem., Int. Ed., 2017, 56, 3177 CrossRef CAS; (d) A. J. Rojas, C. Zhang, E. V. Vinogradova, N. H. Buchwald, J. Reilly, B. L. Pentelute and S. L. Buchwald, Chem. Sci., 2017, 8, 4257 RSC; (e) M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, A. Guerreiro, P. M. S. D. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jiménez-Osés and G. J. L. Bernardes, J. Am. Chem. Soc., 2018, 140, 4004 CrossRef CAS; (f) K. Yamatsugu, M. Furuta, S. Xi, Y. Amamoto, J. Liu, S. A. Kawashima and M. Kanai, Bioorg. Med. Chem., 2018, 26, 5359 CrossRef CAS.
- (a) S. Chen, X. Li and H. Ma, ChemBioChem, 2009, 10, 1200 CrossRef CAS; (b) H. Ban, J. Gav rilyuk and C. F. Barbas, J. Am. Chem. Soc., 2010, 132, 1523 CrossRef CAS; (c) A. M. ElSohly and M. B. Francis, Acc. Chem. Res., 2015, 48, 1971 CrossRef CAS; (d) X. Wu, X. Li, H. Li, W. Shi and H. Ma, Chem. Commun., 2017, 53, 2443 RSC.
- D. W. Romanini and M. B. Francis, Bioconjugate Chem., 2008, 19, 153 CrossRef CAS.
- S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080 CrossRef CAS.
- (a) S. Sato and H. Nakamura, Angew. Chem., Int. Ed., 2013, 52, 8681 CrossRef CAS; (b) S. Sato, K. Hatano, M. Tsushima and H. Nakamura, Chem. Commun., 2018, 54, 5871 RSC.
- (a) H. Ban, M. Nagano, J. Gavrilyuk, W. Hakamata, T. Inokuma and C. F. Barbas, Bioconjugate Chem., 2013, 24, 520 CrossRef CAS; (b) Q.-Y. Hu, M. Allan, R. Adamo, D. Quinn, H. Zhai, G. Wu, K. Clark, J. Zhou, S. Ortiz, B. Wang, E. Danieli, S. Crotti, M. Tontini, G. Brogioni and F. Berti, Chem. Sci., 2013, 4, 3827 RSC; (c) C. M. Madl and S. C. Heilshorn, Bioconjugate Chem., 2017, 28, 724 CrossRef CAS; (d) S. Sato, K. Nakamura and H. Nakamura, ChemBioChem, 2017, 18, 475 CrossRef CAS PubMed; (e) D. Alvarez-Dorta, C. Thobie-Gautier, M. Croyal, M. Bouzelha, M. Mével, D. Deniaud, M. Boujtita and S. G. Gouin, J. Am. Chem. Soc., 2018, 140, 17120 CrossRef CAS.
- (a) J. Gavrilyuk, H. Ban, M. Nagano, W. Hakamata and C. F. Barbas, Bioconjugate Chem., 2012, 23, 2321 CrossRef CAS; (b) M. W. Jones, G. Mantovani, C. A. Blindauer, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 7406 CrossRef CAS; (c) A.-W. Struck, M. R. Bennett, S. A. Shepherd, B. J. C. Law, Y. Zhuo, L. S. Wong and J. Micklefield, J. Am. Chem. Soc., 2016, 138, 3038 CrossRef CAS PubMed; (d) S. Vandewalle, R. De Coen, B. G. De Geest and F. E. Du Prez, ACS Macro Lett., 2017, 6, 1368 CrossRef CAS; (e) N. Ichiishi, J. P. Caldwell, M. Lin, W. Zhong, X. Zhu, E. Streckfuss, H.-Y. Kim, C. A. Parish and S. W. Krska, Chem. Sci., 2018, 9, 4168 RSC; (f) J. Ohata, M. K. Miller, C. M. Mountain, F. Vohidov and Z. T. Ball, Angew. Chem., Int. Ed., 2018, 57, 2827 CrossRef CAS.
- K. L. Seim, A. C. Obermeyer and M. B. Francis, J. Am. Chem. Soc., 2011, 133, 16970 CrossRef CAS.
- (a) J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605 RSC; (b) A. Jutand, Chem. Rev., 2008, 108, 2300 CrossRef CAS; (c) J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef CAS; (d) H.-C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2010, 132, 2839 CrossRef CAS; (e) S. R. Waldvogel and S. Möhle, Angew. Chem., Int. Ed., 2015, 54, 6398 CrossRef CAS; (f) Z.-W. Hou, Z.-Y. Mao, H.-B. Zhao, Y. Y. Melcamu, X. Lu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 9168 CrossRef CAS; (g) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575 CrossRef CAS; (h) K. Liu, S. Tang, P. Huang and A. Lei, Nat. Commun., 2017, 8, 775 CrossRef; (i) N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452 CrossRef CAS; (j) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (k) Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293 CrossRef CAS; (l) H.-B. Zhao, Z.-W. Hou, Z.-J. Liu, Z.-F. Zhou, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 587 CrossRef CAS; (m) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485 CrossRef CAS; (n) K. D. Moeller, Chem. Rev., 2018, 118, 4817 CrossRef CAS; (o) M. Rafiee, F. Wang, D. P. Hruszkewycz and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 22 CrossRef CAS; (p) G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175 CrossRef CAS; (q) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27 CrossRef CAS; (r) S. Tang, S. Wang, Y. Liu, H. Cong and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 4737 CrossRef CAS PubMed; (s) K.-Y. Ye, G. Pombar, N. Fu, G. S. Sauer, I. Keresztes and S. Lin, J. Am. Chem. Soc., 2018, 140, 2438 CrossRef CAS; (t) J.-i. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702 CrossRef CAS.
- (a) S. P. Massie, Chem. Rev., 1954, 54, 797 CrossRef CAS; (b) M. J. Ohlow and B. Moosmann, Drug Discovery Today, 2011, 16, 119 CrossRef CAS; (c) X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411 CrossRef CAS; (d) J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02218j |
| This journal is © The Royal Society of Chemistry 2019 |