
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular azavinyl carbene-triggered rearrangement of furans†
Anton S.
Makarov
a,
Maxim G.
Uchuskin
 *a and
A. Stephen K.
Hashmi
*bc
*a and
A. Stephen K.
Hashmi
*bc
aDepartment of Chemistry, Perm State University, Bukireva 15, 614990 Perm, Russia. E-mail: mu@psu.ru
bInstitut für Organische Chemie, Heidelberg University, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany
cChemistry Department, King Abdulaziz University, Jeddah 21589, Saudi Arabia. E-mail: hashmi@hashmi.de
First published on 26th July 2019
Abstract
An intramolecular rhodium-catalyzed reaction of 1-tosyl-1,2,3-triazoles with furans has been explored. The tosylimino functionality was found to play a significant chemical role participating in the subsequent domino-transformations of a key reaction intermediate. One of the reaction pathways leads to valuable 2-formyl- and 2-acetylpyridine building blocks, which could be obtained in one-pot starting from easily accessible (furan-2-ylmethyl)propargyl amines. An original method for the synthesis of highly functionalized indolizines from the obtained pyridines has been proposed.
Introduction
Ten years ago Gevorgyan and Fokin realized the colossal synthetic potential of metal-intercepted α-diazoimine tautomers1 of sulfonyl triazoles,2 which were easily accessed through copper-catalyzed azide–alkyne cycloaddition (CuAAC).3 The resulting α-iminocarbenoid species possess the characteristic reactivity of carbenes generated by denitrogenative decomposition of conventional diazo compounds; specifically, they undergo cheletropic reactions, X–H and C–X insertions, ylide formation or rearrangements (Fig. 1).4 However, the sulfonyl imine functionality exhibits untypical nucleophilicity, which allows the azavinyl carbenoids to undergo transannulations following the addition of a nucleophile to the carbene centre.5 Eventually, such an upgraded reactivity of rhodium transient azavinyl carbenoid species affords a high level of synthetic flexibility that is to some extent controlled by the nature of a reaction partner.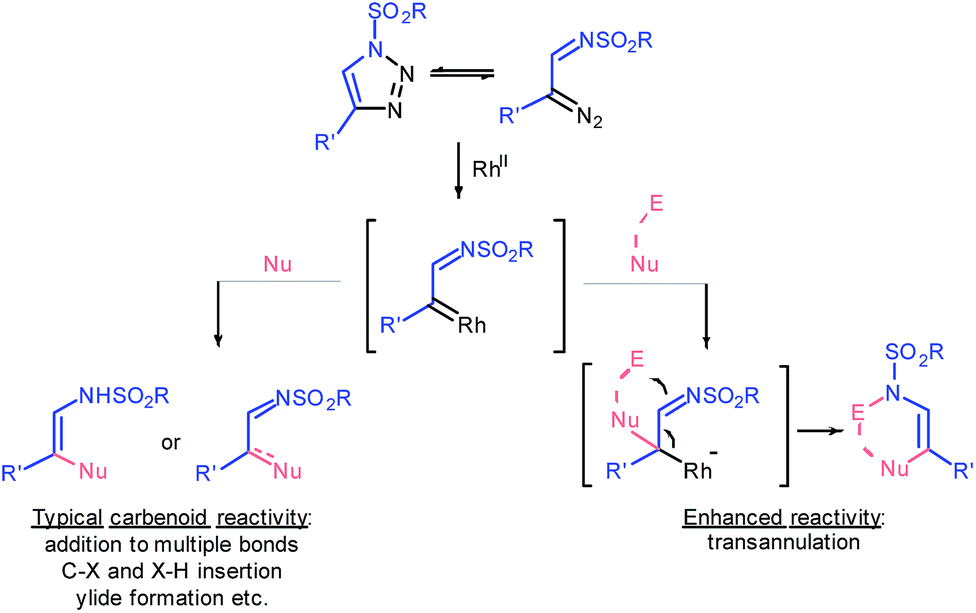 | ||
| Fig. 1 General reactivity patterns of 1-sulfonyl-1,2,3-triazoles. | ||
Furans represent nontrivial biomass-derived building-blocks6 known for their propensity to undergo dearomatization7,8 forming diverse carbo-9 and heterocyclic10 compounds under electrophilic conditions.
Understandably, furans were tested as nucleophiles in a reaction with carbenes derived from diazo compounds. While intermolecular reactions usually result in the formation of cyclopropane and dienone mixtures with poor regiocontrol,11 intramolecular processes afford better chemoselectivity. In particular, the denitrogenative decomposition of various (furan-2-yl)-tethered diazo compounds usually proceeds through the same spiro-intermediate,12,13 and yet leads to products with limited synthetic utility (Fig. 2a). In turn, decomposition of (furan-2-yl)-tethered 1-sulfonyl-1,2,3-triazoles would result in the formation of a common spiro-key intermediate that could be intercepted by an imino group, leading to compounds of type A (Fig. 2b, path I). Alternatively, the key intermediate might rearrange to azatriene B that could further be converted to a variety of nitrogen-containing heterocycles (Fig. 2b, path II). In either scenario there is the possibility for rapid accessing the molecular complexity from rather structurally simple starting materials.
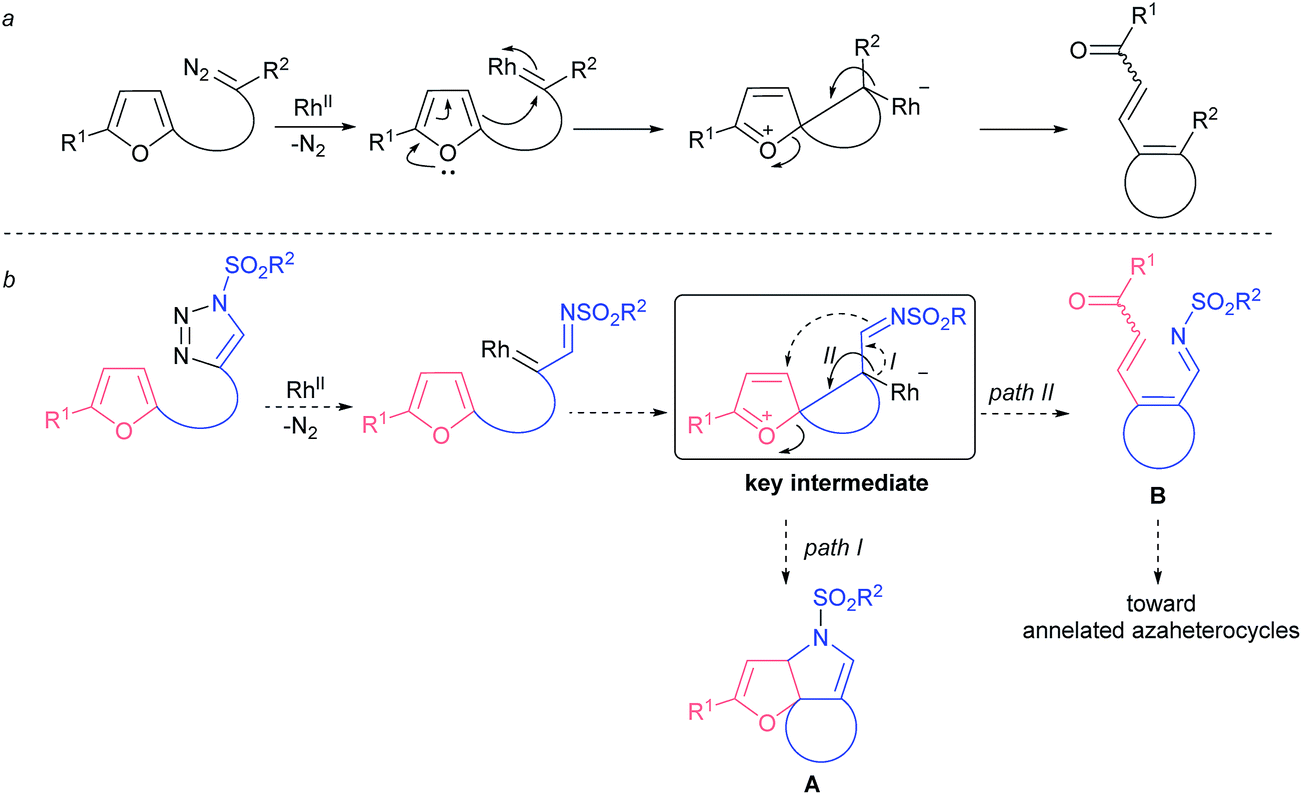 | ||
| Fig. 2 Intramolecular rhodium carbenoid-triggered ring-opening of furans: (a) known reactivity; (b) concept for this work. | ||
To our surprise, we could not find any reports on intermolecular rearrangements of furans initiated by iminocarbene species derived from triazoles, although examples of denitrogenative decomposition of triazoles in the presence of a furanyl substituent within the same molecule are known.14,15 In order to fill the methodological gap, we sought to explore the synthetic prospects of the aforementioned transformation. Very recently, Hashmi et al. described a synthetic method toward functionalized pyrroles based on the rearrangement of a furan ring in the gold(III)-catalyzed reaction of propargylfurfuryl amines with anthranyls.16 The reaction is believed to proceed through the intermediate formation of gold-stabilized iminocarbene species that attack the furan system followed by its dearomatization. The denitrogenative decomposition of furyl-tethered triazoles, in contrast, would provide regioisomeric iminocarbenes, which upon reaction with a furan ring could lead to the formation of valuable products of different architectures. Herein, we report our initial studies of intramolecular azavinyl carbene-triggered rearrangement of furans.
Results and discussion
Our exploratory studies commenced with triazole 2 as a model substrate, which was obtained via CuAAC from propargyl amine 1a (Scheme 1). Treatment of triazole 2 with rhodium acetate dimer in chloroform at 50 °C led to a full conversion of the starting material within 5 h. Careful analysis of the crude reaction mixture revealed that the main component among the formed products appeared to be dihydropyridine 3. However, all attempts to isolate compound 3 by column chromatography on silica gel failed; the only product detected was 2-acetylpyridine 4a. The same outcome was observed when the reaction mixture was treated with a base. Apparently, the formation of 2-acetylpyridine 4a resulted from 6π-azaelectrocyclization17 of initially formed dihydropyrrole II followed by aromatization through elimination of sulfinate.18 Stimulated by the high synthetic value of 2-acylpyridines as intermediates in medicinal19 and material20 chemistry, we decided to study the discovered transformation in more detail. | ||
| Scheme 1 Initial results. | ||
The optimization of reaction conditions revealed that toluene was the solvent of choice for the formation of dihydropyridine 3a (Table 1, entry 1; for more details, see the ESI†). Further screening of rhodium(II) sources led us to identify Rh2(OOct)4 as the optimal catalyst for the first cascade transformation (entries 2–5). As compound 3 appeared to be quite unstable, we continued tuning the reaction conditions towards final acetylpyridine 4a in a one-pot mode.
|
|
||||
|---|---|---|---|---|
| Entry | Rh cat. | Et3N (equiv.) | Yield of 3b, % | Yield of 4ab, % |
| a General considerations: reactions were performed on a 0.05 mmol scale of 2 under dinitrogen; freshly distilled and degassed toluene and Et3N were used. b NMR yields with CH2Br2 as an internal standard. c Reaction time – 10 h. d Reaction was performed on a 0.5 mmol scale; yield of the isolated product is given in parentheses; reaction time for the first step – 16 h. | ||||
| 1 | Rh2(OAc)4 | — | 55 | Trace |
| 2 | Rh2(OCOTr)4 | — | 62 | Trace |
| 3 | Rh2(OPiv)4 | — | 63 | Trace |
| 4 | Rh2(OHex)4 | — | 75 | Trace |
| 5 | Rh2(OOct)4 | — | 90c | Trace |
| 6 | Rh2(OOct)4 | (1) | Trace | 79c |
| 7 | Rh 2 (OOct) 4 | (2) | Trace | 93 (90) |

After some experiments, we found that addition of two equivalents of Et3N resulted in the clean transformation of dihydropyridine 3a into its unsaturated derivative 4a (entry 8).
With the aim of establishing a convenient synthetic method towards 2-acylpyridines, we integrated the CuAAC step into a single one-pot protocol starting from propargyl amines 1. To our delight, the addition of an extra step had almost no effect on the outcome of the model reaction; the yield of pyridine 4a was 88% (Scheme 2). Moreover, on a gram-scale the method afforded pyridine 4a in comparable yield with the use of only 0.5 mol% of rhodium catalyst (see the ESI† for details).
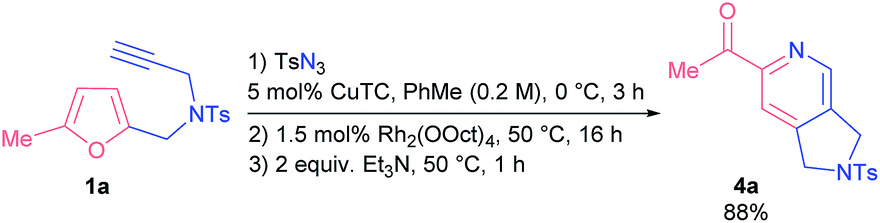 | ||
| Scheme 2 One-pot synthesis of pyridine 4a. | ||
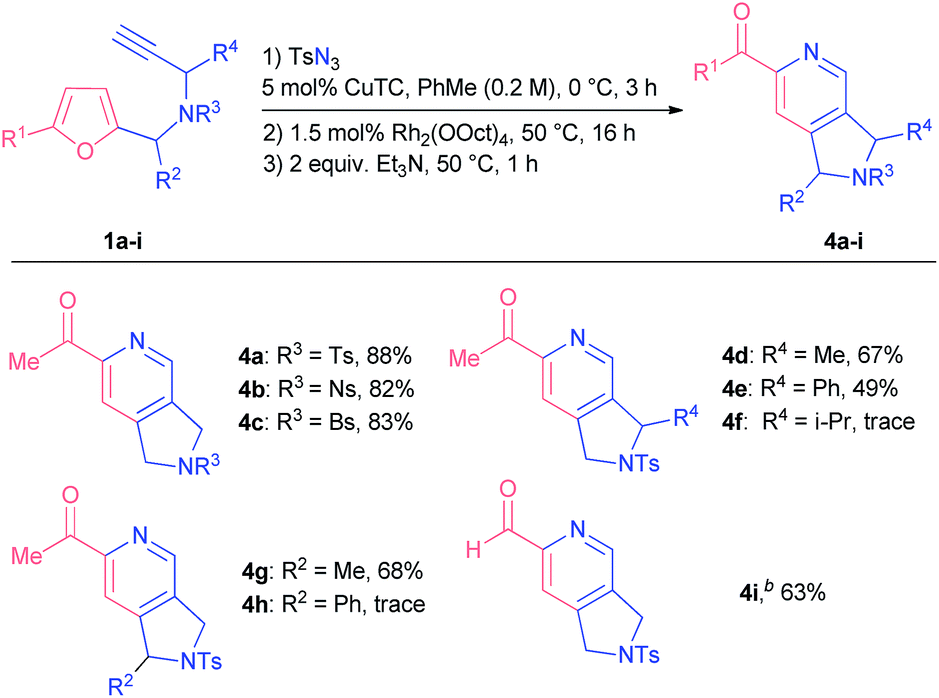
Following the optimization studies, we sought to study the influence of various substituents at different positions of the starting material on the reaction outcome. Substituted propargyl amines 1a-I were screened first (Table 2). Bromo and nitro substituents in other tested sulfonyl protection groups were tolerated well under the optimized conditions (compounds 4b and c). It was possible to introduce substituents at propargylic (compounds 4d and e) and furfuryl positions (compound 4g) of the starting material, although the overall transformation resulted in lower yields of the respective products, probably due to steric reasons. Indeed, more bulky substituents at either position prevented the furan from being attacked by a carbene centre in an initial step: in the cases of starting materials 1f and 1h we observed the formation of corresponding triazoles that decomposed over time. After additional tuning of reaction parameters, we were also able to obtain 2-formylpyridine 4i in 63% yield.
Next, we studied substrates with an extended tether that linked the triazole fragment and a furan ring. Thus, homopropargylamine 5 reacted smoothly under slightly altered reaction conditions to provide tetrahydronaphthiridine 6 (78%) (Scheme 3).
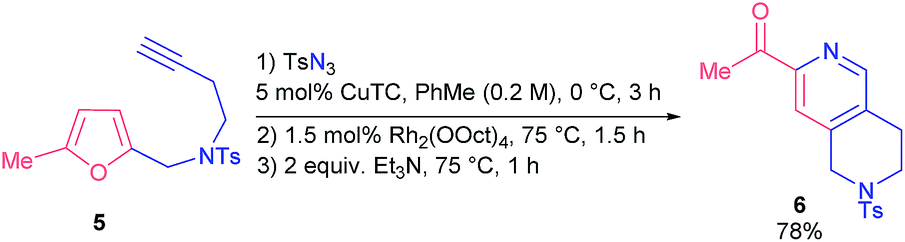 | ||
| Scheme 3 Synthesis of tetrahydronaphthiridine 6. | ||
When we tested homofurfurylamine 7, which was forced to react under quite harsh conditions, we observed the formation of pyrrole 8 as well as dihydropyridine 9 (Scheme 4). Presumably, pyrrole 8 was the product of an alternative reaction where initially formed spiro-intermediate III was trapped intermolecularly with the imino group followed by base-induced ring-opening of dihydrofuran intermediate V (route a). In turn, dihydropyridine 9 was formed via 6π-azaelectrocyclization of azatriene IV following the ring-opening of initial intermediate III (route b). Evidently, the nature of the tether modulates the geometry of an intermediate III at such an elevated reaction temperature, so that it can demonstrate alternative reactivity.
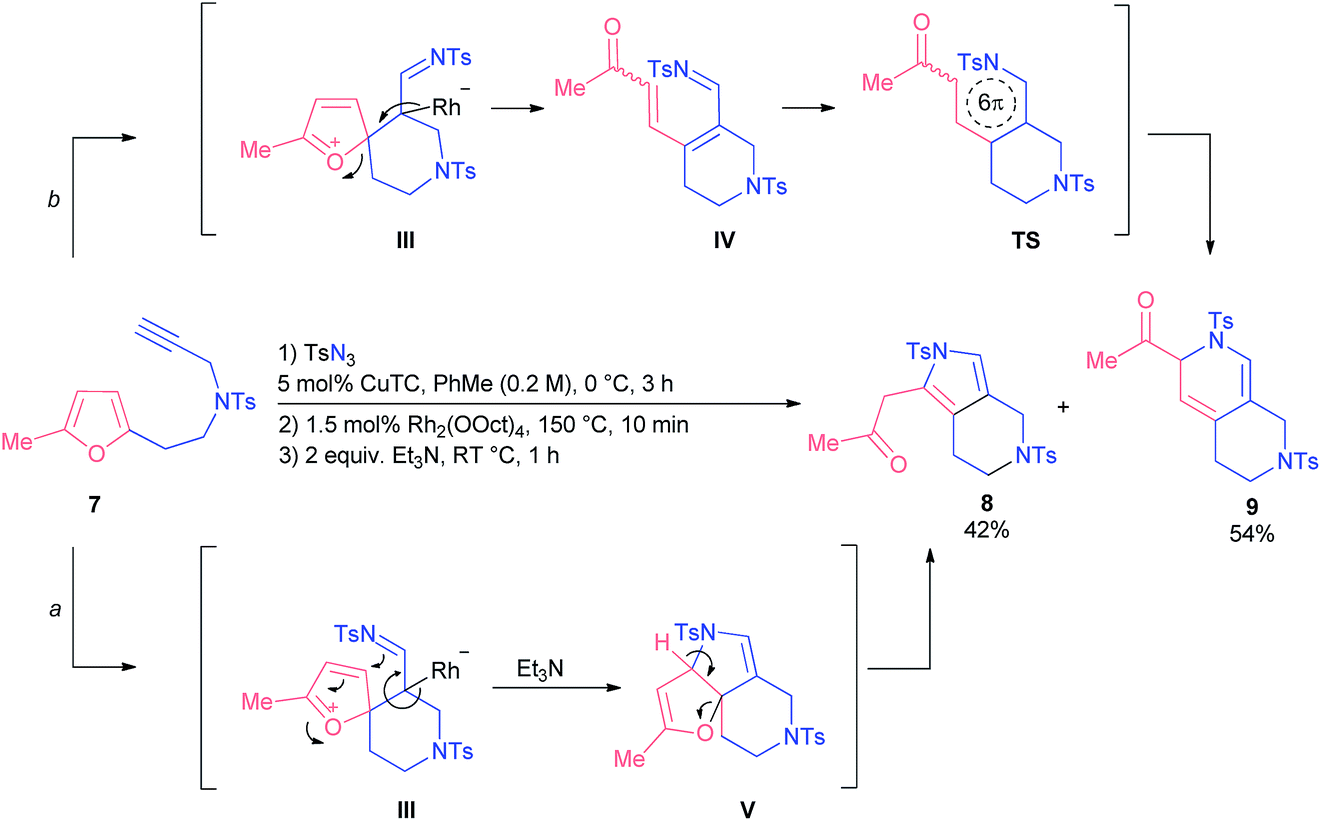 | ||
| Scheme 4 Divergent reactivity of homofurfurylamine 7. | ||
Dihydropyridine 9 was found to be more stable than its analogue, dihydropyridine 3. Conversion of compound 9 into the corresponding pyridine 10 required much harsher reaction conditions as well (Scheme 5).
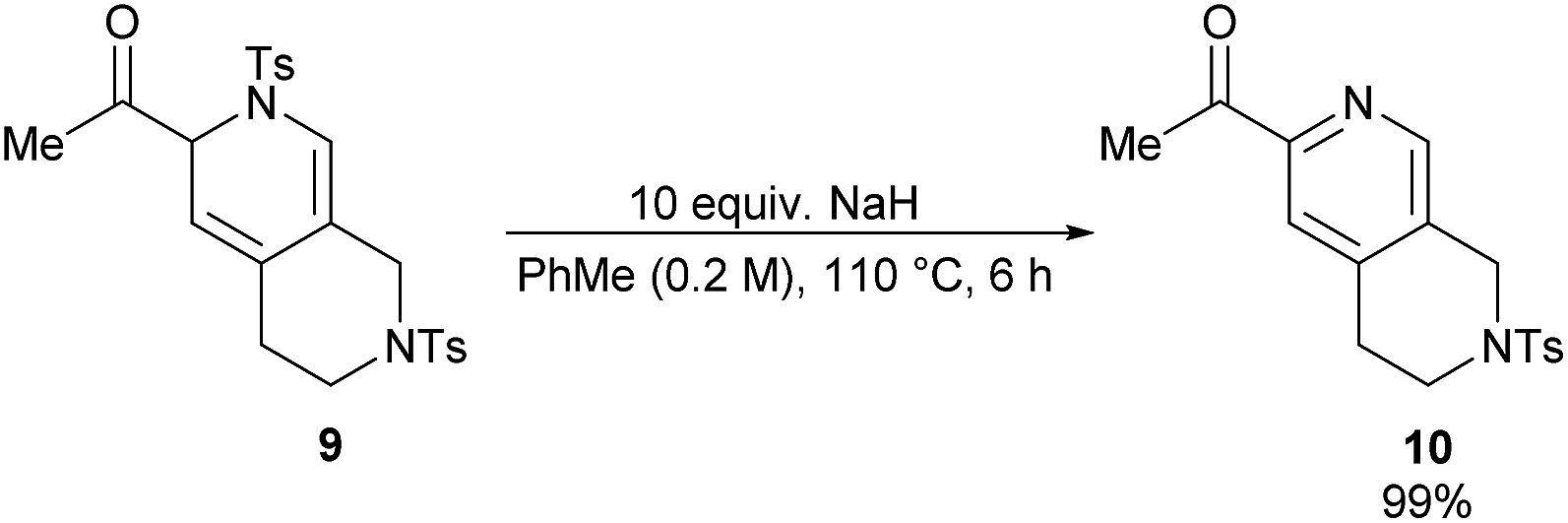 | ||
| Scheme 5 Synthesis of tetrahydronaphthiridine 10. | ||
We examined the reactivity of propargyl ethers 11 that were found to be more reactive than related amines 1 (Table 3). Consequently, they had to be converted under milder conditions in order to avoid undesired side reactions.
| a General considerations: reactions were performed on a 0.5 mmol scale of a propargyl ether 11 under dinitrogen; stepwise addition of catalysts and reagents. For more details, see the ESI. |
|---|
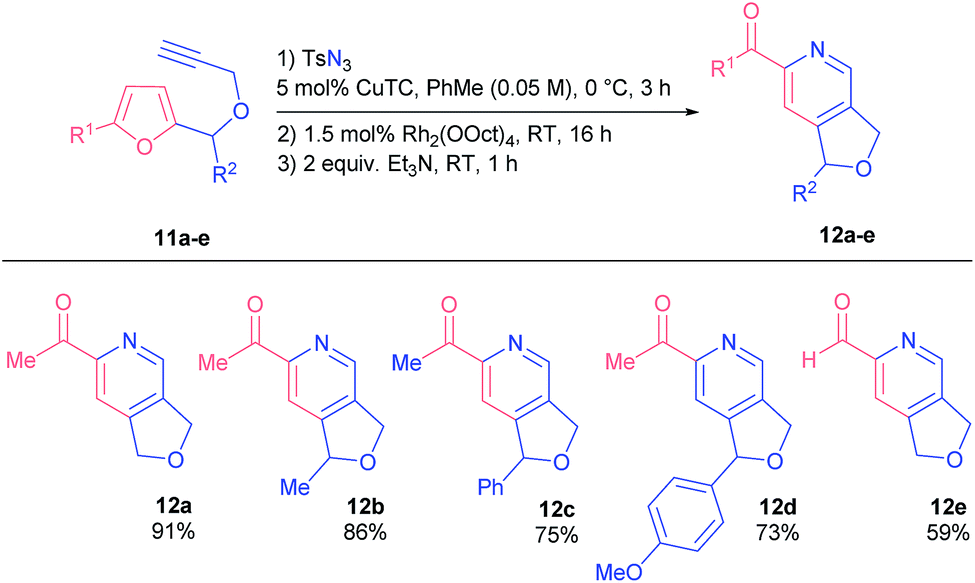
|
Generally, substrates 11 afforded corresponding products 12 in higher yields, even those bearing bulky substituents in the furfuryl position (products 12c and d). Noteworthily, 2-formylpyridine 12e was as well obtained in practical yield.
The reaction of propargyl ethers with voluminous substituents at C(5) of a furan ring (compounds 11f and g) stopped at the formation of Z-azatrienes 13a and b, the products of the ring-opening of the initial spiro-intermediates (Scheme 6). Likely, bulky groups obstruct the electrocyclization of the respective azatrienes. All attempts to force the reactions only led to Z-to-E isomerization of the α,β-unsaturated ketone moiety.
 | ||
| Scheme 6 Reactivity of propargyl ethers 11f and g. | ||
Finally, we probed propargyl ester 14 with a lengthened linker. Under various conditions the only product we always detected was the very unstable enol ether 15, which, apparently, was formed through 1,2-shift of a hydride to a carbene atom. Eventually, we found conditions that provided a nearly quantitative yield of compound 15 (Scheme 7).
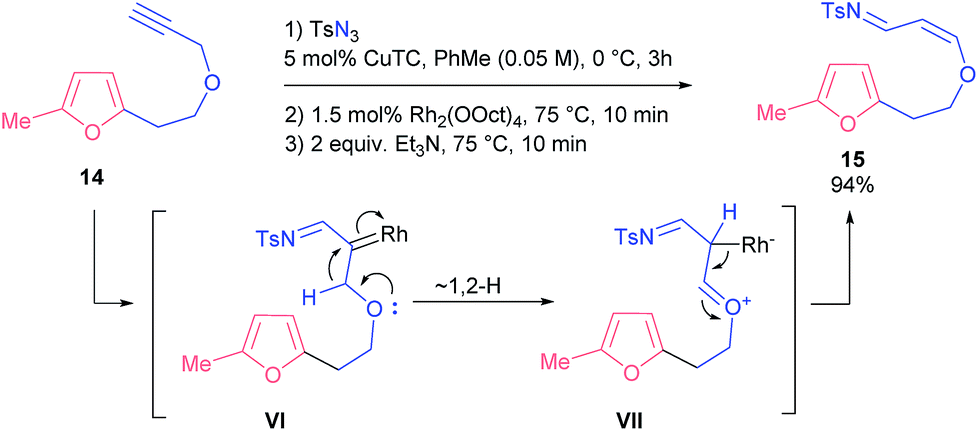 | ||
| Scheme 7 Reaction of propargyl ether 15. | ||
It is known that 1,2-hydride shifts often follow the denitrogenative decomposition of 4-alkyltriazoles, which reduces the yields of the target compounds.21 Possibly, this competing reaction also affects the outcome of the studied processes. It seems that the observed sigmatropic rearrangement is a kinetically controlled reaction in the case of substrate 14: both the longer tether and higher proclivity of the oxygen atom to donate its electron pair (compared to the NTs group) make enol 15 a predominant product.
2-Acylpyridines are known to be largely used for the synthesis22 of indolizines, indolizidines and their close structural analogues, which represent core frameworks for the vast number of alkaloids and biologically active compounds.23 We attempted to further extend the application of the obtained products for the synthesis of functionalized indolizines through trivial functional group manipulations. The addition of (phenylethynyl)lithium to a carbonyl group of 2-acetylpyridine 4a followed by treatment of the resulting propargyl alcohol with molecular iodine afforded indolizinium salt 16 in excellent yield (Scheme 8). The reduction of compound 16 with sodium borohydride directly led to 2-iodoindolizine 17 through the simultaneous dehydration of intermediate dihydroindolizinol. To the best of our knowledge, this synthetic pathway has not been reported previously.
 | ||
| Scheme 8 Synthesis of indolizine 17. Reaction conditions: (i) 1.5 equiv. (phenylethynyl)lithium, 3 equiv. LiBr, Et2O/PhH, RT, and 15 h; (ii) 1.5 equiv. I2, DCM, RT, and 5 h (93% over 2 steps); (iii) 2 equiv. NaBH4, MeOH, RT, and 30 min (99%). | ||
Conclusions
In conclusion, we studied some aspects of previously non-reported intramolecular reactions of azavinyl rhodium carbenoid species with furans. Our findings enabled us to develop a straightforward one-pot protocol toward fused 2-acetyl- and 2-formylpyridines from easily accessible biomass-derived furan building-blocks. We demonstrated that the obtained pyridines could be utilized for the synthesis of highly functionalized indolizines through the original high-yielding protocol. Additionally, we found that rearrangement of furans triggered by rhodium iminocarbenes could possibly proceed via two distinct pathways, thereby providing different heterocyclic frameworks. Our initial results highlight that intermolecular rearrangement of furyl triazoles has high potential as a practical tool for the design of various heterocyclic compounds. Further aspects of the discovered transformations will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ministry of Science and Higher Education of The Russian Federation (project No. 4.5371.2017/8.9) and Russian Foundation for Basic Research (grant No. 19-43-590007 p_a) for the financial support. ASM thanks the Ministry of Science and Higher Education of The Russian Federation for The President academic mobility scholarship.Notes and references
- O. Dimroth, Justus Liebigs Ann. Chem., 1909, 364, 183 CrossRef CAS.
- T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972 CrossRef CAS PubMed.
- (a) M. S. Singh, S. Chowdhury and S. Koley, Tetrahedron, 2016, 72, 5257 CrossRef CAS; (b) L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933 CrossRef CAS; (c) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC.
- (a) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS PubMed; (b) N. R. Candeias, R. Paterna and P. M. Gois, Chem. Rev., 2016, 116, 2937 CrossRef CAS PubMed; (c) X. Zhao, Y. Zhang and J. Wang, Chem. Commun., 2012, 48, 10162 RSC; (d) H. M. Davies and J. R. Denton, Chem. Soc. Rev., 2009, 38, 3061 RSC; (e) H. M. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed.
- (a) Y. Li, H. Yang and H. Zhai, Chem.–Eur. J., 2018, 24, 12757 CrossRef CAS PubMed; (b) Y. Jiang, R. Sun, X. Y. Tang and M. Shi, Chem.–Eur. J., 2016, 22, 17910 CrossRef CAS PubMed; (c) H. M. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151 RSC; (d) P. Anbarasan, D. Yadagiri and S. Rajasekar, Synthesis, 2014, 46, 3004 CrossRef CAS; (e) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862 ( Angew. Chem. , 2012 , 124 , 886 ) CrossRef CAS PubMed.
- (a) Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351 RSC; (b) F. A. Kucherov, L. V. Romashov, K. I. Galkin and V. P. Ananikov, ACS Sustainable Chem. Eng., 2018, 6, 8064 CrossRef CAS; (c) M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827 CrossRef CAS PubMed; (d) R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499 CrossRef CAS PubMed.
- A. T. Balaban, D. C. Oniciu and A. R. Katritzky, Chem. Rev., 2004, 104, 2777 CrossRef CAS PubMed.
- S. P. Roche and J. A. Porco Jr, Angew. Chem., Int. Ed., 2011, 50, 4068 ( Angew. Chem. , 2011 , 123 , 4154 ) CrossRef CAS PubMed.
- (a) N. Sun, X. Xie, H. Chen and Y. Liu, Chem.–Eur. J., 2016, 22, 14175 CrossRef CAS PubMed; (b) D. Lebœuf, M. Gaydou, Y. Wang and A. M. Echavarren, Org. Chem. Front., 2014, 1, 759 RSC; (c) Y. Chen, L. Wang, N. Sun, X. Xie, X. Zhou, H. Chen, Y. Li and Y. Liu, Chem.–Eur. J., 2014, 20, 12015 CrossRef CAS PubMed; (d) N. Huguet, D. Leboeuf and A. M. Echavarren, Chem.–Eur. J., 2013, 19, 6581 CrossRef CAS PubMed.
- (a) E. Y. Zelina, T. A. Nevolina, L. N. Sorotskaja, D. A. Skvortsov, I. V. Trushkov and M. G. Uchuskin, J. Org. Chem., 2018, 83, 11747 CrossRef CAS PubMed; (b) Y. Yang, C. Fei, K. Wang, B. Liu, D. Jiang and B. Yin, Org. Lett., 2018, 20, 2273 CrossRef CAS PubMed; (c) A. S. Makarov, M. G. Uchuskin and V. Gevorgyan, J. Org. Chem., 2018, 83, 14010 CrossRef CAS PubMed; (d) J. Guo, X. Xu, Q. Xing, Z. Gao, J. Gou and B. Yu, Org. Lett., 2018, 20, 7410 CrossRef CAS PubMed; (e) A. S. Hashmi, M. Rudolph, J. Huck, W. Frey, J. W. Bats and M. Hamzic, Angew. Chem., Int. Ed., 2009, 48, 5848 ( Angew. Chem. , 2009 , 121 , 5962 ) CrossRef CAS PubMed.
- (a) V. Lehner, H. M. L. Davies and O. Reiser, Org. Lett., 2017, 19, 4722 CrossRef CAS PubMed; (b) M. Marinozzi, M. C. Fulco, L. Amori, M. Fiumi and R. Pellicciari, Tetrahedron, 2009, 65, 7092 CrossRef CAS; (c) J. Hansen, B. Li, E. Dikarev, J. Autschbach and H. M. Davies, J. Org. Chem., 2009, 74, 6564 CrossRef CAS PubMed; (d) S. J. Hedley, D. L. Ventura, P. M. Dominiak, C. L. Nygren and H. M. Davies, J. Org. Chem., 2006, 71, 5349 CrossRef CAS PubMed; (e) A. Caballero, M. M. Diaz-Requejo, S. Trofimenko, T. R. Belderrain and P. J. Perez, J. Org. Chem., 2005, 70, 6101 CrossRef CAS PubMed.
- (a) H. M. L. Davies and R. L. Calvo, Tetrahedron Lett., 1997, 38, 5623 CrossRef CAS; (b) E. Wenkert, R. Decorzant and F. Näf, Helv. Chim. Acta, 1989, 72, 756 CrossRef CAS; (c) A. Padwa, T. J. Wisnieff and E. J. Walsh, J. Org. Chem., 1989, 54, 299 CrossRef CAS; (d) E. Wenkert, M. Guo, F. Pizzo and K. Ramachandran, Helv. Chim. Acta, 1987, 70, 14298 Search PubMed; (e) A. Padwa, T. J. Wisnieff and E. J. Walsh, J. Org. Chem., 1986, 51, 5036 CrossRef CAS.
- For examples of related intermolecular reactions of furans with a carbenoid generated from a non-diazo precursor, see J. M. Yang, X. Y. Tang and M. Shi, Chem.–Eur. J., 2015, 21, 4534 CrossRef CAS PubMed.
- (a) Z. F. Xu, X. Yu, D. Yang and C. Y. Li, Org. Biomol. Chem., 2017, 15, 3161 RSC; (b) R. Sun, Y. Jiang, X. Y. Tang and M. Shi, Chem.–Eur. J., 2016, 22, 5727 CrossRef CAS PubMed; (c) S. Shin, J. Y. Son, C. Choi, S. Kim and P. H. Lee, J. Org. Chem., 2016, 81, 11706 CrossRef CAS PubMed; (d) B. Seo, W. H. Jeon, J. Kim, S. Kim and P. H. Lee, J. Org. Chem., 2015, 80, 722 CrossRef CAS PubMed; (e) Y. Jiang, X. Y. Tang and M. Shi, Chem. Commun., 2015, 51, 2122 RSC; (f) Y. S. Zhang, X. Y. Tang and M. Shi, Chem. Commun., 2014, 50, 15971 RSC.
- For key examples of intermolecular reactions of furans with azavinyl carbenoids, see (a) Y. Funakoshi, T. Miura and M. Murakami, Org. Lett., 2016, 18, 6284 CrossRef CAS PubMed; (b) B. T. Parr, S. A. Green and H. M. Davies, J. Am. Chem. Soc., 2013, 135, 4716 CrossRef CAS PubMed.
- Z. Zeng, H. Jin, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018, 57, 16549 ( Angew. Chem. , 2018 , 130 , 16787 ) CrossRef CAS PubMed.
- (a) D. F. Vargas, E. L. Larghi and T. S. Kaufman, Eur. J. Org. Chem., 2018, 40, 5605 CrossRef; (b) A. Patel, J. R. Vella, Z. X. Ma, R. P. Hsung and K. N. Houk, J. Org. Chem., 2015, 80, 11888 CrossRef CAS PubMed; (c) M. L. Meketa, S. M. Weinreb, Y. Nakao and N. Fusetani, J. Org. Chem., 2007, 72, 4892 CrossRef CAS PubMed.
- (a) X. Ma, L. Liu, J. Wang, X. Xi, X. Xie and H. Wang, J. Org. Chem., 2018, 83, 14518 CrossRef CAS PubMed; (b) H. Dai, S. Yu, W. Cheng, Z. F. Xu and C. Y. Li, Chem. Commun., 2017, 53, 6417 RSC.
- (a) B. M. Zeglis, V. Divilov and J. S. Lewis, J. Med. Chem., 2011, 54, 2391 CrossRef CAS PubMed; (b) D. R. Richardson, D. S. Kalinowski, V. Richardson, P. C. Sharpe, D. B. Lovejoy, M. Islam and P. V. Bernhardt, J. Med. Chem., 2009, 52, 1459 CrossRef CAS PubMed; (c) J. Easmon, G. Purstinger, K. S. Thies, G. Heinisch and J. Hofmann, J. Med. Chem., 2006, 49, 6343 CrossRef CAS PubMed.
- (a) M. Raeisi, K. Kotturi, I. Del Valle, J. Schulz, P. Dornblut and E. Masson, J. Am. Chem. Soc., 2018, 140, 3371 CrossRef CAS PubMed; (b) M. Wang, C. Wang, X. Q. Hao, X. Li, T. J. Vaughn, Y. Y. Zhang, Y. Yu, Z. Y. Li, M. P. Song, H. B. Yang and X. Li, J. Am. Chem. Soc., 2014, 136, 10499 CrossRef CAS PubMed; (c) C. Wang, X.-Q. Hao, M. Wang, C. Guo, B. Xu, E. N. Tan, Y.-Y. Zhang, Y. Yu, Z.-Y. Li, H.-B. Yang, M.-P. Song and X. Li, Chem. Sci., 2014, 5, 1221 RSC; (d) S. Perera, X. Li, M. Guo, C. Wesdemiotis, C. N. Moorefield and G. R. Newkome, Chem. Commun., 2011, 47, 4658 RSC.
- (a) Y. Z. Zhao, H. B. Yang, X. Y. Tang and M. Shi, Chem.–Eur. J., 2015, 21, 3562 CrossRef CAS PubMed; (b) F. Medina, C. Besnard and J. Lacour, Org. Lett., 2014, 16, 3232 CrossRef CAS PubMed.
- (a) S. T. Heller, T. Kiho, A. R. Narayan and R. Sarpong, Angew. Chem., Int. Ed., 2013, 52, 11129 ( Angew. Chem. , 2013 , 125 , 11335 ) CrossRef CAS PubMed; (b) I. Kim, J. Choi and G. H. Lee, Synlett, 2008, 1243 CrossRef; (c) B. Yan, Y. Zhou, H. Zhang, J. Chen and Y. Liu, J. Org. Chem., 2007, 72, 7783 CrossRef CAS PubMed; (d) C. R. Smith, E. M. Bunnelle, A. J. Rhodes and R. Sarpong, Org. Lett., 2007, 9, 1169 CrossRef CAS PubMed.
- (a) B. Sadowski, J. Klajn and D. T. Gryko, Org. Biomol. Chem., 2016, 14, 7804 RSC; (b) J. P. Michael, Nat. Prod. Rep., 2008, 25, 139 RSC; (c) J. P. Michael, Nat. Prod. Rep., 2005, 22, 603 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures, analytical data, and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c9sc02299f |
| This journal is © The Royal Society of Chemistry 2019 |