
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting trimeric transmembrane domain 5 of oncogenic latent membrane protein 1 using a computationally designed peptide†
Yibo
Wang‡
ab,
Yinghua
Peng‡
c,
Bo
Zhang
a,
Xiaozheng
Zhang
a,
Hongyuan
Li
a,
Andrew J.
Wilson
 de,
Konstantin S.
Mineev
f and
Xiaohui
Wang
*ag
de,
Konstantin S.
Mineev
f and
Xiaohui
Wang
*ag
aLaboratory of Chemical Biology, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, China. E-mail: xiaohui.wang@ciac.ac.cn
bState Key Laboratory of Oncology in South China, Sun Yat-sen University, Guangzhou, Guangdong 510060, China
cState Key Laboratory for Molecular Biology of Special Wild Economic Animals, Institute of Special Animal and Plant Sciences, Chinese Academy of Agricultural Sciences, Changchun, Jilin 130112, China
dSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
eAstbury Centre for Structural Molecular Biology, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
fShemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Moscow, 117997, Russian
gDepartment of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, China
First published on 27th June 2019
Abstract
Protein–protein interactions are involved in diverse biological processes. These interactions are therefore vital targets for drug development. However, the design of peptide modulators targeting membrane-based protein–protein interactions is a challenging goal owing to the lack of experimentally-determined structures and efficient protocols to probe their functions. Here we employed rational peptide design and molecular dynamics simulations to design a membrane-insertable peptide that disrupts the strong trimeric self-association of the fifth transmembrane domain (TMD5) of the oncogenic Epstein–Barr virus (EBV) latent membrane protein-1 (LMP-1). The designed anti-TMD5 peptide formed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 heterotrimers with TMD5 in micelles and inhibited TMD5 oligomerization in bacterial membranes. Moreover, the designed peptide inhibited LMP-1 homotrimerization based on NF-κB activity in EVB positive lymphoma cells. The results indicated that the designed anti-TMD5 peptide may represent a promising starting point for elaboration of anti-EBV therapeutics via inhibition of LMP-1 oligomerization. To the best of our knowledge, this represents the first example of disrupting homotrimeric transmembrane helices using a designed peptide inhibitor.
:2 heterotrimers with TMD5 in micelles and inhibited TMD5 oligomerization in bacterial membranes. Moreover, the designed peptide inhibited LMP-1 homotrimerization based on NF-κB activity in EVB positive lymphoma cells. The results indicated that the designed anti-TMD5 peptide may represent a promising starting point for elaboration of anti-EBV therapeutics via inhibition of LMP-1 oligomerization. To the best of our knowledge, this represents the first example of disrupting homotrimeric transmembrane helices using a designed peptide inhibitor.
Introduction
Membrane proteins make up ∼30% of the human genome, and included in this group are high priority drug targets such as the G protein-coupled receptor (GPCR) family.1 Furthermore, protein–protein interactions in the membrane play vital roles in membrane protein assembly to coordinate a variety of cellular processes.2–4 These interactions within the membrane are therefore important potential targets for the development of chemical probes and therapeutics.5,6 Despite this opportunity, there are intrinsic challenges to study transmembrane domains of membrane proteins, including the low number of experimentally-determined structures and relatively few methods to probe the functions of these domains compared to their water-soluble counterparts.7–11Epstein–Barr virus (EBV) causes a prevalent infection that has been identified as the causative agent of a number of cancers including Burkitt lymphoma, B-cell lymphoma, and Hodgkin's disease.12–15 EBV infects B cells with transformation occurring as a consequence of viral proteins constantly activating signalling cascades for cellular proliferation, differentiation, and survival (Fig. 1).16 Central to this transformation process is the viral protein, latent membrane protein-1 (LMP-1), which constitutively activates NF-κB and other pathways through a direct interaction with tumor necrosis factor receptor-associated factors (TRAFs) via LMP-1's C-terminal domain.17–19 Furthermore, the signalling activity of LMP-1 is contingent on the oligomerization of its six transmembrane helix domains, specifically through transmembrane domain 5 (TMD5).20 Previous studies demonstrated that TMD5 can be trimerized by itself and polar amino acids, such as Asp150, drive transmembrane helix association.20,21
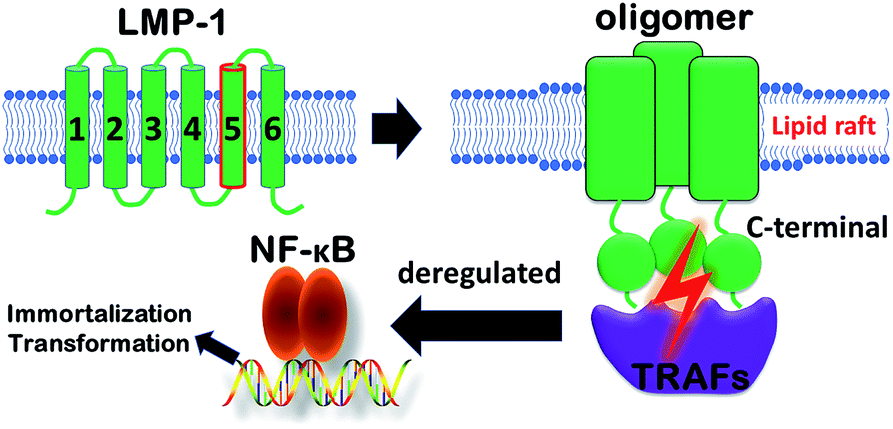 | ||
| Fig. 1 Schematic of transformation of EBV infected B cells. | ||
Recently small molecule inhibitors have been discovered by targeting LMP-1's TMD5 trimerization.21,22 Due to the large interfaces of protein–protein interactions (PPIs), it is challenging to disrupt PPIs at the membrane and the activities of small molecule inhibitors of TMD5 are moderate.23 Compared to small molecule agents, peptides have much larger interacting interfaces. Rational peptide design has been an efficient and reliable method for drugging the “undruggable” transmembrane domains of membrane proteins. Modulation of transmembrane protein dimerization via peptide modulators has been demonstrated in Toll-like receptors (TLRs),24,25 ErbB receptors,26 and integrins.27 However, no peptide inhibitors that target transmembrane protein trimerization have been unveiled. Herein, an anti-TMD5 peptide was designed to target trimeric TMD5 (Fig. 2a) of LMP-1 with better activity than TMD5 small molecule disruptors. Its ability to disrupt the assembly of the homotrimeric TMD5 complex was confirmed and characterized by biophysical and cell-based assays. The designed anti-TMD5 peptide represents a powerful starting point from which to further develop potent peptidomimetics for inhibiting LMP-1 signaling.
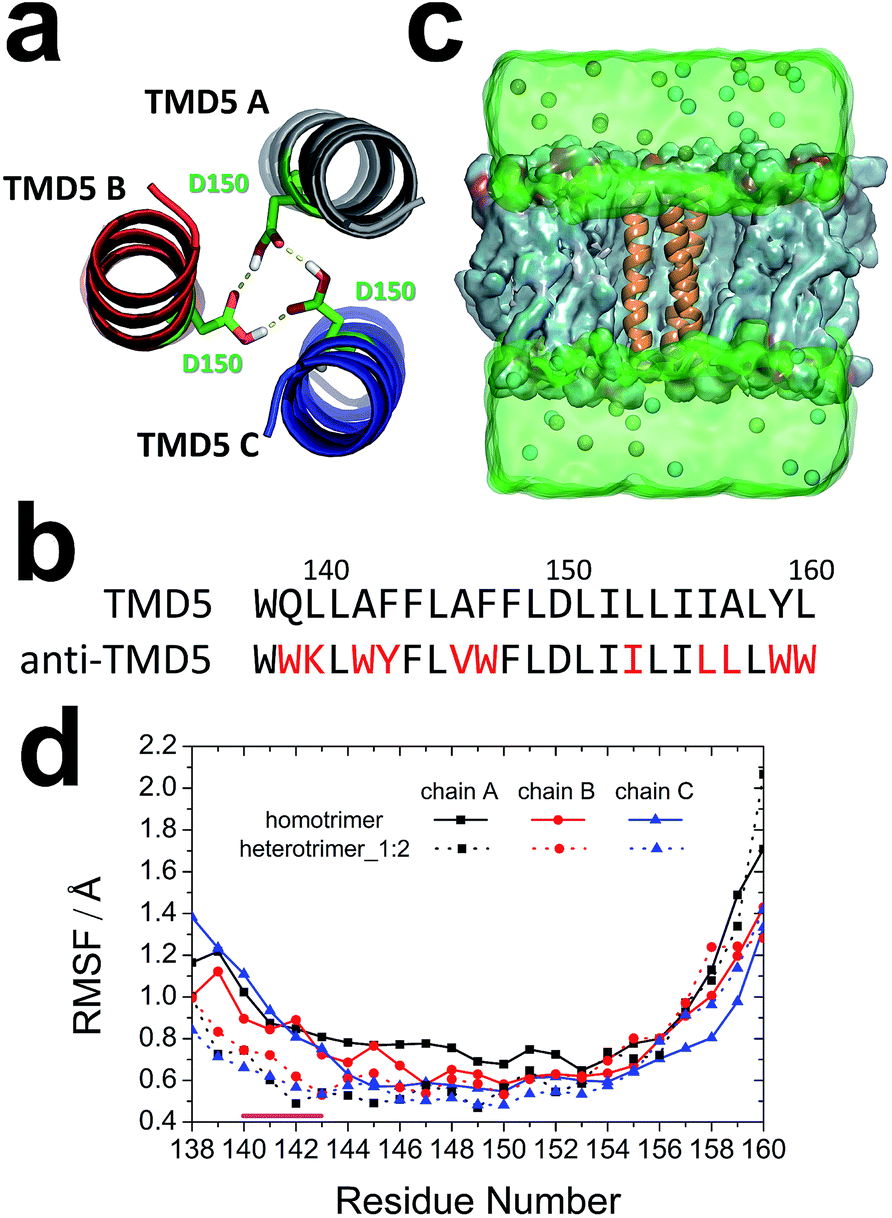 | ||
| Fig. 2 Computational design of anti-TMD5 based on a TMD5 homology model: (a) ribbon representation of homotrimeric TMD5 (chain A: grey, chain B: red, and chain C: blue). The key residues Asp150 are shown in stick representation and the hydrogen bonds between them are represented with yellow dashed lines; (b) sequence alignment of TMD5 and variant TMD5 (anti-TMD5). The variant residues in anti-TMD5 are highlighted in red; (c) representative illustration from an MD simulation; trimeric TMD5s (orange) are embedded in DMPC lipid bilayer (cyan). Chloride and potassium ions are shown as cyan and orange spheres, respectively. The simulated aqueous phase is shown as a green box; (d) RMSF of backbone of homotrimer and heterotrimer_1:2 (one anti-TMD5 and two TMD5s). The significant difference in motion of residues between the two coiled-coils is highlighted by a magenta line (residues 140–143). | ||
Results and discussion
Rosetta rational peptide design
Rational protein design has been applied to design enzymes and peptide modulators.28–31 In this study, 50 designs were first generated using the Rosetta molecular modelling package,32,33 and the best anti-TMD5 sequence (WWKLWYFLVWFLDLIILILLLWW) (Fig. 2b) was selected for further analysis based on the RosettaDesign energy scores.34–37Molecular dynamics simulations
The heterotrimer system (named as heterotrimer_1:2) with one anti-TMD5 (chain A in the trimer) and two TMD5 monomers was built based on the homotrimeric TMD5 (Fig. 2c). The stabilities of the homotrimer and heterotrimer_1:2 in a DMPC membrane were validated via the root-mean-square deviation (RMSD). As shown in Fig. S1,† the homotrimer and heterotrimer_1:2 reached a stable conformation in 10 ns. Hence the last 90 ns of the 100 ns simulation trajectories were employed for subsequent analyses. The backbone root-mean-square fluctuation (RMSF) of each transmembrane helix in the homotrimer and the heterotrimer_1:2 was calculated to unveil the fluctuation of each residue during the last 90 ns of simulation. The RMSF values for residues 140–143 of heterotrimer_1:2 were smaller than those observed for the homotrimer in Fig. 2d, indicating this region of the trimeric coiled-coil became more rigid after the anti-TMD5 replaced a monomer of the homotrimer.
One water molecule was observed to be captured by the homotrimer in 11 ns (Fig. 3a). It has been proven that solvent accessibility decreases the stability of transmembrane coiled-coil trimers in lipid bilayers.38 After the water molecule moved into the lumen from the bulk water, it first localized to a deeper site (location 1 in Fig. 3a) and coordinated with Ala146 and Asp150 for ∼35 ns. Then it jumped to a more stable site (location 2 in Fig. 3a) to interact with Ala146, Phe147 and Asp150 and remained in this location for more than 50 ns. However, no water molecules were found in the lumen of heterotrimer_1:2 because all residues observed to interact with water in the homotrimer were varied for other residues in chain A of the heterotrimer_1:2 except Asp150.
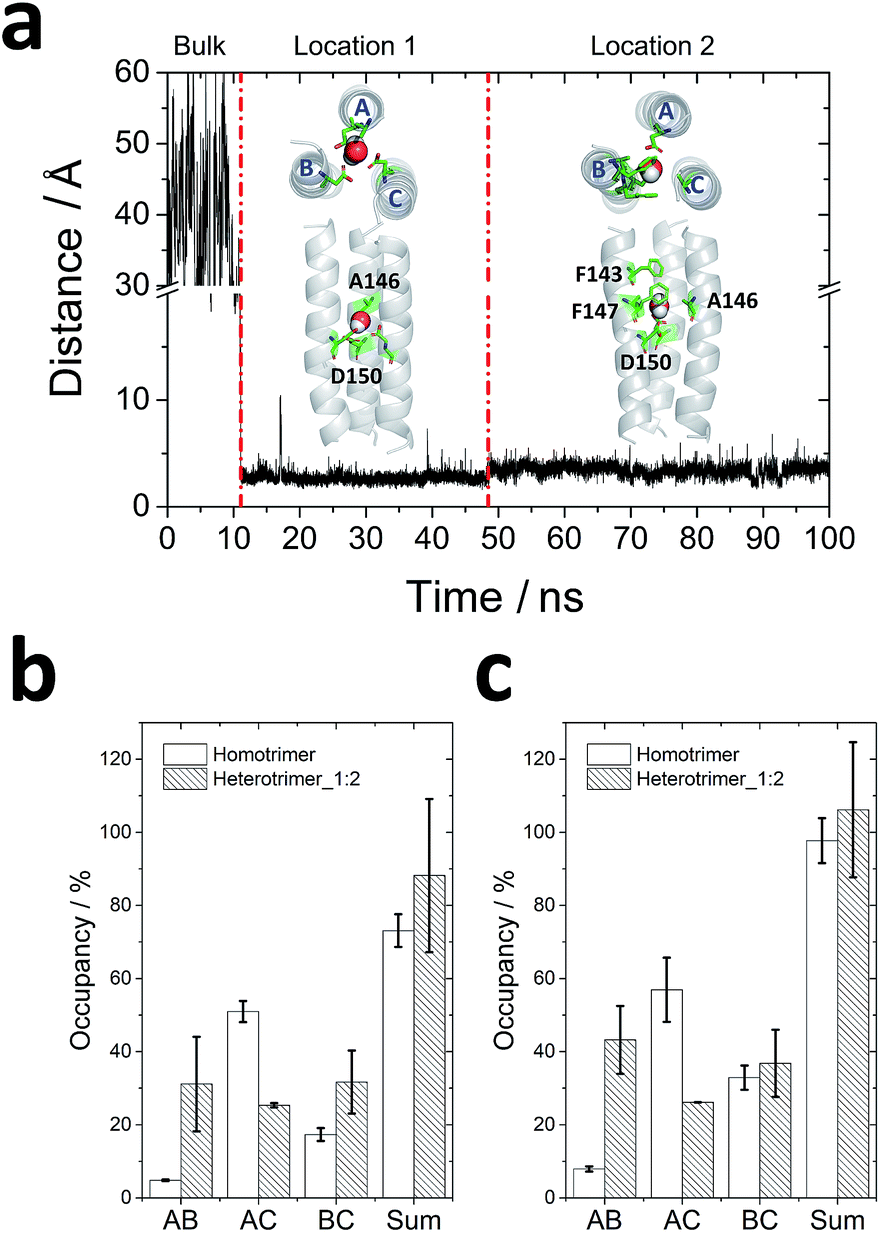 | ||
| Fig. 3 Molecular dynamics simulations for trimeric interactions: (a) water molecule locations in the homotrimer homology model. The distance between the oxygen of the water molecule and the center of mass of the Cα of the homotrimer homology model were calculated. Based on the distance, the water molecule experienced three states: in the bulk water, in location 1 and in location 2. The top view and side view of conformations of water in two locations of homotrimeric TMD5 (grey) are displayed, respectively, with key amino acid residues observed to interact with the water molecule labelled (green sticks); (b and c) hydrogen bond occupancy formed by Asp150 (b) and by all residues (c) between each chain. The order of chains A, B and C is displayed in Fig. 2a and (a). | ||
To analyse the interactions between each of the (two) helices, the occupancy of hydrogen bonds (occupancy = number of hydrogen bonds/total number of frames in the trajectory) between Asp150 of two monomers during the last 50 ns of each trajectory was counted, since one water molecule reached its most stable location in the homotrimer homology model after 50 ns. As shown in Fig. 3b, Asp150 of chain B in the homotrimer barely formed hydrogen bonds with Asp150 from other chains because of the existence of a water molecule. Most hydrogen bonds formed between Asp150s were from chain A and chain C. Therefore, these observations suggest water molecules in the lumen can screen intermolecular interactions within trimers and decrease their stability in the lipid bilayer. Gln139 is another key residue that forms hydrogen bonds stabilizing the homotrimer (occupancy = 24.55 ± 1.73%). It is also a vital residue to capture the water molecule from the bulk according to the molecular dynamics simulations. For the heterotrimer_1:2, hydrogen bonds between the Asp150s were well distributed amongst each chain because no water molecule was observed to compete with these interactions. A more even distribution of interactions fosters greater trimer stability in the heterotrimer_1:2. Since Gln139 in chain A of homotrimer was varied for tryptophan in heterotrimer_1:2 (Fig. S2†), it cannot form hydrogen bonds any more. Instead, Tyr143 of anti-TMD5 (chain A) interacted with Ala142 of chain B or C (Fig. S2†), and the interactions locked the region comprising residues 140–143, accounting for the increased rigidity observed in the simulations (Fig. 2d). The total number of hydrogen bonds observed between each of the two chains (Fig. 3c) follows a similar trend to those observed for Asp150 (Fig. 3b). The occupancy of hydrogen bonds thus indicates that the anti-TMD5 peptide can form stronger interactions with chain B and provide an even distribution of interactions in the heterotrimer_1:2. This may rationalize the stabilizing effect on the whole system observed for anti-TMD5 in silico, providing a basis upon which the designed anti-TMD5 may disrupt the lateral association of transmembrane helices of TMD5 as a peptide inhibitor.
Biophysical chemistry characterization
To validate the effects of anti-TMD5 in vitro, the peptide sequence was synthesized. Four lysine (K) residues were added on both the N- & C-termini of anti-TMD5 to increase water solubility, which did not affect TMD5 trimerization.22 Circular dichroism (CD) spectra of anti-TMD5 in the presence of C14 betaine (3-(N,N-dimethylmyristyl-ammonio)propanesulfonate) micelle exhibited a typical α-helical conformation (76%) with two minima at 208 nm and 220 nm (Fig. S3†).To test whether anti-TMD5 could inhibit TMD5 self-association, fluorescence dequenching experiments were performed. TMD5 forms a homotrimer in the presence of C14 betaine micelles, resulting in coumarin fluorescence self-quenching. Disruption of TMD5 oligomerization dequenches the coumarin dye, leading to fluorescence enhancement.21,22 As shown in Fig. 4a, anti-TMD5 was found to efficiently disrupt TMD5 self-association and reverse the coumarin fluorescence quenching. In contrast, the anti-TMD5 scrambled control peptide, where Asp150 was kept, did not cause the coumarin fluorescence of TMD5 to increase, demonstrating the specific interaction of anti-TMD5 with TMD5. Compared to TMD-5 disruptor NSC259242 (ref. 21) (EC50 = 27.9 ± 3.9 μM), anti-TMD5 (EC50 = 3.3 ± 0.2 μM) showed ∼10-fold increase in disruptor activity.
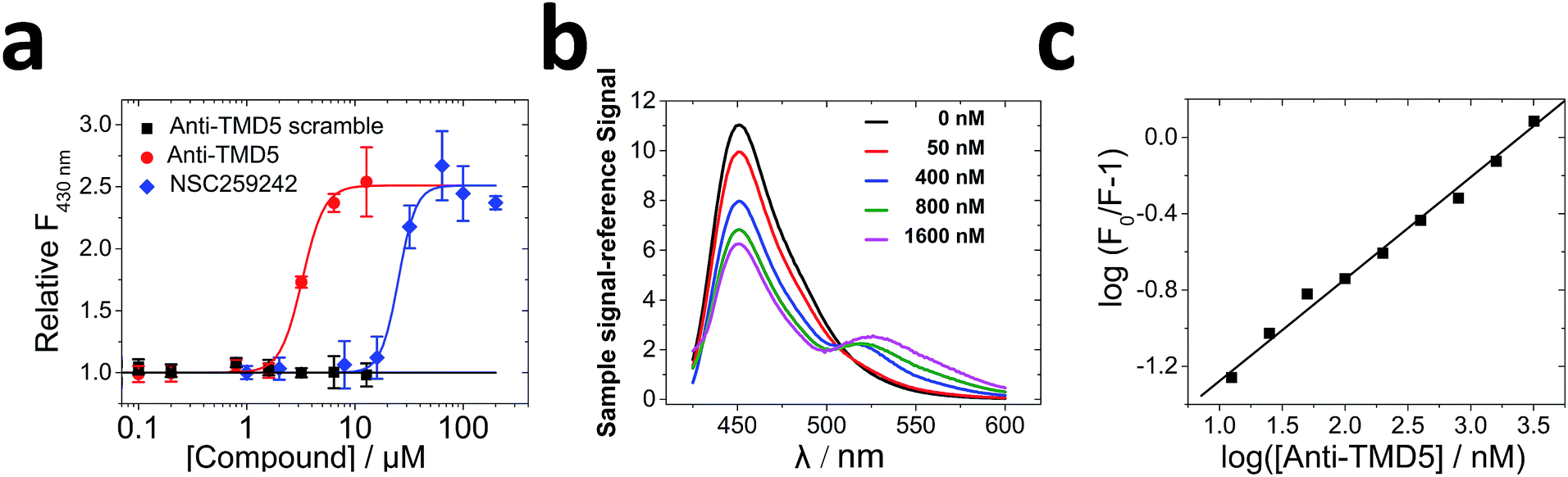 | ||
| Fig. 4 Fluorescence dequenching and FRET assays: (a) anti-TMD5 disrupts LMP-1 TMD5 self-associations in the micelle and causes coumarin-tagged TMD5 fluorescence dequenching. Different concentrations of unlabelled anti-TMD5 scrambled control or anti-TMD5 or NSC259242 were added into 50 nM of coumarin labelled TMD-5 solution (50 mM HEPES, pH = 7.4) with 150 μM C14 betaine. Excitation, 360 nm; emission, 430 nm; (b) fluorescence emission scans of coumarin-labelled TMD-5 (50 nM) in the presence of different concentrations of FITC-tagged anti-TMD-5 in 20 mM HEPES, pH = 7.4, containing 1.0 mM C14 betaine at room temperature. λex was set at 415 nm to selectively excite the coumarin. λex, excitation wavelength; (c) the quenching of coumarin labelled TMD-5 fluorescence by anti-TMD-5 binding. A stoichiometry n = 0.53 ± 0.02 and an apparent Kd of 67.8 ± 7.5 nM were obtained. | ||
Fluorescence resonance energy transfer (FRET) was used to further investigate the association of anti-TMD5 with TMD5 in micelles. The titration of coumarin-tagged TMD5 as a FRET donor, with FITC-tagged anti-TMD5 as a FRET acceptor, resulted in the quenching of the coumarin emission and the appearance of the FITC emission, indicating that the two peptides interacted (Fig. 4b). Compared to anti-TMD5, anti-TMD5 scrambled control peptide showed much less quenching of TMD5-coumarin fluorescence and weaker FRET signal from FITC emission (Fig. S4†). These results agree well with TMD5-coumarin fluorescence dequenching experiments and further support the specificity of anti-TMD5. By analysing the quenching of coumarin-tagged TMD5 fluorescence in the C14 betaine micelle by FITC-tagged anti-TMD5, an apparent dissociation constant (Kd) for the anti-TMD5:TMD5 interaction was determined to be 67.8 ± 7.5 nM (Fig. 4c), and a stoichiometry (n) of 0.53 ± 0.02 was derived. The behaviour of bilayers is different from micelles, which are more dynamic. The interactions of TMD5 with anti-TMD5 was also investigated in bicelles composed of POPC and DHPC (q = 0.3). An apparent dissociation constant of 40.0 ± 6.7 μM and a stoichiometry of 0.44 ± 0.03 were obtained for anti-TMD5:TMD5 interaction in bicelles (Fig. S5†). The results show that anti-TMD5 has slightly stronger binding affininty towards TMD5 in bicelles than in micelles and consistently indicates that two TMD5s interact with one anti-TMD5. To investigate why two anti-TMD5 with one TMD5 (named as heterotrimer_2:1) were not preferred for trimerization, further molecular dynamics simulations were carried out. It was observed that the variant Trp139s in anti-TMD5s fulfilled the function of paddles to trap water molecules in the lumen; four water molecules were captured in the lumen of the heterotrimer_2:1 (Fig. S6†). The occupancy of hydrogen bonds formed between Asp150s of chain A and chain B (two anti-TMD5s) was observed to be less than 1% in these simulations, indicating that two anti-TMD5 and one TMD5 could not form a stable trimer.
To cross validate the biophysical results, the anti-TMD5/TMD5 interaction was investigated using a dominant-negative ToxR assay (Fig. 5a) in bacterial membranes. ToxR is a bacterial cell-based transcription reporter assay that detects transmembrane domain interactions within a cellular membrane.27,39–42 Specifically, a fusion protein consisting of a transmembrane domain flanked at the N-terminus by the DNA binding domain of Vibrio cholerae ToxR protein and at the C-terminus by maltose binding protein (MBP) is expressed in E. coli strain FHK12, which has the ToxR target, the cholera toxin (ctx) promoter, upstream of the β-galactosidase open reading frame. When expressed in E. coli, this fusion protein will localize and orient within the bacterial inner membrane. The ToxR is only active in a dimeric form to cause lacZ transcription with the product of β-galactosidase, and the activity of β-galactosidase expressed in this system is proportional to the strength of oligomerization of the transmembrane domains. In the dominant-negative assay, the interaction of heterocomplexes of TMDs can be observed by using an inactive mutant ToxR fusion (ToxR*) with an active ToxR, and comparing the β-galactosidase activity to the corresponding homotypic interaction. As illustrated in Fig. 5b, integrin αIIb and anti-αIIb27,43 were chosen as the model system to validate the heterotypic interaction assay. The ToxR signal (Miller unit) for the TMD5–ToxR construct was attenuated by co-expression of anti-TMD5–ToxR*, but not by co-expression of anti-TMD5 scramble-ToxR* or poly-Leu-ToxR*. The results showed that anti-TMD5 specifically interacted with TMD5 while anti-TMD5 scramble and poly-Leu controls failed to significantly interact with TMD5.
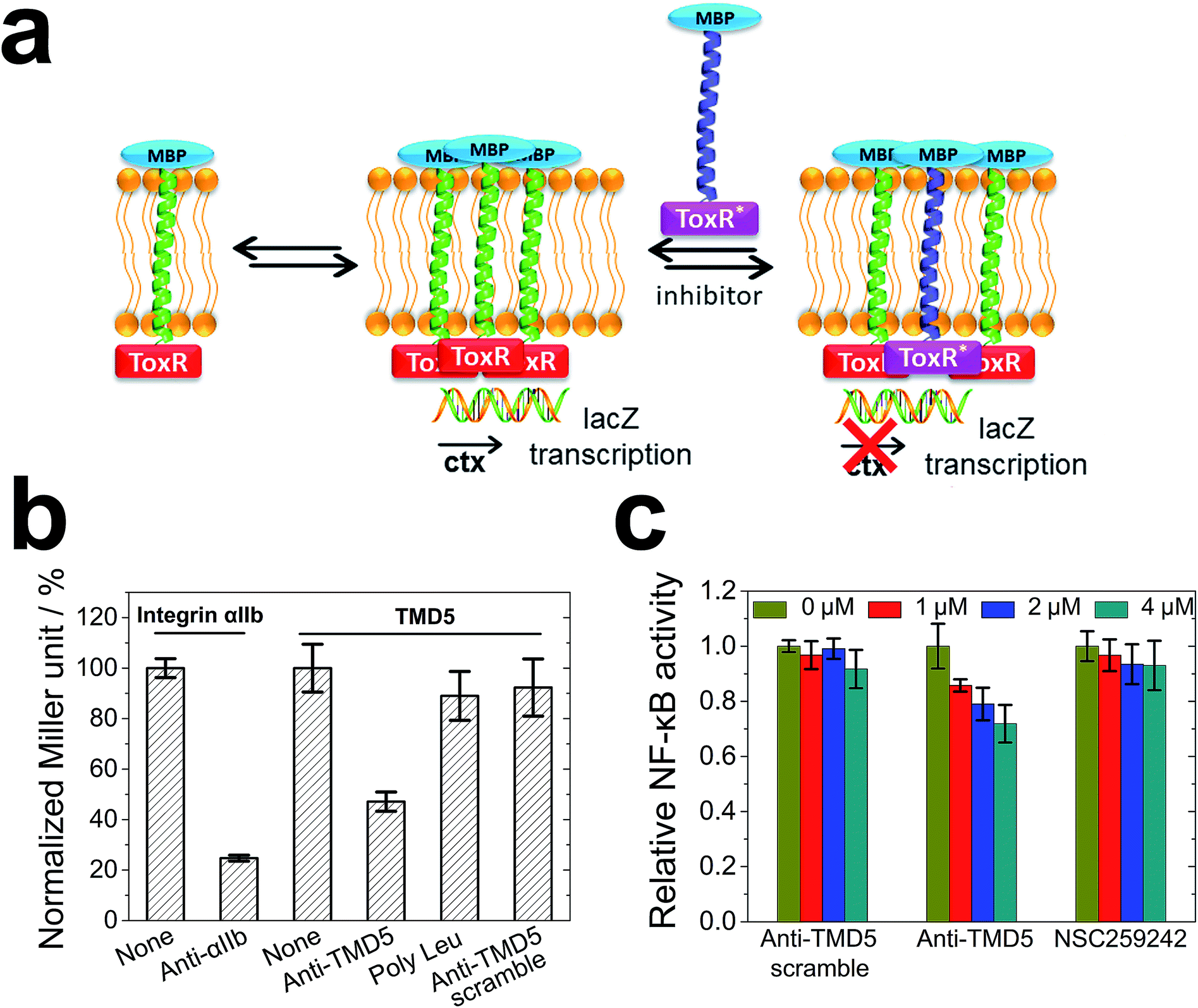 | ||
| Fig. 5 ToxR and NF-κB assays: (a) schematic representation of the ToxR assay used for testing LMP-1 TMD5 disruptors; (b) dominant-negative ToxR assay. The anti-TMD5 reduced the TMD5 Miller unit of the TMD5/TMD5 interaction by over 50%, whereas the controls (anti-TMD5 scramble and poly-Leu) did not inhibit the TMD5 Miller unit; (c) anti-TMD5 inhibited NF-κB activity in EBV positive B721 cells. Following 24 h treatment, the constitutive LMP-1 NF-κB activity in B721 cells was determined by a Steady-Glo Luciferase Assay System. The NF-κB activity of the untreated cells was normalized as 1. | ||
TMD-5 trimerization is essential for LMP-1 signalling NF-κB activation.20 In order to evaluate whether anti-TMD5 could inhibit LMP-1 signalling, an NF-κB signalling assay was performed. Naive B cells are the target of EBV infection in vivo,44,45 and thus an immortalized EBV positive B721 NF-κB reporter cell line was selected for investigating the effect of anti-TMD5 on LMP-1 signalling. As shown in Fig. 5c, anti-TMD5 inhibited NF-κB activity in EBV positive B721 cells in a concentration dependent manner, while anti-TMD5 scramble and NSC259242, a TMD5 small molecule inhibitor with an IC50 of 29.6 ± 6.6 μM in inhibiting B721 cell NF-κB activity as a control,21 did not significantly affect the NF-κB activity at the tested concentration range of 0–4 μM. The effects of anti-TMD5 on cell viability were also assessed in WST-1 assay. Anti-TMD5 and anti-TMD5 scramble showed no apparent cellular toxicity (Fig. S7†), which eliminates the possibility that the observed NF-κB inhibition is due to the artifact of growth inhibition.
Conclusions
Membrane proteins play vital roles in a tremendous number of crucial biological functions. The transmembrane domains have been deemed “undruggable” compared to their water soluble counterparts. In this study, we have exemplified a strategy to design a peptide to inhibit the homotrimeric self-association of TMD5 by formation of an asymmetric heterotrimer by the combination of computational and experimental methods. A peptide inhibitor was first designed in silico, and, the stability of the trimer was investigated via molecular dynamics simulations when one or two of three TMD5s were replaced by the designed anti-TMD5 peptide. A single anti-TMD5 peptide inhibitor elicited strong ability to stabilize a heterotrimer assembly by obscuring water molecules from the lumen and forming evenly distributed intermolecular interactions. The TMD5 targeting and inhibiting properties of anti-TMD5 were further cross validated and characterized by biophysical, biochemical and cellular assays. The results established the anti-TMD5 peptide inhibitor as a promising starting point to elaborate drug candidates for treatment of EBV infection. More broadly, this study provides a proof of concept for the development of peptides that exert a biological effect by targeting membrane-based protein–protein interactions to modulate the supramolecular assembly of membrane proteins.Data availability
The datasets generated and/or analysed during the current study are available from the corresponding authors on request.Author contributions
Y. W., Y. P., and X. W. designed research; Y. W., Y. P., B. Z., and X. Z. performed research; Y. W., Y. P., B. Z., X. Z., H. L., A. J. W., K. S. M. and X. W. analysed data; and Y. W., Y. P., B. Z., X. Z., H. L., A. J. W., K. S. M. and X. W. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21602216, 21877106, 21807098), the 100 Talents Program of Chinese Academy of Sciences, the Young Talents Program of Chinese Academy of Agricultural Sciences, the Central Public-Interest Scientific Institution Basal Research Fund (NO. 1610342016013), the Natural Science Foundation of Jilin Province (20180520059JH, 20180101021JC), the Open Funds of State Key Laboratory of Oncology in South China (HN2018-07) and the Royal Society-Newton Advanced Fellowship (NA170152). Computing time was supported by the Network and Computing Center, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, the National Supercomputer Center in Guangzhou and the Computing Center of Jilin Province. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.Notes and references
- M. Ahram, Z. I. Litou, R. Fang and G. Al-Tawallbeh, In Silico Biol., 2006, 6, 379–386 CAS.
- T. A. Stone and C. M. Deber, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 577–585 CrossRef CAS PubMed.
- D. M. Hershey, X. F. Ren, R. A. Melnyk, P. J. Browne, E. Ozyamak, S. R. Jones, M. C. Y. Chang, J. H. Hurley and A. Komeili, PLoS Biol., 2016, 14, e1002402 CrossRef PubMed.
- R. A. Brown, V. Diemer, S. J. Webb and J. Clayden, Nat. Chem., 2013, 5, 853–860 CrossRef CAS PubMed.
- C. M. Grison, J. A. Miles, S. Robin, A. J. Wilson and D. J. Aitken, Angew. Chem., Int. Ed. Engl., 2016, 55, 11096–11100 CrossRef CAS PubMed.
- A. J. Wilson, Chem. Soc. Rev., 2009, 38, 3289–3300 RSC.
- H. Yin, Angew. Chem., Int. Ed. Engl., 2008, 47, 2744–2752 CrossRef CAS PubMed.
- T. X. Zhao, A. J. Martinko, V. H. Le, J. Zhao and H. Yin, Curr. Pharm. Des., 2010, 16, 1055–1062 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- S. H. White, Protein Sci., 2004, 13, 1948–1949 CrossRef CAS PubMed.
- M. De Poli, W. Zawodny, O. Quinonero, M. Lorch, S. J. Webb and J. Clayden, Science, 2016, 352, 575–580 CrossRef CAS PubMed.
- G. Brady, G. J. MacArthur and P. J. Farrell, J. Clin. Pathol., 2007, 60, 1397–1402 CAS.
- G. Henle, W. Henle and V. Diehl, Proc. Natl. Acad. Sci. U. S. A., 1968, 59, 94–101 CrossRef CAS PubMed.
- Z. M. Zheng, Int. J. Biol. Sci., 2010, 6, 730–755 CrossRef CAS PubMed.
- K. J. Flavell and P. G. Murray, Mol. Pathol., 2000, 53, 262–269 CrossRef CAS PubMed.
- B. Sugden, Cell, 1989, 57, 5–7 CrossRef CAS PubMed.
- U. Dirmeier, B. Neuhierl, E. Kilger, G. Reisbach, M. L. Sandberg and W. Hammerschmidt, Cancer Res., 2003, 63, 2982–2989 CAS.
- K. M. Kaye, K. M. Izumi and E. Kieff, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 9150–9154 CrossRef CAS PubMed.
- D. Wang, D. Liebowitz and E. Kieff, Cell, 1985, 43, 831–840 CrossRef CAS PubMed.
- D. W. Sammond, C. Joce, R. Takeshita, S. E. McQuate, N. Ghosh, J. M. Martin and H. Yin, Biopolymers, 2011, 95, 772–784 CAS.
- X. H. Wang, J. P. Saludes, T. X. Zhao, A. Csakai, Z. Fiorini, S. A. Chavez, J. Li, G. I. Lee, K. Varga and H. Yin, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 2282–2289 CrossRef CAS PubMed.
- X. H. Wang, Z. Fiorini, C. Smith, Y. N. Zhang, J. Li, L. R. Watkins and H. Yin, PLoS One, 2012, 7, e47703 CrossRef CAS PubMed.
- X. Zeng, P. Wu, C. Yao, J. Liang, S. Zhang and H. Yin, Biochemistry, 2017, 56, 2076–2085 CrossRef CAS PubMed.
- A. Fink, E. M. Reuven, C. J. Arnusch, L. Shmuel-Galia, N. Antonovsky and Y. Shai, J. Immunol., 2013, 190, 6410–6422 CrossRef CAS PubMed.
- L. Shmuel-Galia, T. Aychek, A. Fink, Z. Porat, B. Zarmi, B. Bernshtein, O. Brenner, S. Jung and Y. Shai, EMBO J., 2016, 35, 685–698 CrossRef CAS PubMed.
- A. Bennasroune, M. Fickova, A. Gardin, S. Dirrig-Grosch, D. Aunis, G. Cremel and P. Hubert, Mol. Biol. Cell, 2004, 15, 3464–3474 CrossRef CAS PubMed.
- H. Yin, J. S. Slusky, B. W. Berger, R. S. Walters, G. Vilaire, R. I. Litvinov, J. D. Lear, G. A. Caputo, J. S. Bennett and W. F. DeGrado, Science, 2007, 315, 1817–1822 CrossRef CAS PubMed.
- S. Q. Zhang, H. Huang, J. J. Yang, H. T. Kratochvil, M. Lolicato, Y. X. Liu, X. K. Shu, L. J. Liu and W. F. DeGrado, Nat. Chem. Biol., 2018, 14, 870–875 CrossRef CAS PubMed.
- F. Findeisen, M. Campiglio, H. Jo, F. Abderemane-Ali, C. H. Rumpf, L. Pope, N. D. Rossen, B. E. Flucher, W. F. DeGrado and D. L. Minor, ACS Chem. Neurosci., 2017, 8, 1313–1326 CrossRef CAS PubMed.
- S. P. Ho and W. F. Degrado, J. Am. Chem. Soc., 1987, 109, 6751–6758 CrossRef CAS.
- B. Kuhlman, G. Dantas, G. C. Ireton, G. Varani, B. L. Stoddard and D. Baker, Science, 2003, 302, 1364–1368 CrossRef CAS PubMed.
- A. Leaver-Fay, M. Tyka, S. M. Lewis, O. F. Lange, J. Thompson, R. Jacak, K. Kaufman, P. D. Renfrew, C. A. Smith, W. Sheffler, I. W. Davis, S. Cooper, A. Treuille, D. J. Mandell, F. Richter, Y. E. Ban, S. J. Fleishman, J. E. Corn, D. E. Kim, S. Lyskov, M. Berrondo, S. Mentzer, Z. Popovic, J. J. Havranek, J. Karanicolas, R. Das, J. Meiler, T. Kortemme, J. J. Gray, B. Kuhlman, D. Baker and P. Bradley, Methods Enzymol., 2011, 487, 545–574 CAS.
- N. Koga, R. Tatsumi-Koga, G. Liu, R. Xiao, T. B. Acton, G. T. Montelione and D. Baker, Nature, 2012, 491, 222–227 CrossRef CAS PubMed.
- V. D. Sood and D. Baker, J. Mol. Biol., 2006, 357, 917–927 CrossRef CAS PubMed.
- R. Jacak, A. Leaver-Fay and B. Kuhlman, Proteins, 2012, 80, 825–838 CrossRef CAS PubMed.
- B. Kuhlman and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10383–10388 CrossRef CAS PubMed.
- C. A. Rohl, C. E. M. Strauss, K. M. S. Misura and D. Baker, Methods Enzymol., 2004, 383, 66–93 CAS.
- A. Piai, J. Dev, Q. Fu and J. J. Chou, J. Am. Chem. Soc., 2017, 139, 18432–18435 CrossRef CAS PubMed.
- E. Lindner and D. Langosch, Proteins, 2006, 65, 803–807 CrossRef CAS PubMed.
- D. Langosch, B. Brosig, H. Kolmar and H. J. Fritz, J. Mol. Biol., 1996, 263, 525–530 CrossRef CAS PubMed.
- W. P. Russ and D. M. Engelman, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 863–868 CrossRef CAS PubMed.
- C. Joce, A. Wiener and H. Yin, J. Visualized Exp., 2011, 51, e2721 Search PubMed.
- S. J. Shandler, I. V. Korendovych, D. T. Moore, K. B. Smith-Dupont, C. N. Streu, R. I. Litvinov, P. C. Billings, F. Gai, J. S. Bennett and W. F. DeGrado, J. Am. Chem. Soc., 2011, 133, 12378–12381 CrossRef CAS PubMed.
- D. A. Thorley-Lawson, Nat. Rev. Immunol., 2001, 1, 75–82 CrossRef CAS PubMed.
- R. Kuppers, Nat. Rev. Immunol., 2003, 3, 801–812 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02474c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |