
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Borazatruxenes†
Simone
Limberti‡
,
Liam
Emmett‡
,
Anamaria
Trandafir
,
Gabriele
Kociok-Köhn
and
G. Dan
Pantoş
 *
*
Department of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: g.d.pantos@bath.ac.uk
First published on 28th August 2019
Abstract
We report the synthesis and characterization of a series of arene-borazine hybrids called borazatruxenes. These molecules are BN-isosteres of truxene whereby the central benzene core has been replaced by a borazine ring. The straightforward three step synthesis, stability and their chiroptical and electronic properties recommend them as new scaffolds for BN–carbon hybrid materials. Computational studies at DFT level, closely matching the experimental data, provided insights in the electronic structure of these molecules.
Introduction
The interest in polycyclic aromatic hydrocarbons (PAHs) has grown over the past decade due to their stability, tuneable properties and versatility.1–3 This has made them ideal candidates for use as semiconductors in the next generation of organic electronic components.4,5 Moreover, the electronic properties of PAHs are intimately related to their structure; therefore small modifications in their scaffold can lead to vastly different optical and electronic properties.6–14Truxene is a C3-symmetric PAH that has been intensively studied for the past 15–20 years.15 In particular, it has been used as a precursor to polyarenes,16,17 liquid crystals18,19 and hemi-fullerenes,20,21 all of which have potential to be used in organic electronics.22 Many truxene derivatives have been synthesised23–25 and utilized for their light emitting and light harvesting properties26–28 while other analogues have found applications in dendrimers,29–31 gels,32,33 photoresists,34,35 fluorescent probes,36,37 and two photon absorbers.38,39
The B–N bond is quasi-isosteric and isoelectronic with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond,40,41 except that it is more polarized42 due to the difference in electronegativity between the B and N atoms. Therefore, replacing a CC with a B–N in a PAH will lead to quasi-identical geometries but drastically different electronic structures.43 This concept has been at the core of the development of BN doped graphenes and BN-PAHs. Borazine containing PAHs are prone to hydrolytic decomposition which delayed the development of this field.43 In most cases, this reactivity is mitigated by steric bulk,44,45 introduction of electron rich substituents or structural confinement of the borazine ring.45–52
C bond,40,41 except that it is more polarized42 due to the difference in electronegativity between the B and N atoms. Therefore, replacing a CC with a B–N in a PAH will lead to quasi-identical geometries but drastically different electronic structures.43 This concept has been at the core of the development of BN doped graphenes and BN-PAHs. Borazine containing PAHs are prone to hydrolytic decomposition which delayed the development of this field.43 In most cases, this reactivity is mitigated by steric bulk,44,45 introduction of electron rich substituents or structural confinement of the borazine ring.45–52
Results and discussion
Herein we introduce a new class of borazine-PAHs that are moisture stable§ and easy to synthesise. Borazatruxenes are truxene analogues in which the central benzene core has been replaced by borazine. The B atom is directly linked to the phenylene rings which are connected via a methylene bridge to the N atom of the borazine core. This particular arrangement is responsible for the borazatruxenes' hydrolytic stability. Borazatruxenes 1–3 were synthesised according to the synthetic pathway illustrated in Scheme 1.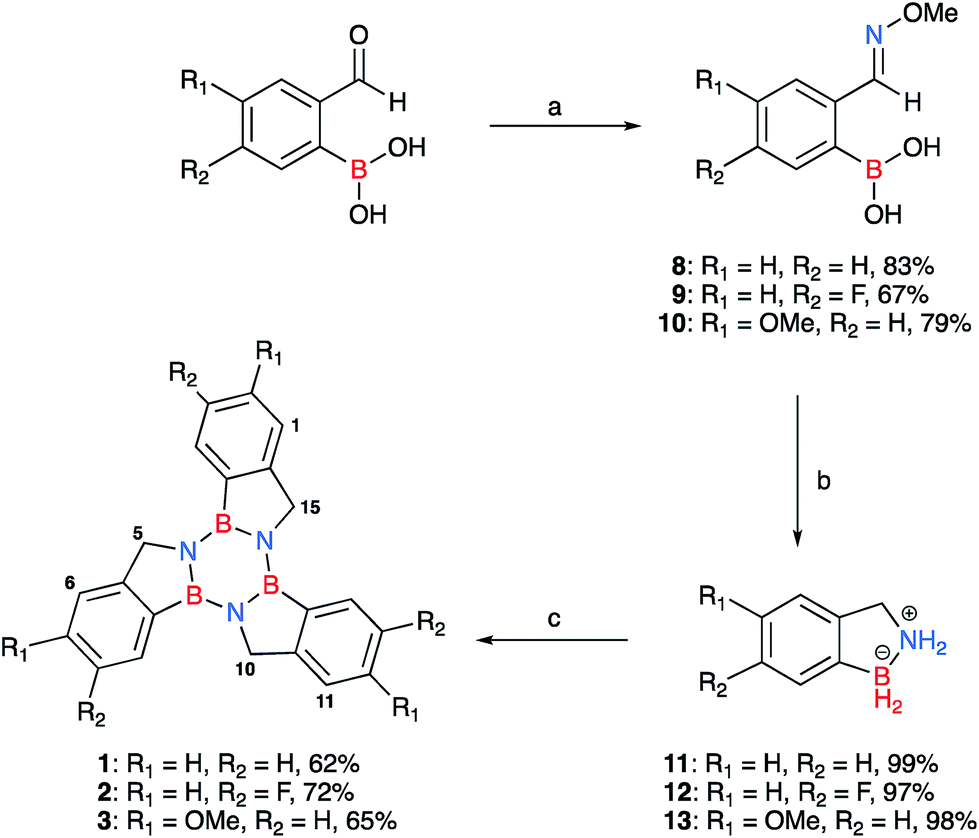 | ||
| Scheme 1 Synthesis of borazatruxenes 1–3 from substituted 2-formylphenylboronic acids; the isolated yields for each derivative are given after their descriptor. (a) (i) H2O, 45 °C, 15 min, (ii) NH2OMe·HCl (1.3 equiv.), 10% NaOH (w/w%) pH 7, (iii) reflux, 15 min; (b) LiAlH4 (3 equiv.) THF, −78 °C, then room temperature, followed by heating to reflux, 3 h; (c) PhMe, μW irradiation, 2 h, 180 °C. The numbering scheme adopted for the borazatruxenes is also shown for compounds 1–3. | ||
Commercially available meta- and para- substituted 2-formylbenzeneboronic acids were reacted with methoxyamine hydrochloride under refluxing conditions for 15 minutes in water (pH 7) to produce compounds 8–10 in high yields (70–90%). LiAlH4 in THF was added dropwise to a solution of phenylboronic acid derivatives 8–10 in THF at −78 °C, the mixture was allowed to warm to room temperature followed by heating to reflux for 3 hours. In all cases, the amine-borane53 product 11–13, a BN analogue of indane, was isolated in excellent yields (85–90%). Trimerisation54 of 11–13 under microwave-assisted conditions afforded the desired borazatruxenes 1–3 in 56–65% yields as white solids after a simple filtration/washing protocol.
Due to the limited number of commercially available 2-formylphenylboronic acids and their relatively lengthy syntheses, a second pathway was devised in order to obtain functionalised borazatruxenes (Scheme 2). 2-Cyanobenzene-boronic esters 14–17 were synthesised from meta- and para- substituted benzonitriles. The benzonitriles were reacted with lithium tetramethylpiperidine (LTMP) and B(OiPr)3 at −78 °C, followed by slow warming to room temperature (20 °C) overnight to afford the respective ortho- substituted 2-cyanophenylboronic acid. The reaction was quenched using AcOH (2.2 equiv.) followed by an in situ protection of the boronic acid with 1,3-propanediol. The use of a protecting group is key for isolation of 14–17, and the choice of 1,3-propanediol allows the reduction to the corresponding amine-borane products 18–21 in very good yields (65–95%), without requiring a further deprotection step (Scheme 2 and ESI†). Using more robust protecting groups, such as pinacol or neopentyl glycol, results in the reduction of the CN group to the corresponding 1° amine while the protected boronic acid remains intact under the reduction conditions utilised. Trimerisation of amine-boranes 18–21, using microwave assisted synthesis in dry toluene at 180 °C for 2 hours, provided borazatruxenes 4–7 in 50–70% yields. Two additional borazatruxenes, 8 and 9 (Scheme 3), have been synthesised in order to expand the scope of the reaction to larger PAHs and chiral derivatives (vide infra), respectively.
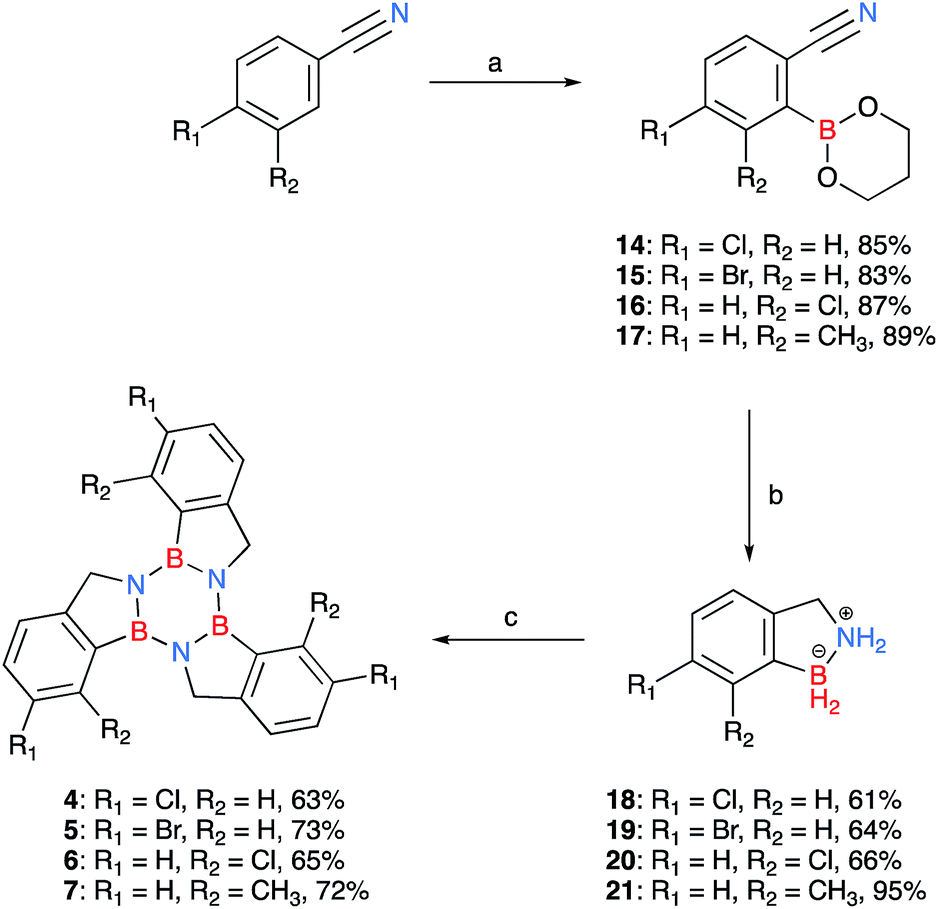 | ||
| Scheme 2 Synthesis of borazatruxenes 4–7 from substituted benzonitriles; the isolated yields for each derivative are given after their descriptor. (a) (i) LTMP (in situ: n-BuLi 2.5 M in n-hexane (1.5 equiv.) and TMP (1.5 equiv.)), THF, −10 °C, (ii) B(OiPr)3 (2.0 equiv.), −78 °C, benzonitrile (1.0 equiv.) then room temperature, 16 h, (iii) AcOH (2.2 equiv.), 1,3-propanediol (6.0 equiv.); (b) LiAlH4 (5.0 equiv.) THF, −78 °C, then room temperature, followed by μW irradiation, 1.5 h, 90 °C; (c) PhMe, μW irradiation, 2 h, 180 °C. | ||
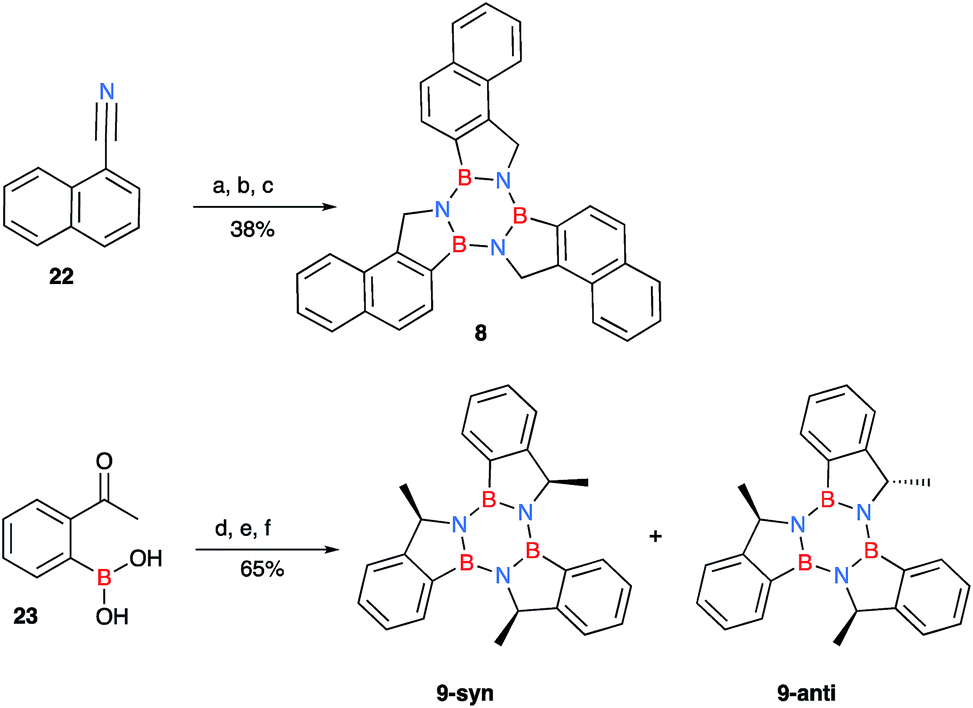 | ||
| Scheme 3 Synthesis of naphthylborazatruxene 8 from 1-naphthonitrile and the chiral trimethylborazatruxene 9, the reported yields are over three steps. (a) (i) LTMP (in situ: n-BuLi 2.5 M in n-hexane (1.5 equiv.) and TMP (1.5 equiv.)), THF, −10 °C, (ii) B(OiPr)3 (2.0 equiv.), −78 °C, benzonitrile (1.0 equiv.), then room temperature, o.n, (iii) AcOH (2.2 equiv.), 1,3-propanediol (6.0 equiv.); (b) LiAlH4 (3.0 equiv.) THF, −78 °C, then room temperature, followed by reflux, 48 h; (c) PhMe, μW irradiation, 2 h, 180 °C; (d) NH2OMe·HCl (1.2 equiv.), MeOH, 36 °C, 16 h; (e) LiAlH4 (5.0 equiv.) THF, −78 °C, then room temperature, followed by μW irradiation, 1.5 h, 90 °C; (f) PhMe, μW irradiation, 2 h, 180 °C. | ||
Borazatruxene 1 is soluble in organic solvents of medium polarity. This is in stark contrast to the parent truxene, which has very poor solubility in most common solvents.15 The halogenated borazatruxene derivatives have significantly lower solubility compared to 1, which is likely due to their increased molecular weight. The 1H-NMR of 1 (CDCl3, 300 MHz) indicates that the aromatic protons closest to the borazine rings are most deshielded (8.03–8.05 ppm) while the external ones experience a lower influence of the BN anisotropy with chemical shifts in the range of 7.42–7.59 ppm. The 11B NMR spectrum (CDCl3, 96 MHz) displays a peak resonating at 37.2 ppm which is in good agreement with a typical 11B chemical shift of a substituted borazine. The UV-vis spectrum of 1 shows a very intense and broad absorbance centered at 250 nm followed by three distinct peaks of lower intensity at 279.5, 272.0 and 265.0 nm. These peaks are blue shifted compared to corresponding truxene ones, thus highlighting that introducing BN bonds into the all-carbon system increases the HOMO–LUMO gap (Fig. 3). The borazatruxenes have higher quantum yields than the corresponding all-carbon derivatives. Borazatruxenes 5 and 8 aggregate at concentrations higher than 0.05 mM, while compounds 3, 5 and 6 are non-emissive. The variable temperature UV-vis and emission spectra, as well as the excitation spectra for the molecules that exhibited emission, are collated in the ESI, Fig. S10–S18.† The extinction coefficients, quantum yields and the excitation and emission wavelengths are collated in Table 1.
| Borazatruxene | λ ex (nm) | ε (L mol−1 cm−1) | λ em (nm) | Φ (A) |
|---|---|---|---|---|
a HPLC grade CHCl3.
b Determined using anthracene standard.
c Not determined due to aggregation.
d The solvent was n-hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2-propanol 9:1. :2-propanol 9:1.
|
||||
| 1 | 279.5 | 10650 |
294 | 0.065 |
| 2 | 285.5 | 10280 |
301 | 0.104 |
| 3 | 287.5 | 12950 |
380 | –c |
| 4 | 290 | 6740 | 305 | 0.099 |
| 5 | 290 | 2880 | 294 | –c |
| 6 | 289 | 4410 | 298 | –c |
| 7 | 278 | 6563 | 301 | 0.039 |
| 8 | 298.5 | 7930 | 304 | –c |
| 9 | 272 | 3177 | 293 | 0.038d |
There are four possible optical isomers of borazatruxene 9 despite having three stereogenic centres. This is because the molecule has C3 symmetry which makes the (R,R,S) isomer identical with the (R,S,R) and (S,R,R) isomers. Therefore, the only possible isomers are the enantiomeric pairs syn: (R,R,R) and (S,S,S) with all three methyl groups located on the same side of the borazatruxenes plane, and anti: (R,R,S) and (S,S,R), where one methyl is on the opposite side of the plane with respect to the other two (Scheme 3). The syn and anti enantiomeric pairs can be readily separated because the syn isomer is more soluble in an 8:2 n-hexane:CH2Cl2 mixture and thus it can be isolated through recrystallisation. Furthermore, all four stereoisomers can be separated using a Chiralpak OD chiral column (details in the ESI†). The selectivity factor for the syn enantiomers is 1.92 (98:2 n-hexane:propan-2-ol, 0.5 ml min−1) while the anti enantiomers are separated with a selectivity factor of 1.17 (99:1 n-hexane:propan-2-ol, 0.5 ml min−1). The Circular Dichroism (CD) spectra of the two enantiomeric pairs syn (R,R,R and S,S,S) and anti (R,R,S and R,S,S) of chiral derivative 9 display strong Cotton effects on both the arene and borazine absorbance. The identification of all four isomers was possible by comparing the experimental (Fig. 1) with the computed (vide infra) CD spectra.
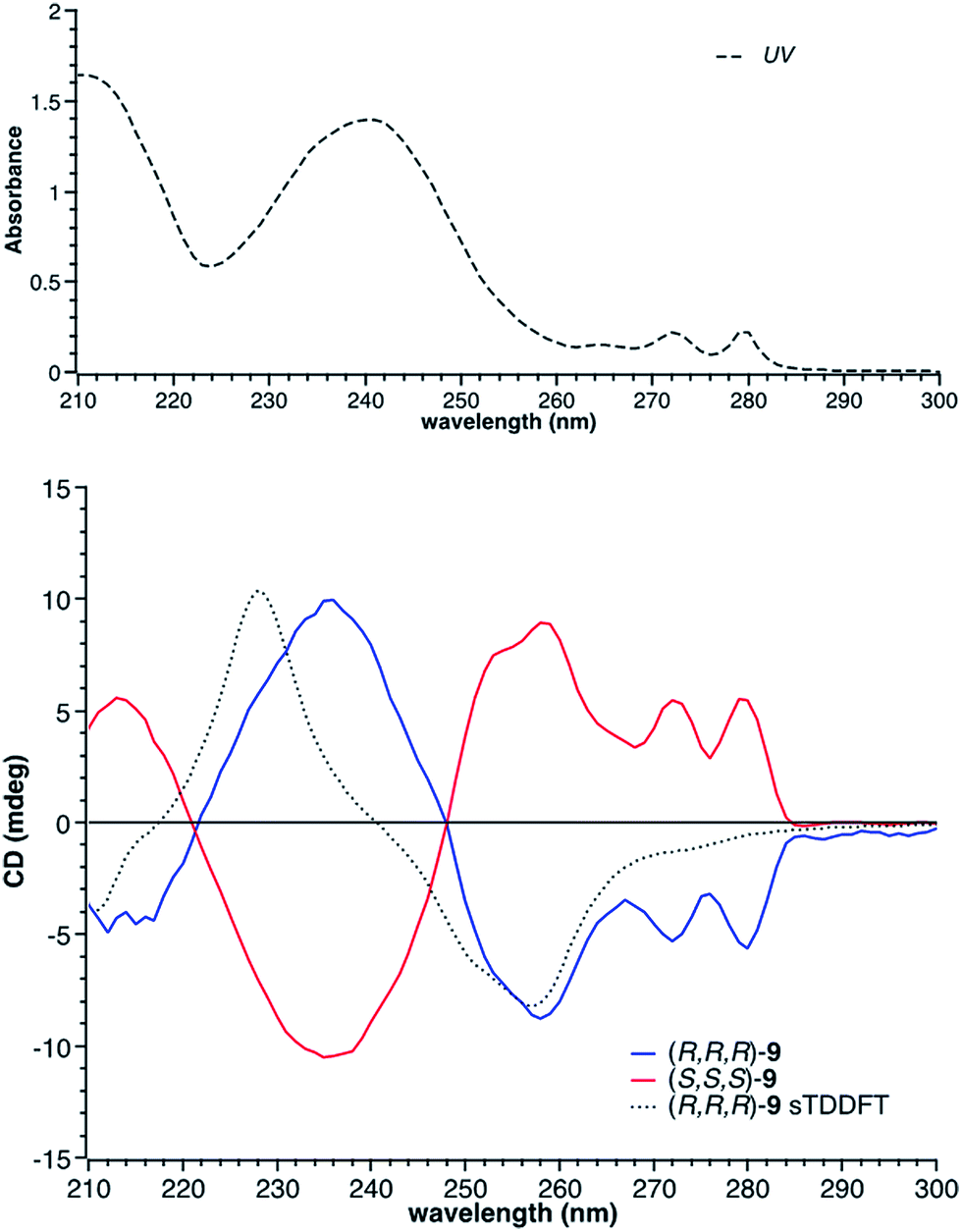 | ||
Fig. 1 Top: UV spectrum of (R,R,R)-9 ( ); bottom: the CD spectra of the two enantiomers of the syn isomer of 9, along with the computed CD spectrum (sTDDFT) of the (R,R,R)-9. ); bottom: the CD spectra of the two enantiomers of the syn isomer of 9, along with the computed CD spectrum (sTDDFT) of the (R,R,R)-9. | ||
The molecular structure of BN-indane 11 (Fig. 2) was obtained from the X-ray analysis of single crystals grown by slow evaporation from an ethyl acetate solution. This compound crystallised in the centrosymmetric monoclinic space group P21/n with four molecule units per unit cell packed in staggered array motif.
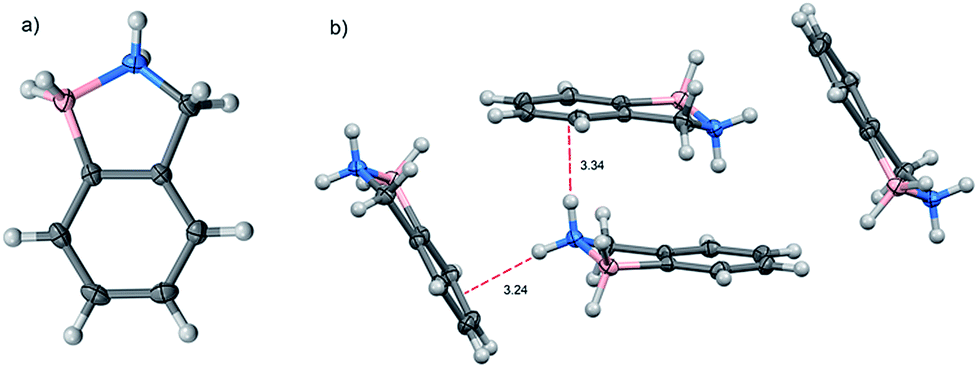 | ||
| Fig. 2 (a) Molecular structure and (b) unit cell of BN-indane 11. | ||
The distance between boron and nitrogen atom is 1.637 Å consistent with an N → B Lewis-type interaction. There are two weak NH⋯π intermolecular interactions between the amine group with the aromatic rings of two neighbouring molecules: N⋯Ph(C2–C7) distance of 3.239 Å and N⋯Ph(C3, C4, C5) distance of 3.339 Å.
Structural proof of borazatruxene 1 came from an X-ray diffraction analysis of single crystals grown by slow evaporation from a CH2Cl2 solution. Fig. 3A shows a top view of the X-ray structure of 1, which crystallises in a P21/c space group. This compound forms a staggered L-shaped configuration in the unit cell (Fig. 3C) where weak CH⋯π bonding is seen between C6 and C19 with the close contact distance being 3.801 Å. The π surfaces are also aligned parallel to one another with the closest distance between B2 and C16 being 3.561 Å.
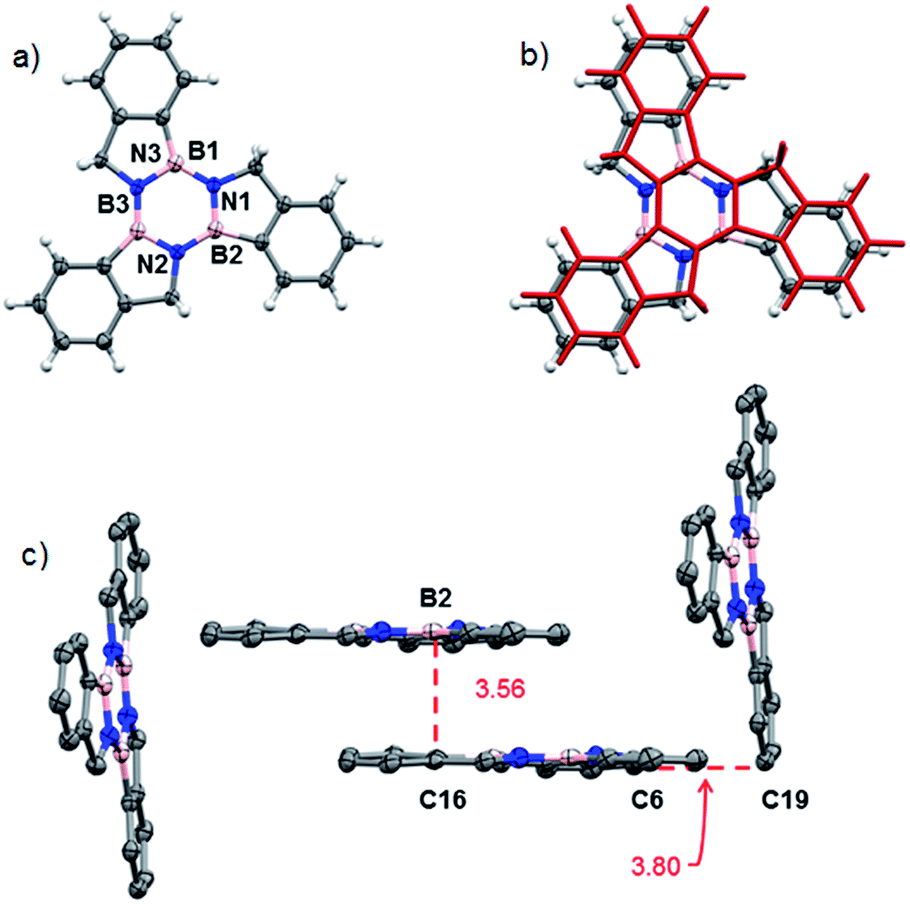 | ||
| Fig. 3 (a) Top view of the molecular structure of 1;44 (b) the molecular structure of 1, as determined by X-ray crystallography, overlaid (RMSD = 0.0603) with the computed structure for 1 in red, translated to (x + 1/2, y, z + 1/2) with respect to the X-ray coordinates; (c) packing structure highlighting the CH⋯π and aromatic π⋯π interactions. | ||
Single crystals of the syn isomer of 9 were obtained from a 9:1 hexane:2-propyl alcohol mixture. The X-ray diffraction revealed that the (R,R,R)-9 and (S,S,S)-9 co-crystallised in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group. In this structure the two enantiomers adopt a 60° rotated face-on stacked arrangement with an intermolecular B–N distance of 3.749 Å and the Me groups pointing outwards (Fig. 4).
space group. In this structure the two enantiomers adopt a 60° rotated face-on stacked arrangement with an intermolecular B–N distance of 3.749 Å and the Me groups pointing outwards (Fig. 4).
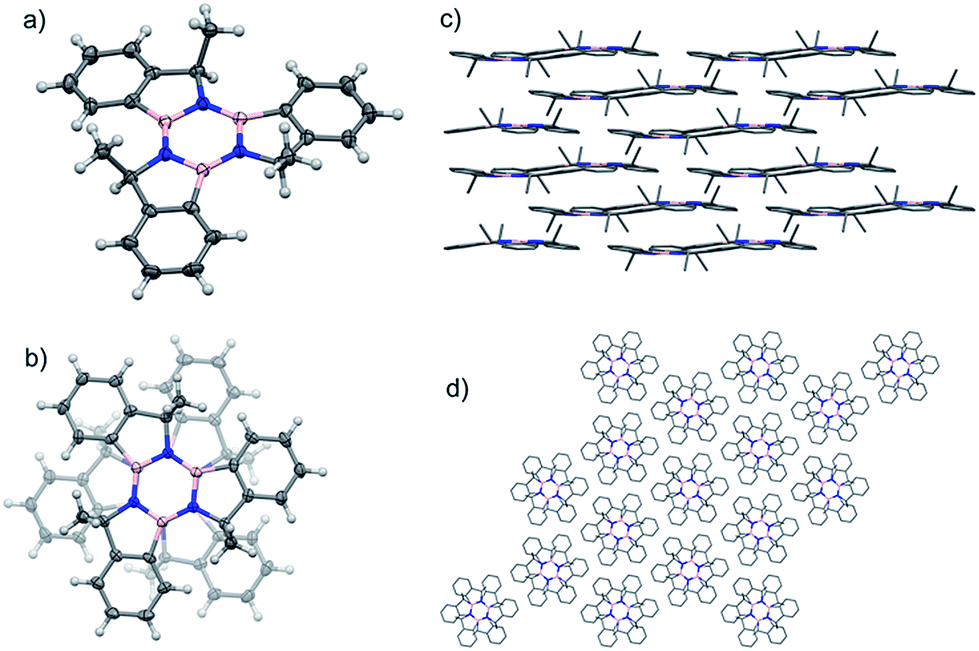 | ||
| Fig. 4 (a) Angled view of the molecular structure of syn-9;44 (b) stacking detail of the two enantiomers present in the structure. (c) Packing structure highlighting the layers and columns (d) present in the crystal structure. | ||
The molecules pack in an off-set columnar arrangement with a distance of 1.981 Å between two sequential planes of distinct columns.
The X-ray determined structure of 1 allowed us to validate a series of molecular modelling parameters used to predict the geometry of borazatruxenes 1–9. Fig. 3b shows the translated (x + 1/2, y, z + 1/2) overlay of the experimentally determined (X-ray diffraction) co-ordinates and the computed ones. Geometry optimisations were performed using M06-2X,55 M11,56 M11-L57 or B3LYP58,59 exchange-correlation functionals and various basis sets as implemented in the Gaussian 16 software. Also, TD-DFT60,61 (sTDDFT62) methods were employed to calculate the UV-vis and CD spectra with the above mentioned functionals (Fig. S8 and S21†), the best prediction for the CD spectra being obtained by M06-2X, while the closest agreement for the UV-vis spectra being given by the M11-L functional (Table S11†). Similarly the AICD properties (Fig. S22†) of borazatruxenes can be predicted at the M06-2X/6-311G level. A short study of the required geometry optimization time versus the agreement to experimental X-ray data (Table S10, Fig. S20†) yields M06-2X and M11 functionals along with 6-31G and 6-31G(d,p) basis sets as excellent choices for the modelling of borazatruxenes geometries while using reasonable computation resources.
Conclusions
In summary, we have developed the synthesis of a new class of BN-PAHs that incorporate a borazine unit in place of the central benzene core of a truxene. These derivatives have higher solubilities than the parent all-carbon derivatives and can be synthesised in three steps from commercially available starting materials. These derivatives are air and moisture stable which is in contrast to the majority of non-sterically hindered borazines. We have synthesised and separated the first chiral derivatives of these molecules which also represents a premiere in the larger truxene family. The borazatruxenes molecules are likely to become important materials in molecular electronic devices due to their unique structure which combines areas of electron-conductance with electron-insulating domains. Further investigations into the synthesis of new borazatruxenes analogues are currently in progress in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the EPSRC (DTA SL, LME, AT) and the University of Bath. X-ray diffraction and NMR facilities were provided through the Chemical Characterisation and Analysis Facility (CCAF) at the University of Bath, while the mass spectrometry data were acquired at the EPSRC National Mass Spectrometry Facility at Swansea University.Notes and references
- V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868–4884 CrossRef CAS PubMed.
- X. Feng, W. Pisula and K. Müllen, Pure Appl. Chem., 2009, 81, 2203–2224 CAS.
- L. Dössel, L. Gherghel, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 2540–2543 CrossRef PubMed.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS.
- F. Chen and N. J. Tao, Acc. Chem. Res., 2009, 42, 429–438 CrossRef CAS PubMed.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS.
- J. Wu and K. Müllen, All-benzenoid Polycyclic Aromatic Hydrocarbons: Synthesis, Self-assembly and Applications in Organic Electronics, WILEY-VCH Verlag GmbH & Co. KGaA, 1st edn, 2006 Search PubMed.
- Z. Liu, J. S. A. Ishibashi, C. Darrigan, A. Dargelos, A. Chrostowska, B. Li, M. Vasiliu, D. A. Dixon and S.-Y. Liu, J. Am. Chem. Soc., 2017, 139, 6082–6085 CrossRef CAS PubMed.
- A. Chrostowska, S. Xu, A. Mazière, K. Boknevitz, B. Li, E. R. Abbey, A. Dargelos, A. Graciaa and S.-Y. Liu, J. Am. Chem. Soc., 2014, 136, 11813–11820 CrossRef CAS.
- J. S. A. Ishibashi, J. L. Marshall, A. Mazière, G. J. Lovinger, B. Li, L. N. Zakharov, A. Dargelos, A. Graciaa, A. Chrostowska and S.-Y. Liu, J. Am. Chem. Soc., 2014, 136, 15414–15421 CrossRef CAS.
- S. Tsuchiya, H. Saito, K. Nogi and H. Yorimitsu, Org. Lett., 2019, 21, 3855–3860 CrossRef CAS.
- C. Zhang, L. Zhang, C. Sun, W. Sun and X. Liu, Org. Lett., 2019, 21, 3476–3480 CrossRef CAS PubMed.
- M. Müller, C. Maichle-Mössmer and H. F. Bettinger, Angew. Chem., Int. Ed., 2014, 53, 9380–9383 CrossRef PubMed.
- A. Abengózar, P. García-García, D. Sucunza, D. Sampedro, A. Pérez-Redondo and J. J. Vaquero, Org. Lett., 2019, 21, 2550–2554 CrossRef.
- F. Goubard and F. Dumur, RSC Adv., 2014, 5, 3521–3551 RSC.
- K. Shi, J.-Y. Wang and J. Pei, Chem. Rec., 2015, 15, 52–72 CrossRef CAS.
- M. M. Boorum, Science, 2001, 294, 828–831 CrossRef CAS PubMed.
- C. Destrade, H. Gasparoux, A. Babeau, N. H. Tinh and J. Malthete, Mol. Cryst. Liq. Cryst., 1981, 67, 37–47 CrossRef CAS.
- K. Isoda, T. Yasuda and T. Kato, Chem.–Asian J., 2009, 4, 1619–1625 CrossRef CAS.
- L. T. Scott, Science, 2002, 295, 1500–1503 CrossRef CAS.
- A. H. Abdourazak, Z. Marcinow, A. Sygula, R. Sygula and P. W. Rabideau, J. Am. Chem. Soc., 1995, 117, 6410–6411 CrossRef CAS.
- C. Ruiz, E. M. García-Frutos, G. Hennrich and B. Gómez-Lor, J. Phys. Chem. Lett., 2012, 3, 1428–1436 CrossRef CAS.
- Ó. de Frutos, B. Gómez-Lor, T. Granier, Á. Monge, E. Gutiérrez-Puebla and A. M. Echavarren, Angew. Chem., Int. Ed., 1999, 38, 204–207 CrossRef.
- B. Gómez-Lor, Ó. de Frutos, P. A. Ceballos, T. Granier and A. M. Echavarren, Eur. J. Org. Chem., 2001, 2001, 2107–2114 CrossRef.
- E. González-Cantalapiedra, M. Ruiz, B. Gómez-Lor, B. Alonso, D. García-Cuadrado, D. J. Cárdenas and A. M. Echavarren, Eur. J. Org. Chem., 2005, 2005, 4127–4140 CrossRef.
- Z. Yang, B. Xu, J. He, L. Xue, Q. Guo, H. Xia and W. Tian, Org. Electron., 2009, 10, 954–959 CrossRef CAS.
- J. Huang, B. Xu, J.-H. Su, C. H. Chen and H. Tian, Tetrahedron, 2010, 66, 7577–7582 CrossRef CAS.
- K. M. Omer, A. L. Kanibolotsky, P. J. Skabara, I. F. Perepichka and A. J. Bard, J. Phys. Chem. B, 2007, 111, 6612–6619 CrossRef CAS PubMed.
- X.-Y. Cao, W.-B. Zhang, J.-L. Wang, X.-H. Zhou, H. Lu and J. Pei, J. Am. Chem. Soc., 2003, 125, 12430 CrossRef CAS PubMed.
- L. Wang, Y. Jiang, J. Luo, Y. Zhou, J. Zhou, J. Wang, J. Pei and Y. Cao, Adv. Mater., 2009, 21, 4854–4858 CrossRef CAS PubMed.
- J.-M. Koenen, S. Jung, A. Patra, A. Helfer and U. Scherf, Adv. Mater., 2012, 24, 681 CrossRef CAS PubMed.
- K.-P. Tseng, M.-T. Kao, T. W. T. Tsai, C.-H. Hsu, J. C. C. Chan, J.-J. Shyue, S.-S. Sun and K.-T. Wong, Chem. Commun., 2012, 48, 3515 RSC.
- S. Gomez-Esteban, M. Pezella, A. Domingo, G. Hennrich and B. Gómez-Lor, Chem.–Eur. J., 2013, 19, 16080–16086 CrossRef CAS.
- S. Hattori, A. Yamada, S. Saito, K. Asakawa, T. Koshiba and T. Nakasugi, J. Photopolym. Sci. Technol., 2009, 22, 609 CrossRef CAS.
- B. Guilhabert, N. Laurand, J. Herrnsdorf, Y. Chen, A. R. Mackintosh, A. L. Kanibolotsky, E. Gu, P. J. Skabara, R. A. Pethrick and M. D. Dawson, J. Opt., 2010, 12, 035503 CrossRef.
- A.-M. Haughey, B. Guilhabert, A. L. Kanibolotsky, P. J. Skabara, M. D. Dawson, G. A. Burley and N. Laurand, Biosens. Bioelectron., 2014, 54, 679–686 CrossRef CAS PubMed.
- M. S. Yuan, Q. Wang, W. Wang, D. E. Wang, J. Wanga and J. Wang, Analyst, 2014, 139, 1541–1549 RSC.
- C.-H. Tao, H. Yang, N. Zhu, V. W.-W. Yam and S.-J. Xu, Organometallics, 2008, 27, 5453–5458 CrossRef CAS.
- C. K. M. Chan, C.-H. Tao, H.-L. Tam, N. Zhu, V. W.-W. Yam and K.-W. Cheah, Inorg. Chem., 2009, 48, 2855–2864 CrossRef CAS PubMed.
- M. Müller, S. Behnle, C. Maichle-Mössmer and H. F. Bettinger, Chem. Commun., 2014, 50, 7821 RSC.
- Z. Liu and T. B. Marder, Angew. Chem., Int. Ed., 2008, 47, 242–244 CrossRef CAS PubMed.
- M. Côté, P. Haynes and C. Molteni, Phys. Rev. B, 2001, 63, 125207 CrossRef.
- D. Bonifazi, F. Fasano, M. M. Lorenzo-Garcia, D. Marinelli, H. Oubaha and J. Tasseroul, Chem. Commun., 2015, 51, 15222–15236 RSC.
- S. Kervyn, N. Kalashnyk, M. Riello, B. Moreton, J. Tasseroul, J. Wouters, T. S. Jones, A. De Vita, G. Costantini and D. Bonifazi, Angew. Chem., Int. Ed., 2013, 52, 7410–7414 CrossRef CAS.
- D. Marinelli, F. Fasano, B. Najjari, N. Demitri and D. Bonifazi, J. Am. Chem. Soc., 2017, 139, 5503–5519 CrossRef CAS PubMed.
- S. Biswas, M. Müller, C. Tönshoff, K. Eichele, C. Maichle-Mössmer, A. Ruff, B. Speiser and H. F. Bettinger, Eur. J. Org. Chem., 2012, 2012, 4634–4639 CrossRef CAS.
- M. Müller, C. Maichle-Mössmer, P. Sirsch and H. F. Bettinger, ChemPlusChem, 2013, 78, 988–994 CrossRef.
- F. Ciccullo, A. Calzolari, I. Píš, S.-A. Savu, M. Krieg, H. F. Bettinger, E. Magnano, T. Chassé and M. B. Casu, J. Phys. Chem. C, 2016, 120, 17645–17651 CrossRef CAS.
- J. A. Snyder, P. Grüninger, H. F. Bettinger and A. E. Bragg, J. Phys. Chem. A, 2017, 121, 5136–5146 CrossRef CAS PubMed.
- J. Dosso, J. Tasseroul, F. Fasano, D. Marinelli, N. Biot, A. Fermi and D. Bonifazi, Angew. Chem., Int. Ed., 2017, 56, 4483–4487 CrossRef CAS PubMed.
- M. Krieg, F. Reicherter, P. Haiss, M. Ströbele, K. Eichele, M.-J. Treanor, R. Schaub and H. F. Bettinger, Angew. Chem., Int. Ed., 2015, 54, 8284–8286 CrossRef CAS PubMed.
- C. Sánchez-Sánchez, S. Brüller, H. Sachdev, K. Müllen, M. Krieg, H. F. Bettinger, A. Nicolaï, V. Meunier, L. Talirz, R. Fasel and P. Ruffieux, ACS Nano, 2015, 9, 9228–9235 CrossRef PubMed.
- M. Beguerie, C. Faradji, L. Vendier, S. Sabo-Etienne and G. Alcaraz, ChemCatChem, 2017, 9, 3303–3306 CrossRef CAS.
- W. Luo, P. G. Campbell, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2011, 133, 19326–19329 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. Peverati and D. G. Truhlar, J. Phys. Chem. Lett., 2011, 2, 2810–2817 Search PubMed.
- R. Peverati and D. G. Truhlar, J. Phys. Chem. Lett., 2012, 3, 117–124 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- C. Bannwarth and S. Grimme, Comput. Theor. Chem., 2014, 1040–1041, 45–53 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic protocols, modelling and analytical data. CCDC 1843607, 1843608 and 1865474. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02489a |
| ‡ These authors have made equal contribution to this work. |
| § The borazatruxenes are stable in solid state for at least one year if kept under N2 in close capped vials; they are stable in solution for at least two weeks with minimal decomposition. |
| This journal is © The Royal Society of Chemistry 2019 |