
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalyst-controlled regiodivergent ring-opening C(sp3)–Si bond-forming reactions of 2-arylaziridines with silylborane enabled by synergistic palladium/copper dual catalysis†
Youhei
Takeda
 *a,
Kaoru
Shibuta
a,
Shohei
Aoki
a,
Norimitsu
Tohnai
b and
Satoshi
Minakata
*a
*a,
Kaoru
Shibuta
a,
Shohei
Aoki
a,
Norimitsu
Tohnai
b and
Satoshi
Minakata
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Yamadaoka 2-1, Suita, Osaka 565-0871, Japan. E-mail: takeda@chem.eng.osaka-u.ac.jp; minakata@chem.eng.osaka-u.ac.jp
bDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, Yamadaoka 2-1, Suita, Osaka 565-0871, Japan
First published on 31st July 2019
Abstract
A catalyst-controlled regiodivergent and stereospecific ring-opening C(sp3)–Si cross-coupling of 2-arylaziridines with silylborane enabled by synergistic Pd/Cu dual catalysis has been developed. Just by selecting a suitable combination of catalysts, the regioselectivity of the coupling is completely switched to efficiently provide two regioisomers of β-silylamines (i.e., β-silyl-α-phenethylamines and β-silyl-β-phenethylamines) in good to high yields. Furthermore, a slight modification of the reaction conditions caused a drastic change in reaction pathways, leading to a tandem reaction to produce another regioisomer of silylamine (i.e., α-silyl-β-phenethylamines) in an efficient and selective manner.
Introduction
Alkylsilanes are ubiquitous motifs in industrial products, and they serve as useful synthetic building blocks.1 A wide variety of synthetic methods for alkylsilanes including silyl-SN2 reaction,2 silyl-Negishi and silyl-Kumada-Tamao reactions of silyl halides,3 hydrosilylation of alkenes,4 C–H silylation of alkanes,5 the addition of Si–E (e.g., E = Si, B) bonds across C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds,6 Cu-catalyzed nucleophilic substitution of alkyltriflates,7 and radical-mediated C(sp3)–Si coupling of unactivated alkylhalides8a,c and carboxylate derivatives8b,c with silylboranes have been developed. Also, more recently, transition-metal (Ni and Pd)-catalyzed C(sp3)–Si cross-coupling of an alkyl electrophile with a silyl (pro)nucleophile (Si–M, M = Zn, B) has been emerging as a powerful synthetic method for alkylsilanes because of high chemoselectivity and functional group tolerance.9–12 Nevertheless, to date, alkyl electrophiles that are applicable to coupling are limited to branch-type alkyl halides,9 benzyl halides,10,11 and benzyl ethers.12 Therefore, the development of C(sp3)–Si cross-coupling using hitherto unexplored alkyl electrophiles with a silyl (pro)nucleophile holds great promise for widening the diversity of available alkylsilanes.
C double bonds,6 Cu-catalyzed nucleophilic substitution of alkyltriflates,7 and radical-mediated C(sp3)–Si coupling of unactivated alkylhalides8a,c and carboxylate derivatives8b,c with silylboranes have been developed. Also, more recently, transition-metal (Ni and Pd)-catalyzed C(sp3)–Si cross-coupling of an alkyl electrophile with a silyl (pro)nucleophile (Si–M, M = Zn, B) has been emerging as a powerful synthetic method for alkylsilanes because of high chemoselectivity and functional group tolerance.9–12 Nevertheless, to date, alkyl electrophiles that are applicable to coupling are limited to branch-type alkyl halides,9 benzyl halides,10,11 and benzyl ethers.12 Therefore, the development of C(sp3)–Si cross-coupling using hitherto unexplored alkyl electrophiles with a silyl (pro)nucleophile holds great promise for widening the diversity of available alkylsilanes.
Aziridines have emerged as relatively new alkyl electrophiles in transition-metal-catalyzed regioselective ring-opening C–C13–18 and C–B19,20 cross-couplings with organoboron and organozinc nucleophiles to give β-organo- and borylated alkylamines, respectively. In conjunction with the fact that alkylamines bearing a C(sp3)–Si bond have been recognized as unique bioisosteres of pharmacological agents in medicinal chemistry,21 regiodivergent ring-opening C(sp3)–Si cross-coupling of aziridines with a silyl (pro)nucleophile would open up a new avenue to the preparation of a set of regioisomeric β-silyl-alkylamines. A seminal work on regioselective ring opening of aziridines with a silyl nucleophile was reported by Fleming, where silyllithium was applied as a nucleophile (Scheme 1a).22 The reaction exclusively gives one regioisomer of silylamine (β-silyl-α-substituted ethylamines); however, the regiochemistry (opening at the 3-position) seems to be governed by the combination of reagents, and it requires excess amounts of silyllithium (3 equivalents) that can deteriorate electron-withdrawing functionalities.22 During the preparation of this manuscript, as a related study to our present work, a Cu-catalyzed reagent-controlled regiodivergent nucleophilic ring opening of 2-arylaziridines with silyl Grignard reagents has been reported by the Oestreich group (Scheme 1b).23 Although an example using a silylborane (Me2PhSi–Bpin) as a pronucleophile was also shown in the same paper to give 3-position selective silylative ring opening coupling,23 as is generally the case, the reagent-controlled approach quite limits the diversity of products. To the best of our knowledge, catalyst-controlled regiodivergent ring-opening C(sp3)–Si cross-coupling of aziridines has not been achieved yet. Herein, we disclose a catalyst-controlled regiodivergent and stereospecific ring-opening C(sp3)–Si cross-coupling of 2-arylaziridines with a silylborane to selectively give two different regioisomers of silylphenethylamines enabled by Pd/Cu synergistic dual catalysis (the upper and middle equations, Scheme 1c).24 Notably, a catalytic tandem reaction to give another regioisomer of silylamines was also discovered (the bottom equation, Scheme 1c).
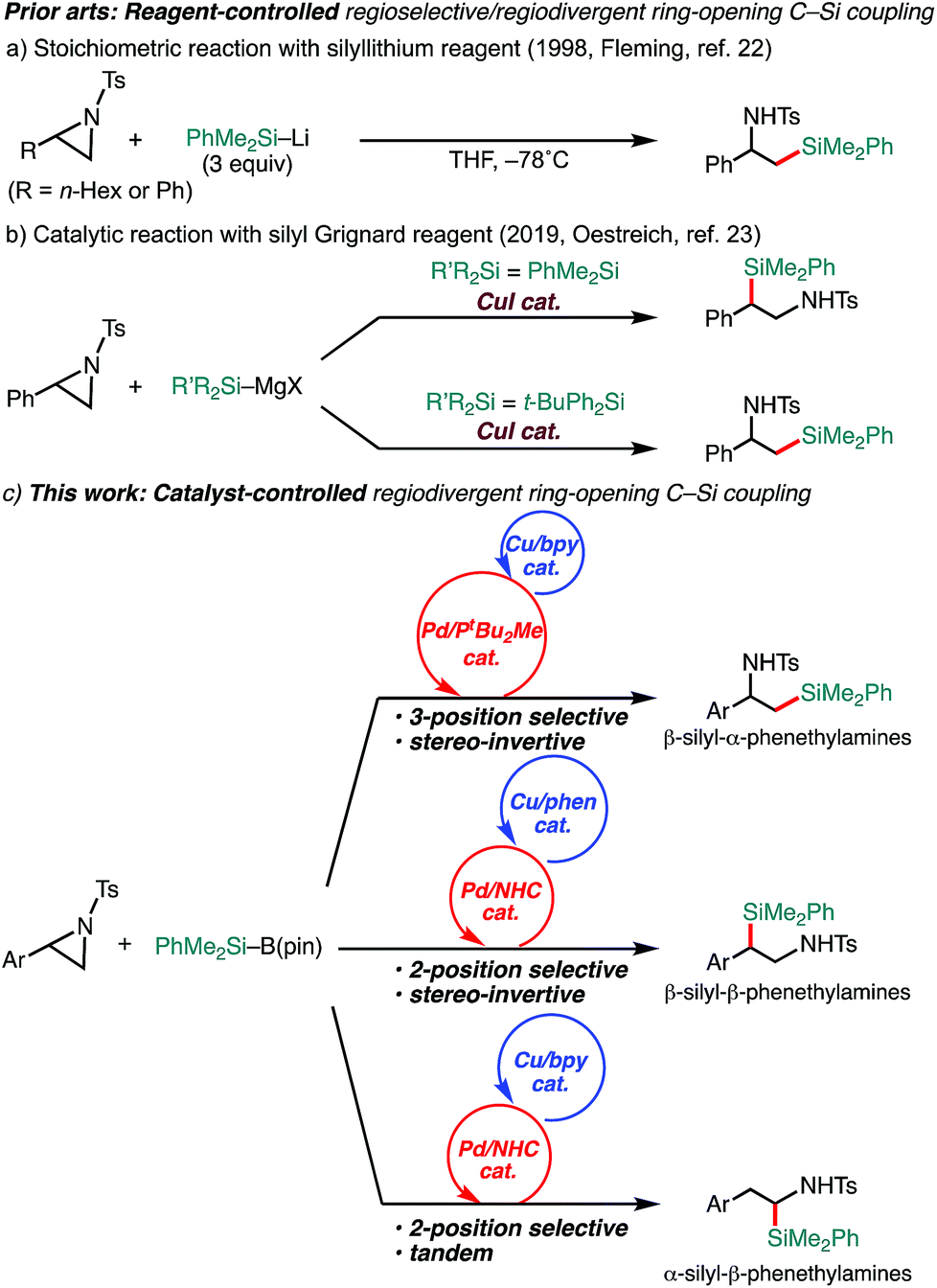 | ||
| Scheme 1 Regioselective C(sp3)–Si bond-forming reactions of 2-arylaziridines with silylboranes. | ||
Results and discussion
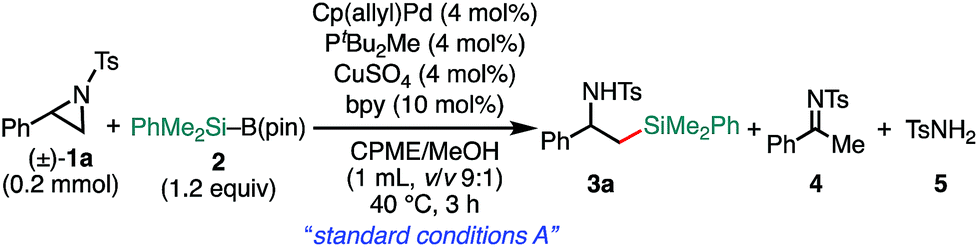
We previously reported that NHC/Pd and PtBu2Me/Pd catalysts allow for regioselective and stereospecific (stereo-invertive) ring-opening Suzuki–Miyaura arylation and borylation of 2-arylaziridines13a at the 2- and 3-positions, respectively.19 Theoretical calculations on the mechanisms suggested that the regioselectivity and stereospecificity are determined in an SN2-type oxidative addition step through the interaction between the catalyst and aziridine.13b,19 Based on the borylation conditions,19 as an initial attempt, racemic aziridine 1a was treated with silylborane (Me2PhSi–Bpin, 2) (1.2 equiv.) in the presence of Cp(allyl)Pd/PtBu2Me and bipyridine (bpy) catalysts in a CPME/H2O co-solvent. Although the expected coupled product 3a was formed, the chemical yield was very low even at an elevated temperature (Table 1, entry 2), suggesting that the transmetalation between Si–B and Pd–OMe species would be energetically much higher than that of the C–B coupling. At a temperature higher than 60 °C, β-hydride elimination from the oxidative adduct was accelerated,19 indicating the difficulty of identifying suitable reaction conditions for the coupling with a single metallic catalytic system (for details, see Tables S1–S8, ESI†). We envisaged that the addition of a Cu salt could allow for stepwise transmetalation (i.e., Si–B → Si–Cu → Si–Pd) to promote the coupling, and the effect of Cu additives was investigated (Table S4, ESI†). Importantly, the addition of CuSO4 drastically improved the chemical yield of 3a up to 89% (Table 1, entry 1 vs. entry 2). Other copper salts such as CuF2 and Cu(OH)2 gave 3a in comparable yields (Table 1, entries 3 and 4), while the addition of CuCl resulted in a lower yield (Table 1, entry 5), possibly due to the poorer solubility of the salt in the solvents. Without the Pd complex and phosphine ligand, no reaction occurred (Table 1, entries 6 and 7), excluding the possibility of direct nucleophilic ring-opening substitution with Cu–Si species.23 Furthermore, bpy should play an important role in the coupling, because coupled product 3a was not produced at all in the absence of bpy (Table 1, entry 8). In addition, β-methoxy-β-phenethylamine 7 was produced in a substantial yield, which should be produced through the nucleophilic ring opening of 1a with MeOH under the electrophilic activation of 1 by the Cu Lewis acid,25 implying that bpy adjusts the Lewis acidity of the Cu additive to suppress the background side reaction. Another possible role is to enhance the solubility of the Cu-species by coordination to increase the concentration of the active Cu species. A proton source was required for the reaction to smoothly proceed (Table 1, entries 9 and 10), indicating that the M–OMe species (M = Pd, Cu) generated in situ would promote stepwise transmetalation (Si–B → Si–Cu → Si–Pd) through Lewis acid–base interactions. The C(sp3)–Si coupling proceeds smoothly under neutral conditions without adding any explicit strong Lewis bases, which are usually required to activate silylboranes to transfer the silyl unit.7,8a,b,10,11 Other silylboranes such as Ph2MeSi–Bpin, Ph2tBuSi–Bpin, and Et3Si–Bpin did not give any silylated products.|
|
|||||
|---|---|---|---|---|---|
| Entry | Variations from the “standard conditions A” | Yield (%) | Recovery of 1a (%) | ||
| 3a | 4 | 5 | |||
a The “standard conditions A”: 1a (0.2 mmol), 2 (0.24 mmol), Cp(allyl)Pd (8 μmol), PtBu2Me (8 μmol), CuSO4 (8 μmol), and bpy (20 μmol) were stirred in CPME/MeOH (1 mL, v/v 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 40 °C for 3 h.
b The reaction was conducted at 60 °C.
c Phenethylamines 6 and 7 were obtained in 12% and 51%, respectively. :1) at 40 °C for 3 h.
b The reaction was conducted at 60 °C.
c Phenethylamines 6 and 7 were obtained in 12% and 51%, respectively.
|
|||||
| 1 | None | 89 | 6 | 0 | 0 |
| 2b | w/o CuSO4 | 13 | Trace | 39 | 46 |
| 3 | CuF2 in place of CuSO4 | 83 | 0 | 5 | 0 |
| 4 | Cu(OH)2 in place of CuSO4 | 83 | 0 | 7 | 0 |
| 5 | CuCl | 35 | 0 | 4 | 55 |
| 6 | w/o Cp(ally)Pd and PtBu2Me | 0 | 0 | 0 | 99 |
| 7 | w/o Cp(allyl)Pd | 0 | 0 | 0 | 99 |
| 8c | w/o bpy | 0 | 23 | 0 | 13 |
| 9 | H2O in place of MeOH | 75 | 4 | 0 | 8 |
| 10 | w/o MeOH | Trace | 0 | 0 | 93 |

|
|||||
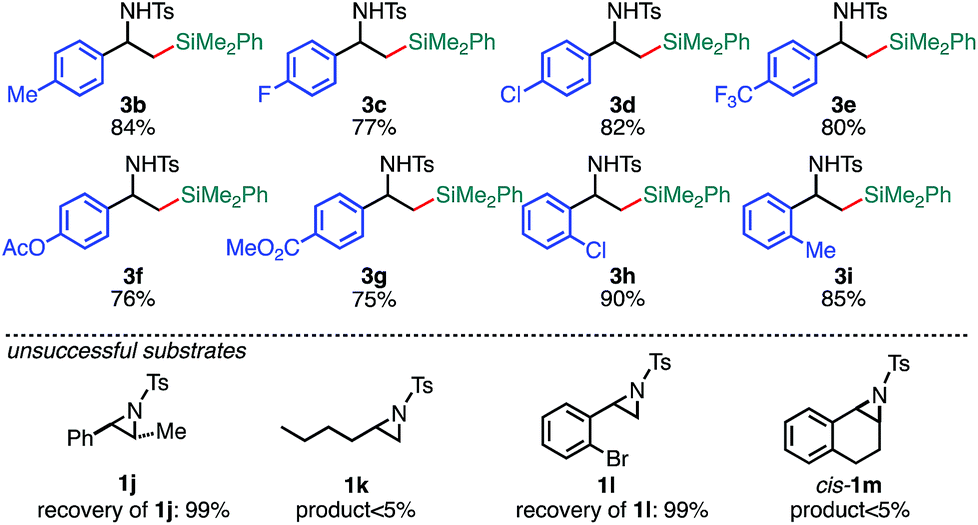
With the optimized reaction conditions in hand, the substrate scope of aziridines was investigated (Table 2). A variety of 2-arylaziridines 1 having a functional group on the aromatic ring were applicable, giving β-silyl-α-phenethylamines 3 in a regioselective manner in good to high yields. In particular, it is noted that chlorine and ester functionalities tolerated the reaction conditions to give the corresponding coupling products (3d, 3f, 3g, and 3h). On the other hand, the reactions with 2,3-disubstituted (1j), 2-alkylated (1k), 2-(o-Br–C6H4)–substituted (1l), and cyclic (cis-1m) aziridines were not successful (Table 2). Since N-tosyl-2-(p-Br–C6H4)–aziridine was also not applicable to the reaction conditions (recovery of aziridine 87%), the reason why the coupling reaction using 1l did not proceed would be the electronic effect rather than the steric effect of the o-Br substituent. The preference of the oxidative addition of the C–Br bond to the Pd(0) complex might be the side reaction which inhibits the desired reaction.
|
|
|---|
| a The reaction was conducted at 0.50 mmol (1) scale under “standard conditions A”. |
|
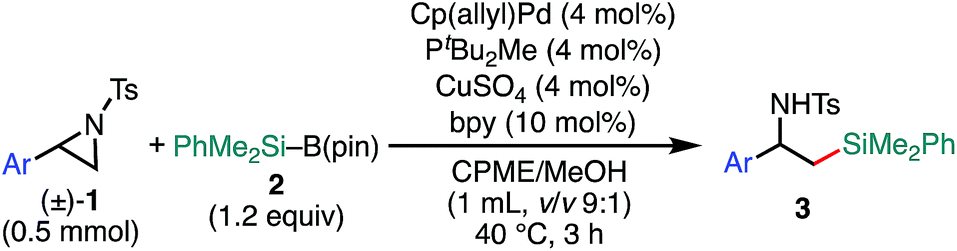
To gain stereochemical information about the reaction, a deuterated aziridine (cis-1a-d1) was coupled with silylborane under the standard conditions A [eqn (1)]. Derivatization of the coupled product 3a-d1 (for detailed procedures to determine the relative stereochemistry, see Scheme S1, ESI†) revealed that 3a-d1 has the trans-configuration, indicating that the C(sp3)–Si cross-coupling proceeds in a stereo-invertive manner. This indicates that the regio- and stereospecificity-determining step would be an SN2-type oxidative addition even in the dual catalytic system.
 | (1) |
Delighted with the validity of the Pd/Cu dual catalysis in the 3-position-selective C(sp3)–Si coupling, we turned our attention to switching the regioselectivity of the ring-opening silylative coupling. Although NHC-ligated Pd(II) complexes are suitable pre-catalysts for Suzuki–Miyaura arylation of 1a,13a the attempts with those Pd(II) complexes failed only to recover the aziridine, indicating the weaker nucleophilic ability of the silylborane to reduce Pd(II) to Pd(0) than arylboronic acid.13b To circumvent the reduction process, we applied NHC-ligated Pd(0) complexes26 as Pd pre-catalysts and found them to be successful: a coupling reaction proceeded exclusively at the 2-position to give the regioisomeric coupled product (β-silyl-β-phenethylamine) (for detailed results, see Tables S9–S16, ESI†). Among the tested, a catalytic system comprising SIPr–Pd(0)–PPh3, CuF2, and 1,10-phenanthroline (phen) was found to be suitable for the 2-position-selective ring-opening C(sp3)–Si coupling to give 8a in a high yield (standard conditions B) [eqn (2)]. Intriguingly, along with 8a, 11% of α-silyl-β-phenethylamine 9a, which is another regioisomer of 8a, was produced [eqn (2)]. Although the detailed mechanism leading to 9a should await further study, a putative pathway involves tandem processes comprising (i) a Pd-catalyzed isomerization of 1a into aldimines through the oxidative addition of 1a into the Pd(0) complex at the 2-position followed by β-hydride elimination/tautomerization27 and (ii) a subsequent nucleophilic attack of the resulting aldimines by the Cu–Si species generated in situ (vide infra).28 Delighted with the discovery of this hitherto unknown reaction, we surveyed the effect of reaction parameters on the product distribution (for details, see the ESI†). It turned out that the tandem reaction was drastically promoted by simply adding extra PPh3 and changing the additive from phen to bpy (Tables S10 and S16, ESI†), which led to exclusive and quantitative formation of 9a (standard conditions C) [eqn (3)].
 | (2) |
 | (3) |
The scope of the 2-position-selective ring-opening C(sp3)–Si cross-coupling was investigated (Table 3). The cross-coupling of enantiopure aziridine (R)-1a (>99% ee) under the standard conditions B proceeded regioselectively and enantiospecifically to give enantiopure product 8a in 89% yield (99% ee). The absolute configuration of 8a was unambiguously determined to be S by the single crystal X-ray diffraction analysis (Table 3, the inset figure; for detailed crystallographic data, see Table S17, ESI†). This indicates that the coupling proceeds with stereo-inversion, which is fully consistent with an SN2-type oxidative addition of aziridine.13a The coupling conditions were applicable to 2-arylaziridines bearing a variety of functional groups, giving rise to the corresponding coupled products in good to high yields in a regioselective and stereospecific manner (Table 3). Again, the reactions using aziridines 1j–1m were not successful.
|
|
|---|
| a The reaction was conducted at 0.50 mmol (1) scale under the “standard conditions B”. b The reaction was run for 3 h. c es (enantiospecificity) = ee (8)/ee (1) × 100; ee (enantiomeric excess) was determined by chiral HPLC analysis. d The reaction was conducted with the MeIPr–Pd–PPh3 catalyst at 60 °C. e The reaction was conducted at room temperature. |

|
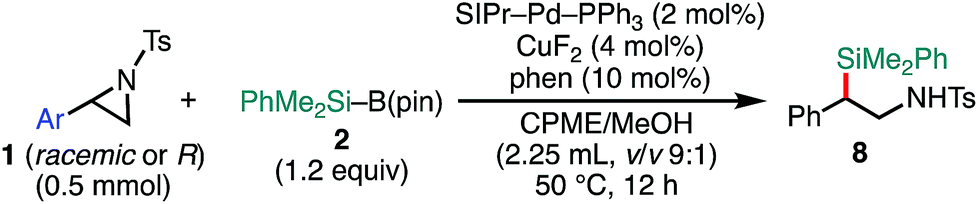
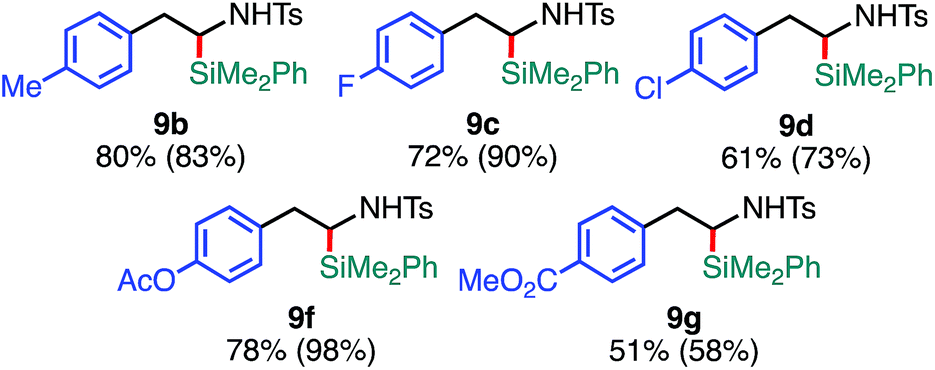
The substrate scope of the tandem C(sp3)–Si bond-forming reaction was also investigated (Table 4). A variety of 2-arylaziridines with a functional group were efficiently converted into the corresponding α-silyl-β-phenethylamine products, which are often found in bioisosteres of protease.21
|
|
|---|
| a The reaction was conducted at 0.20 mmol (1) scale under the “standard conditions C”. |
|

A proposed reaction mechanism of the 2-position-selective C(sp3)–Si coupling and the tandem reaction are illustrated in Scheme 2. The catalytic cycle would start from the pre-coordination of SIPr–Pd(0) to the aryl moiety of 1 to stabilize the complex (Scheme 2a),13b which then undergoes oxidative addition from the backside of the C(2)–N(1) bond with stereo-inversion (Scheme 2b). The regioselectivity for the ring opening would be determined by the pre-coordination of the Ar moiety to the Pd center to gain large interaction energy (INT), which was reasonably suggested by the energy decomposition analysis (EDA) of the computed oxidative addition process of the aziridines into a Pd(0)–NHC complex.13b In sharp contrast, in the case of the phosphine-Pd catalyst system, the oxidative addition takes place on the opposite side (i.e., C(3)–N(1)) in a regioselective fashion. This would be caused by the larger deformation energy (DEF) than INT gained when the V-shaped bisphosphine–Pd complex [(tBu)2MeP–Pd(0)–PMe(tBu)2] approaches from the backside of the C(2)–N(1) bond.19 Therefore, we can conclude that the regioselectivity of the ring opening would be mainly governed by the balance between (i) how large interactions between the Pd catalyst and aziridine operate and (ii) how large deformations the Pd catalyst and aziridine experience when they approach each other. The resulting zwitterionic oxidative adduct A can be protonated with MeOH to form alkoxide complex B (Scheme 2c).19 This intermediate would then undergo transmetalation with the PhMe2Si–Cu(phen) complex D, giving an alkyl(silyl)Pd complex E (Scheme 2e), where silylcopper D could be generated from the transmetalation between Cu–OMe species C with silylborane (Scheme 2d).29 The reactions of 1a with the silylborane in the presence of Cu catalysts in different oxidation states [(phen)CuIF30a and (phen)CuIIF230b] gave 8a in almost the same yields (see Table S15, ESI†), implying that the oxidation level of the actual Cu species under the optimized conditions would be either CuI or CuII. To probe the oxidation level of the actual Cu active species, the reaction mixtures starting with a CuI and CuII additive were monitored with an electron paramagnetic resonance (EPR) technique. From the results, the concentration of CuII species was found to be significantly decreased under the optimized conditions (see Fig. S1 in the ESI†), indicating that the reduction of CuII to CuI occurs in situ. Taken together, we assume that the putative Cu active species in Cu catalysis is (phen)CuI–L (L = F or OMe). The reductive elimination from E should produce 8 and regenerate the Pd(0) catalyst (Scheme 2f). Almost the same dual catalysis would be involved in the C–Si cross-coupling at the 3-position, except for the 3-position-selective oxidative addition.19 A possible pathway to 9 would involve (i) a PPh3/Pd-catalyzed isomerization of 1 into aldimine Fvia β-hydride elimination and tautomerization (Scheme 2g)27 and (ii) the following nucleophilic addition of Si–Cu species D to the imine (Scheme 2h).28 Since the treatment of separately prepared aldimine F with silylborane 2 under similar reaction conditions (“standard conditions C”) gave 9 in a moderate yield (when Ar = Ph, 31% of 9a was obtained with no recovery of the aldimine substrate; for details, see Scheme S2, ESI†), this scenario is likely to occur.
 | ||
| Scheme 2 A proposal of dual catalysis in the 2-position-selective C(sp3)–Si cross-coupling and tandem reaction. | ||
Conclusions
In conclusion, we have succeeded in developing catalyst-controlled highly regiodivergent ring-opening C–Si bond formation reactions to selectively provide three regioisomers of silylamines via synergistic Pd/Cu dual catalysis. It is noted that the balance between the efficiency of Pd and Cu catalysis should be the origin of the discovery of the tandem reaction, and this knowledge would provide us with insights for designing more intricate and sophisticated transformations in the future. Detailed catalytic reaction mechanisms are investigated experimentally and theoretically in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research on Innovative Areas “Precisely Designed Catalysts with Customized Scaffolding” (KAKENHI, JP16H01023, to YT) and “Coordination Asymmetry” (KAKENHI, JP19H04580, to NT). We also acknowledge Professors Yuma Morimoto and Shinobu Ito for the assistance with EPR measurements and fruitful discussions.Notes and references
- (a) M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley-Interscience, New York, 2000 Search PubMed; (b) N. Anner and J. Weis, Organosilicon Chemistry V, Wiley-VCH Verlag GmbH, Weinheim, 2003 CrossRef.
- P. J. Lennon, D. P. Mack and Q. E. Thompson, Organometallics, 1989, 8, 1121–1122 CrossRef CAS.
- (a) A. P. Cinderella, B. Vulovic and D. A. Watson, J. Am. Chem. Soc., 2017, 139, 7741–7744 CrossRef CAS PubMed; (b) B. Vulovic, A. P. Cinderella and D. A. Watson, ACS Catal., 2017, 7, 8113–8117 CrossRef CAS PubMed.
- For selected reviews on hydrosilylation of alkenes, see: (a) D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS; (b) X. Du and Z. Huang, ACS Catal., 2017, 7, 1227–1243 CrossRef CAS.
- For a review on C–H silylation of alkanes, see: C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed.
- For a review on the addition of a Si–E bond to CC double bonds, see: M. Suginome and Y. Ito, Chem. Rev., 2000, 100, 3221–3256 CrossRef CAS PubMed.
- (a) J. Scharfbier and M. Oestreich, Synlett, 2016, 27, 1274–1276 CrossRef CAS; (b) J. Scharfbier, H. Hazrati, E. Irran and M. Oestrich, Org. Lett., 2017, 19, 6562–6565 CrossRef CAS PubMed.
- (a) W. Xue, Z.-W. Qu, S. Grimme and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 14222–14225 CrossRef CAS PubMed; (b) W. Xue and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 11649–11652 CrossRef CAS PubMed; (c) W. Xue, R. Shishido and M. Oestreich, Angew. Chem., Int. Ed., 2018, 57, 12141–12145 CrossRef CAS PubMed.
- (a) C. K. Chu, Y. Liang and G. C. Fu, J. Am. Chem. Soc., 2016, 138, 6404–6407 CrossRef CAS PubMed; (b) H. Yi, W. Mao and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 3575–3578 CrossRef CAS PubMed.
- Z.-D. Huang, R. Ding, P. Wang, Y.-H. Xu and T. P. Loh, Chem. Commun., 2016, 52, 5609–5612 RSC.
- B. Cui, S. Jia, E. Tokunaga and N. Shibata, Nat. Commun., 2018, 9, 4393 CrossRef PubMed.
- (a) C. Zarate and R. A. Martin, J. Am. Chem. Soc., 2014, 136, 2236–2239 CrossRef CAS PubMed; (b) C. Zarate, M. Nakajima and R. A. Martin, J. Am. Chem. Soc., 2017, 139, 1191–1197 CrossRef CAS PubMed.
- (a) Y. Takeda, Y. Ikeda, A. Kuroda, S. Tanaka and S. Minakata, J. Am. Chem. Soc., 2014, 136, 8544–8547 CrossRef CAS PubMed; (b) A. K. Sharma, W. M. C. Sameera, Y. Takeda and S. Minakata, ACS Catal., 2019, 9, 4582–4592 CrossRef CAS.
- (a) M. L. Duda and F. E. Michael, J. Am. Chem. Soc., 2013, 135, 18347–18349 CrossRef CAS PubMed; (b) W. P. The and F. E. Michael, Org. Lett., 2017, 19, 1738–1740 CrossRef PubMed.
- (a) C.-Y. Huang and A. G. Doyle, J. Am. Chem. Soc., 2012, 134, 9541–9544 CrossRef CAS PubMed; (b) D. K. Nielsen, C.-H. Huang and A. G. Doyle, J. Am. Chem. Soc., 2013, 135, 13605–13609 CrossRef CAS PubMed; (c) K. L. Jensen, E. A. Standley and T. F. Jamison, J. Am. Chem. Soc., 2014, 136, 11145–11152 CrossRef CAS PubMed; (d) C.-Y. Huang and A. G. Doyle, J. Am. Chem. Soc., 2015, 137, 5638–5641 CrossRef CAS PubMed.
- B. P. Woods, M. Orlandi, C.-Y. Huang, M. S. Sigman and A. G. Doyle, J. Am. Chem. Soc., 2017, 139, 5688–5691 CrossRef CAS PubMed.
- J. Kjellgren, J. Aydin, O. A. Wallner, I. V. Saltanova and K. J. Szabó, Chem.–Eur. J., 2005, 11, 5260–5268 CrossRef CAS PubMed.
- J. Yin, T. Mekelburg and C. Hyland, Org. Biomol. Chem., 2014, 12, 9113–9115 RSC.
- Y. Takeda, A. Kuroda, W. M. C. Sameera, K. Morokuma and S. Minakata, Chem. Sci., 2016, 7, 6141–6152 RSC.
- (a) S. Sebelius, V. J. Olsson and K. J. Szabó, J. Am. Chem. Soc., 2005, 127, 10478–10479 CrossRef CAS PubMed; (b) J. Zhao and K. J. Szabó, Angew. Chem., Int. Ed., 2016, 55, 1502–1506 CrossRef CAS PubMed.
- For selected reviews on aminoalkylsilanes, see: (a) G. K. Min, D. Hernéndez and T. Skrydstrup, Acc. Chem. Res., 2013, 46, 457–470 CrossRef CAS PubMed; (b) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed; (c) E. Rémond, C. Martin, J. Martinez and F. Cavelier, Chem. Rev., 2016, 116, 11654–11684 CrossRef PubMed.
- I. Fleming, J. Frackenpohl and H. Ila, J. Chem. Soc., Perkin Trans. 1, 1998, 1229–1235 RSC.
- H. Yi and M. Oestreich, Chem.–Eur. J., 2019, 25, 6505–6507 CrossRef CAS PubMed.
- For selected examples of Pd/Cu synergetic cocatalysis in organic synthetic reactions, see: (a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef; (b) K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 7567–7570 CrossRef CAS PubMed; (c) M. Takeda, K. Yabushita, S. Yasuda and H. Ohmiya, Chem. Commun., 2018, 54, 6776–6779 RSC; (d) S. D. Friis, M. T. Pirnot and S. L. Buchwald, J. Am. Chem. Soc., 2016, 138, 8372–8375 CrossRef CAS PubMed , and references cited therein.
- M. K. Ghorai, D. Shukla and A. Bhattacharyya, J. Org. Chem., 2012, 77, 3740–3753 CrossRef CAS PubMed.
- S. Fantasia and S. P. Nolan, Chem.–Eur. J., 2008, 14, 6987–6993 CAS.
- J. P. Wolfe and J. E. Ney, Org. Lett., 2003, 5, 4607–4610 CrossRef CAS PubMed.
- D. J. Vyas, R. Fröhlich and M. Oestreich, Org. Lett., 2011, 13, 2094–2097 CrossRef CAS PubMed.
- (a) C. Kleeberg, M. S. Cheung, Z. Lin and T. Marder, J. Am. Chem. Soc., 2011, 133, 19060–19063 CrossRef CAS PubMed; (b) Y. Tani, T. Fujihara, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 17706–17709 CrossRef CAS PubMed; (c) J. Plotzitzka and C. Kleeberg, Inorg. Chem., 2017, 56, 6671–6680 CrossRef CAS PubMed.
- (a) M. Ohashi, N. Ishida, K. Ando, Y. Hashimoto, A. Shigaki, K. Kikushima and S. Ogoshi, Chem.–Eur. J., 2018, 24, 9794–9798 CrossRef CAS PubMed; (b) P. R. Gibbs, P. R. Graves and D. J. Gulliver, Inorg. Chim. Acta, 1980, 45, L207–L208 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for synthesis and screening, spectroscopic data of new compounds, X-ray crystallographic data, EPR spectra copies of 1H and 13C NMR charts, and HPLC analysis data. CCDC 1911669. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02507c |
| This journal is © The Royal Society of Chemistry 2019 |