
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOctahedral iron(IV)–tosylimido complexes exhibiting single electron-oxidation reactivity†
Gerard
Sabenya
a,
Ilaria
Gamba
a,
Laura
Gómez
a,
Martin
Clémancey
b,
Jonathan R.
Frisch
c,
Eric J.
Klinker
c,
Geneviève
Blondin
b,
Stéphane
Torelli
b,
Lawrence
Que
Jr
 *c,
Vlad
Martin-Diaconescu
*ad,
Jean-Marc
Latour
*b,
Julio
Lloret-Fillol
*ade and
Miquel
Costas
*a
*c,
Vlad
Martin-Diaconescu
*ad,
Jean-Marc
Latour
*b,
Julio
Lloret-Fillol
*ade and
Miquel
Costas
*a
aInstitut de Química Computacional i Catàlisi (IQCC), Departament de Química, Universitat de Girona, Campus Montilivi, E17071 Girona, Spain. E-mail: miquel.costas@udg.edu
bUniv. Grenoble-Alpes, CNRS, CEA, IRIG, DIESE, CBM, Grenoble 38000, France
cDepartment of Chemistry, University of Minnesota, Pleasant Str 207, Minneapolis, Minnesota, USA
dInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Avinguda Països Catalans 16, 43007 Tarragona, Spain. E-mail: jlloret@iciq.es
eCatalan Institution for Research and Advanced Studies (ICREA), Passeig Lluïs Companys, 23, 08010, Barcelona, Spain
First published on 20th August 2019
Abstract
High valent iron species are very reactive molecules involved in oxidation reactions of relevance to biology and chemical synthesis. Herein we describe iron(IV)–tosylimido complexes [FeIV(NTs)(MePy2tacn)](OTf)2 (1(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NTs) and [FeIV(NTs)(Me2(CHPy2)tacn)](OTf)2 (2(IV)NTs), (MePy2tacn = N-methyl-N,N-bis(2-picolyl)-1,4,7-triazacyclononane, and Me2(CHPy2)tacn = 1-(di(2-pyridyl)methyl)-4,7-dimethyl-1,4,7-triazacyclononane, Ts = Tosyl). 1(IV)NTs and 2(IV)NTs are rare examples of octahedral iron(IV)–imido complexes and are isoelectronic analogues of the recently described iron(IV)–oxo complexes [FeIV(O)(L)]2+ (L = MePy2tacn and Me2(CHPy2)tacn, respectively). 1(IV)NTs and 2(IV)NTs are metastable and have been spectroscopically characterized by HR-MS, UV-vis, 1H-NMR, resonance Raman, Mössbauer, and X-ray absorption (XAS) spectroscopy as well as by DFT computational methods. Ferric complexes [FeIII(HNTs)(L)]2+, 1(III)–NHTs (L = MePy2tacn) and 2(III)–NHTs (L = Me2(CHPy2)tacn) have been isolated after the decay of 1(IV)NTs and 2(IV)NTs in solution, spectroscopically characterized, and the molecular structure of [FeIII(HNTs)(MePy2tacn)](SbF6)2 determined by single crystal X-ray diffraction. Reaction of 1(IV)NTs and 2(IV)NTs with different p-substituted thioanisoles results in the transfer of the tosylimido moiety to the sulphur atom producing sulfilimine products. In these reactions, 1(IV)NTs and 2(IV)NTs behave as single electron oxidants and Hammett analyses of reaction rates evidence that tosylimido transfer is more sensitive than oxo transfer to charge effects. In addition, reaction of 1(IV)NTs and 2(IV)NTs with hydrocarbons containing weak C–H bonds results in the formation of 1(III)–NHTs and 2(III)–NHTs respectively, along with the oxidized substrate. Kinetic analyses indicate that reactions proceed via a mechanistically unusual HAT reaction, where an association complex precedes hydrogen abstraction.
NTs) and [FeIV(NTs)(Me2(CHPy2)tacn)](OTf)2 (2(IV)NTs), (MePy2tacn = N-methyl-N,N-bis(2-picolyl)-1,4,7-triazacyclononane, and Me2(CHPy2)tacn = 1-(di(2-pyridyl)methyl)-4,7-dimethyl-1,4,7-triazacyclononane, Ts = Tosyl). 1(IV)NTs and 2(IV)NTs are rare examples of octahedral iron(IV)–imido complexes and are isoelectronic analogues of the recently described iron(IV)–oxo complexes [FeIV(O)(L)]2+ (L = MePy2tacn and Me2(CHPy2)tacn, respectively). 1(IV)NTs and 2(IV)NTs are metastable and have been spectroscopically characterized by HR-MS, UV-vis, 1H-NMR, resonance Raman, Mössbauer, and X-ray absorption (XAS) spectroscopy as well as by DFT computational methods. Ferric complexes [FeIII(HNTs)(L)]2+, 1(III)–NHTs (L = MePy2tacn) and 2(III)–NHTs (L = Me2(CHPy2)tacn) have been isolated after the decay of 1(IV)NTs and 2(IV)NTs in solution, spectroscopically characterized, and the molecular structure of [FeIII(HNTs)(MePy2tacn)](SbF6)2 determined by single crystal X-ray diffraction. Reaction of 1(IV)NTs and 2(IV)NTs with different p-substituted thioanisoles results in the transfer of the tosylimido moiety to the sulphur atom producing sulfilimine products. In these reactions, 1(IV)NTs and 2(IV)NTs behave as single electron oxidants and Hammett analyses of reaction rates evidence that tosylimido transfer is more sensitive than oxo transfer to charge effects. In addition, reaction of 1(IV)NTs and 2(IV)NTs with hydrocarbons containing weak C–H bonds results in the formation of 1(III)–NHTs and 2(III)–NHTs respectively, along with the oxidized substrate. Kinetic analyses indicate that reactions proceed via a mechanistically unusual HAT reaction, where an association complex precedes hydrogen abstraction.
Introduction
High valent iron compounds are of interest in biological and chemical sciences because their high reactivity makes them competent at performing challenging oxidative transformations in living organisms and in chemical synthesis.1 Iron–oxo species receive major interest in reactions entailing oxygenation of organic molecules.2–4 Comparable in synthetic relevance but somewhat less explored are high valent iron–imidos and iron mediated N-transfer reactions.5–37 Interestingly, the electronic structure of iron–imido complexes appears to be subtler than that of iron–oxos, and some formal iron(IV)–imidos are best described as ferric imido radical complexes.38–40 Iron–imido complexes described to date generally contain metal centres with low coordination numbers, with trigonal and tetrahedral geometries as the most common.32,41–45 On the other hand, octahedral complexes are very rare. The most thoroughly studied compound [FeIV(NTs)(N4Py)]2+, 3(IV)NTs, (N4Py, N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine),30,46 exhibits two-electron N-transfer and HAT reactivity. The later reaction is initiated by an electron transfer (ET) from the substrate forming a ferric–imido intermediate, rapidly followed by hydrogen atom transfer from the alkyl cation radical.46,47 More recent work by Fukuzumi and Nam comparing the reactivity of 3(IV)NTs and its oxo analogue [FeIV(O)(N4Py)]2+, (3(IV)O), concluded that 3(IV)NTs exhibits a greatly enhanced electron transfer reactivity arising from a much more positive one electron reduction potential and a smaller reorganization energy during electron transfer.46 These studies suggest that iron(IV)–imidos may exhibit a yet unexplored rich and distinctive chemistry from their better known iron(IV)–oxo counterparts.
Herein we describe two novel octahedral iron(IV)–tosylimido complexes using pentadentate ligands based on the 1,4,7-triazacyclononane macrocycle. The reactivity of these complexes in model N-transfer and HAT reactions has been explored, putting forward novel mechanistic scenarios, distinct from those previously established for analogous iron(IV)–oxo complexes and 3(IV)NTs complexes. Overall, the current study discloses unprecedented single electron mechanistic scenarios for high-valent iron compounds.
Results and discussion
Synthesis and characterization of [FeIV(NTs)(MePy2tacn)](OTf)2 (1(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) NTs) and [FeIV(NTs)(Me2(CHPy2)tacn)](OTf)2 (2(IV)NTs)
NTs) and [FeIV(NTs)(Me2(CHPy2)tacn)](OTf)2 (2(IV)NTs)
Ferrous complexes [FeII(CH3CN)(L)](OTf)2 (1(II)-OTf, L = MePy2tacn = N-methyl-N,N-bis(2-picolyl)-1,4,7-triazacyclononane, and 2(II)-OTf, L = Me2(CHPy2)tacn = 1-(di(2-pyridyl)methyl)-4,7-dimethyl-1,4,7-triazacyclononane; Scheme 1) have previously been described.48,49 Both complexes contain aminopyridine ligands based on a triazacyclononane moiety, providing a pentadentate donor set, leaving a sixth site available for an external labile ligand S (acetonitrile for 1(II) and 2(II)). In 2(II), the two pyridine rings are in the same plane as the Fe–S axis, while in 1(II), one of the pyridines is parallel and the second perpendicular to this axis. Reaction of 1(II) and 2(II) with 1.2 equivalents of N-tosyliminophenyliodinane (PhINTs) in anhydrous CH3CN at 25 °C, produce [FeIV(NTs)(MePy2tacn)]2+ (1(IV)NTs) and [FeIV(NTs)(Me2(CHPy2)tacn)]2+ (2(IV)NTs), (Scheme 1). The reactions can be conveniently monitored by UV-vis spectroscopy; reaction of 1(II) with PhINTs produces within minutes 1(IV)NTs, characterized by a low energy feature at λmax = 750 nm (ε = 200 M−1 cm−1) and a prominent absorption at λmax = 455 nm (ε = 4500 M−1 cm−1). On the other hand, reaction of 2(II) with PhINTs is slower, and formation of 2(IV)NTs (λmax = 456 nm; ε = 3600 M−1 cm−1, and 740 nm; ε = 170 M−1 cm−1) is mixed with its decay. However, use of acetone instead of acetonitrile leads to fast and quantitative formation of 2(IV)NTs (λmax = 455 nm; ε = 4000 M−1 cm−1, and 730 nm; ε = 230 M−1 cm−1), with good levels of purity (vide infra). Of notice, the near-IR low absorption bands observed are characteristic of d–d transitions of high-valent iron(IV) species with a S = 1 state.2 Complexes 1(IV)NTs and 2(IV)NTs are metastable and decompose over time in acetonitrile solution (at 20 °C complex 1(IV)NTs has a t1/2 of about 40 min while for complex 2(IV)NTs the t1/2 is on the order of a few hours) to form iron(III) species (vide infra).
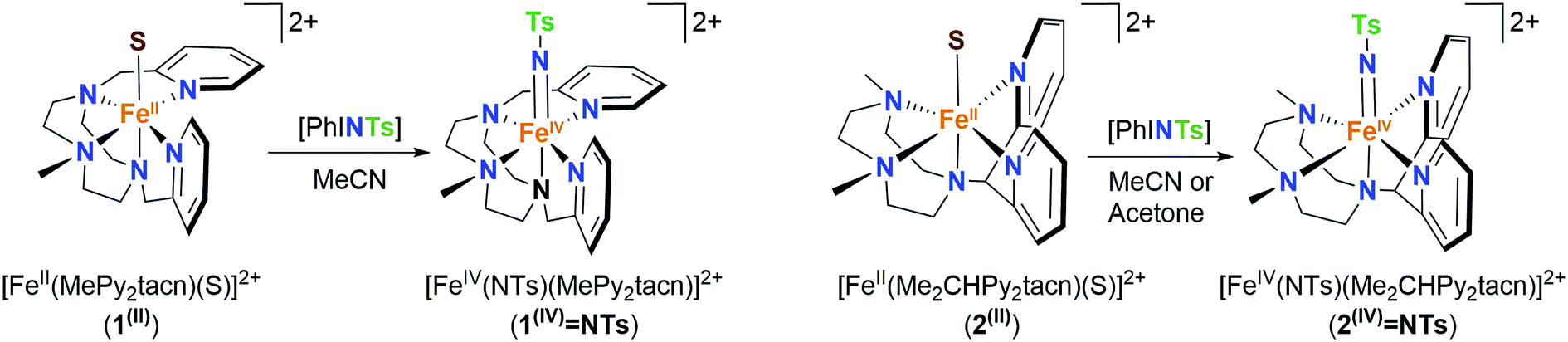 | ||
| Scheme 1 Chemical strategy for the generation of 1(IV)NTs and 2(IV)NTs from iron(II) precursors. 1(II) and 2(II) (S = MeCN, CF3SO3). | ||
High resolution mass spectra (HR-MS) evidence the proposed formulation of 1(IV)NTs and 2(IV)NTs, and both show a major peak at m/z = 275.09, with an isotopic pattern that corresponds to [FeIV(NTs)(L)]2+, (L = MePy2tacn and Me2(CHPy2)tacn) (Fig. 1 and S1†). The spectra also show a lower intensity peak at m/z = 699.13, which corresponds to monocationic [[FeIV(NTs)(L)](OTf)]+. The later peaks shift by one mass unit upon formation of 1(IV)NTs and 2(IV)NTs with PhI(15N)Ts. Close inspection of these peaks reveals small contributions of the respective [FeIII(HNTs)(L)]2+ species (∼10%). Finally, a minor peak at m/z = 550.18 and its isotopic pattern are consistent with a [FeIII(NTs)(L)]+ formulation.
 | ||
| Fig. 1 Top, Mössbauer spectrum recorded at 80 K in zero-field of 1(IV)NTs (left) in acetonitrile and 2(IV)NTs (right) prepared in acetone. Experimental data are hatched bars and solid lines represent the contributions of 1(IV)NTs and 2(IV)NTs. Centre, UV-vis spectrum of oxidation of 1(II) to 1(IV)NTs in CH3CN; kinetic trace shows the evolution of the λmax = 750 nm feature. Below, HR-MS spectrum of 1(IV)NTs prepared in CH3CN exhibiting a base peak at m/z 275.09; inset show amplification of major peak for [FeIV(NTs)(MePy2tacn)]2+ species. Proper simulation of the peak requires inclusion of ≈10% of [FeIII(NHTs)(MePy2tacn)]2+ in the isotopic pattern corresponding to [FeIV(NTs)(MePy2tacn)]2+. | ||
Spectroscopic characterization of 1(IV)NTs and 2(IV)NTs
Complexes 1(IV)NTs and 2(IV)NTs were characterized by Mössbauer spectroscopy using 57Fe enriched samples. The spectrum of a frozen sample prepared after maximum accumulation of 1(IV)NTs (followed by UV-vis spectroscopy) is shown in Fig. 1. The spectrum recorded at 80 K under zero applied magnetic field, is dominated by a quadrupole doublet which accounts for 87% of the total iron content. Its simulation yields spin hamiltonian parameters (isomer shift δ = 0.05 mm s−1, quadrupole splitting ΔEQ = 1.09 mm s−1), consistent with an iron(IV) centre in a low-spin (S = 1) configuration.1,50 A frozen acetonitrile sample prepared after maximum accumulation of 2(IV)NTs according to the UV-vis provides a more complex spectrum, showing only 48% of high-valent iron(IV) species, with δ = 0.04 mm s−1 and ΔEQ = 0.74 mm s−1 (Fig. S2†). However, when acetone was used as a solvent, 2(IV)NTs was obtained in higher purity (81%, δ = 0.06 mm s−1, ΔEQ = 0.73 mm s−1, Fig. 1). Obtained Mössbauer values are reminiscent of the previously reported [FeIV(NTs)(N4Py)]2+, 3(IV)NTs (δ = 0.02 mm s−1 and ΔEQ = 0.98 mm s−1).30
The 1H-NMR spectra of 1(IV)NTs and 2(IV)NTs in CD3CN or D6-acetone show paramagnetically shifted signals between −75 and 50 ppm, with the signals of the pyridine moiety as the most distinctive features due to their relative sharpness (Fig. 2). These could be identified by comparison with the spectra of the parent iron(IV)–oxo complexes, and also with that of [FeIV(X)(N4Py)]2+3(IV)X (X = O, NTs) and [FeIV(O)(Bn-TPEN)]2+, 4(IV)O (Bn-TPEN, N-benzyl-N,N′,N′-tris(2-pyridylmethyl)-1,2-diaminoethane).30,51 Moreover, the presence of broad signals for both 1(IV)NTs and 2(IV)NTs in the high-field region of the spectra, which are not present in the spectrum of 3(IV)NTs, suggests that they belong to the N-methyl and N-methylene protons on the triazacyclononane macrocycle. A bidimensional COSY spectrum of 2(IV)NTs (inset of Fig. 2), allowed the assignment of pyridine hydrogens to the signals at 20.87 (β), 9.70 (γ) and −6.56 (β′) ppm. Of note, the signal pattern closely resembles that of 3(IV)X (X = O, NTs), where all the pyridine rings are also parallel to the FeO/N axis. On the other hand, 1(IV)NTs presents a more complex spectrum, in accordance with its reduced symmetry, where one pyridine is parallel to the FeN axis and the other pyridine ring perpendicular to the FeN axis of this complex. The 1H-NMR spectrum and corresponding COSY data identify two distinct sets of pyridine protons. Subset “a” is assigned to the pyridine parallel to the FeN axis, showing the same pattern as observed for 2(IV)NTs and 3(IV)NTs, with the βα proton signal downfield shifted to 41.0 ppm, the 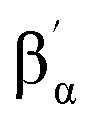 proton signal upfield shifted to −13.3 ppm, and the γa signal at 12.7 ppm. However, subset “b” belongs to the pyridine perpendicular to the FeN axis and shows a distinct pattern, with both βb and
proton signal upfield shifted to −13.3 ppm, and the γa signal at 12.7 ppm. However, subset “b” belongs to the pyridine perpendicular to the FeN axis and shows a distinct pattern, with both βb and 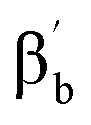 proton signals upshifted to −5.6 and −6.9 ppm and the γb proton at 6.0 ppm, consistent with the pattern observed for the perpendicular pyridine previously reported for 4(IV)O.51
proton signals upshifted to −5.6 and −6.9 ppm and the γb proton at 6.0 ppm, consistent with the pattern observed for the perpendicular pyridine previously reported for 4(IV)O.51
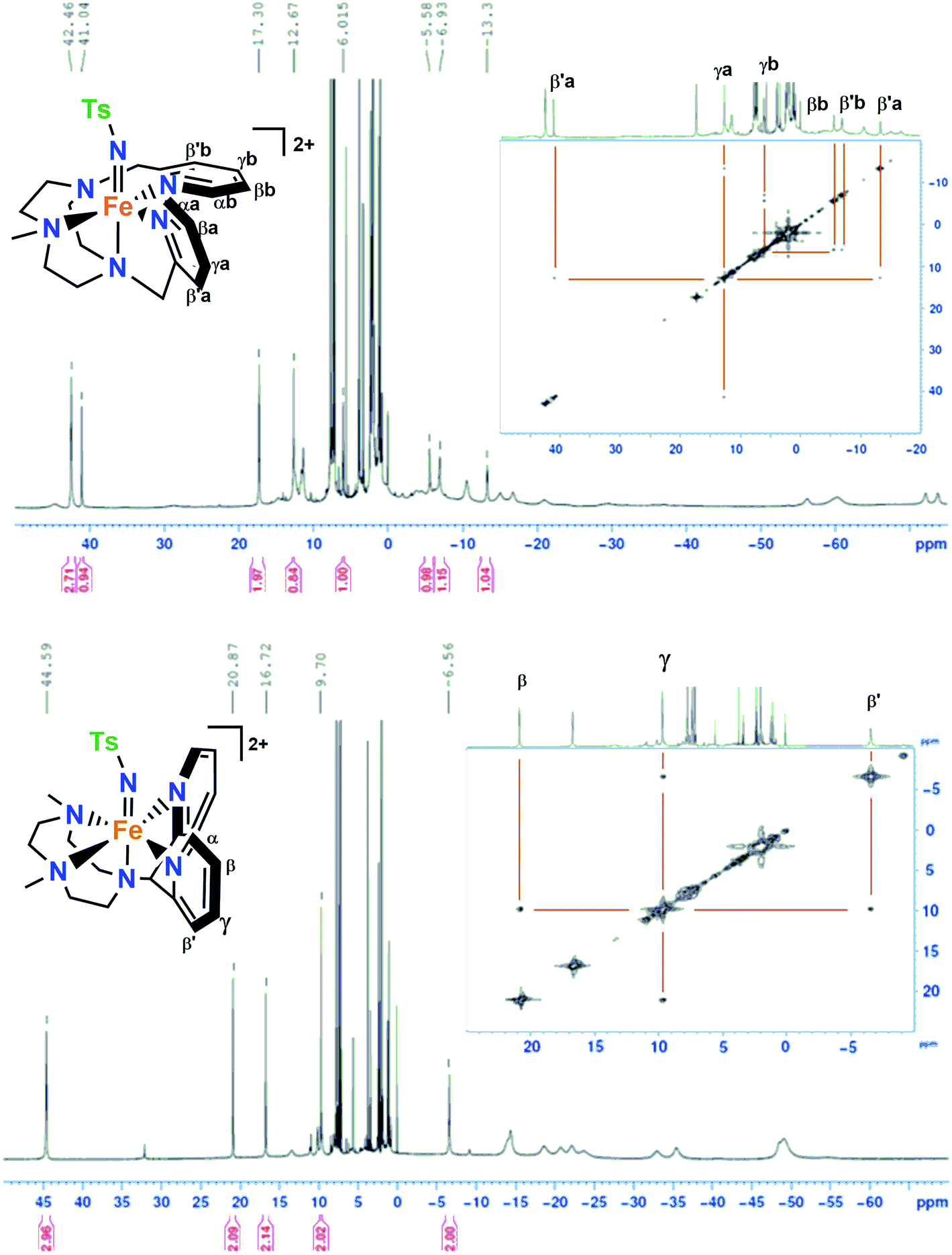 | ||
| Fig. 2
1H-NMR and COSY spectra of 1(IV)NTs (top) in acetone-d6 at 0 °C and 2(IV)NTs (bottom) in acetone-d6 at 25 °C; small peaks at 32 and −9 ppm belong to <5% of 2(IV)O. | ||
The range of chemical shifts observed for the pyridine protons in this pair of complexes reflects differences in the orientation of the pyridine ring relative to the FeNTs axis. These shifts can be understood within the context of trends recently identified by Rasheed et al. in a survey of NMR data for a group of nonheme iron(IV)–oxo complexes, in which the paramagnetic shifts observed are found to be inversely dependent on the size of the torsion angle between the pyridine plane and the FeIVO unit (Table 1).52
X complexes
| Py ring to FeX angle |
β | γ | β′ | Ref. | |
|---|---|---|---|---|---|
| a Torsion angles based on the crystal structure of the corresponding iron(II) complex.53 b Torsion angles obtained from the crystal structures of [FeIV(O)(N4Py)]2+ and [FeIV(O)(Py5Me2)]2+.51,52 c Torsion angles in parentheses from the crystal structure of [FeIV(O)(5Me2N4Py)]2+.52 | |||||
|
1(IV)NTs
|
41.0 (34) | 12.7 (5.7) | −13.3 (−20.3) | This work | |
| −5.6 (−12.6) | 6.0 (−1.0) | −6.9 (−13.9) | |||
|
2(IV)NTs
|
20.9 (13.9) | 9.7 (2.7) | −6.6 (−13.6) | This work | |
| [FeIV(O)-(BnTPEN)]2+a | 6° | 43 (36) | 10.6 (3.6) | −15 (−22) | 51 |
| 16° | 40 (33) | 10.0 (3.0) | −15.5 (−22.5) | ||
| 91° | −0.3 (−7.3) | 8.2 (1.2) | −1.2 (−8.2) | ||
| [FeIV(O)-(N4Py)]2+b | 13.9° (6.8°) c | 44 (37) | 9.5 (2.5) | −17 (−24) | 51 |
| 21.1° (30.4°) c | 30 (23) | 8.3 (1.3) | −11 (−18) | ||
| [FeIV(O)-(Py5Me2)]2+b | Ave 36.5° | 27 (20) | 3.5 (−3.5) | −12 (−19) | 52 |
The largest shifts are found for the pyridines parallel to the FeO unit, while the smallest shifts are found for the pyridines perpendicular to the FeO unit (Table 1). A perusal of this table indicates that this pattern can also be used to interpret the NMR data for 1(IV)NTs and 2(IV)NTs. There are two sets of pyridine peaks in the spectrum of 1(IV)NTs, because one pyridine is nearly parallel to the FeN bond while the other is almost perpendicular to the FeN bond. On the other hand, 2(IV)NTs only exhibits one set of pyridine-derived signals because of the plane of symmetry relating the two pyridines and the paramagnetic shifts observed for the pyridine protons have intermediate values that are between the two extremes found in 1(IV)NTs because of the intermediate angle found for 2(IV)NTs.
Furthermore, complex 1(IV)NTs exhibits a broad resonance enhanced Raman feature at ca. 984 cm−1 (Fig. 3) obtained with 488.0 nm laser excitation, which can be assigned to a vibration arising from the FeN moiety on the basis of the 24 cm−1 downshift observed upon formation of 1(IV)NTs with PhI15NTs. Similarly, the Raman spectrum of compound 2(IV)NTs shows a broad feature around 1016 cm−1 that shifts ca. 22 cm−1 upon formation of 2(IV)NTs with PhI15NTs. The observed downshifts approximate the 27 cm−1 shift calculated for a diatomic FeN oscillator, but the frequencies observed are 200 cm−1 higher than found for corresponding FeIVO units, which have shorter FeO distances than the FeN distances found for FeNTs complexes.2
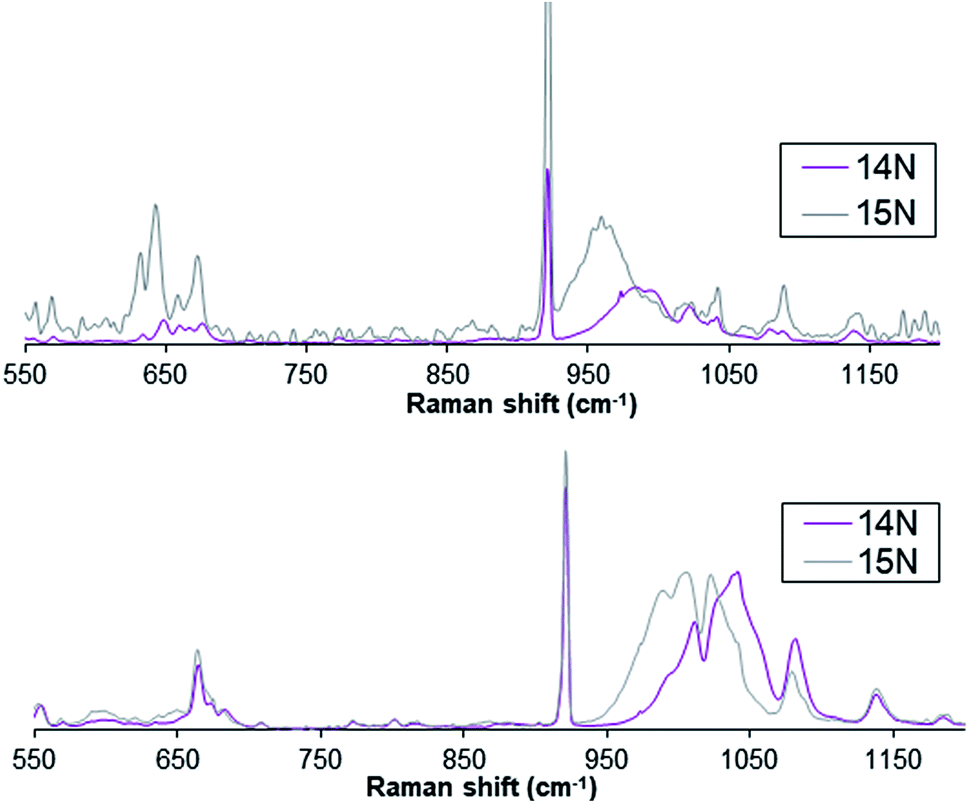 | ||
| Fig. 3 Resonance enhanced Raman spectra for complex 1(IV)NTs (top) and 2(IV)NTs (bottom) upon excitation at 488 nm. | ||
A rationale for the higher frequencies observed for 1(IV)NTs and 2(IV)NTs complexes reported here can be found from a comparison of the vibrational spectra of other FeNR complexes. For example, analogous six-coordinate FeIV(NTs) complexes supported by other pentadentate N5 ligands, 3(IV)NTs and 4(IV)NTs, where N5 = N4Py and BnTPEN, respectively, also exhibit vibrations at 998 and 984 cm−1 that respectively downshift to 975 and 957 cm−1 upon 15N labelling.54 Related tetrahedral FeNR complexes give rise to 15N-sensitive vibrations with frequencies in the 1100 cm−1 range.55 The higher frequencies observed for these complexes are rationalized by the coupling of the FeN stretching mode to the higher-frequency N–S or N–C stretching mode of the NTs or NR moiety to account for the upshift in vibrational frequency. This coupling interaction increases as the FeN and N–(S or C) bonds approach linearity. For example, DFT calculations on 6-coordinate [FeIV(NTs)(N4Py)]2+ predict an FeN–S angle of 161°,54 while an FeN–C angle of 179° is found crystallographically for the tetrahedral [(PhBP3)FeIII(NtBu)] (PhBP3 = [PhB(CH2PPh2)3]−) complex.
One exception to the vibrational trends observed in the FeNTs complexes discussed above is the recently reported [FeV(NTs)(TAML)]− complex (TAML = TetraAmido Macrocyclic Ligand), which exhibits a ν(FeN) mode at 817 cm−1,31 which falls in the range of values reported for FeO vibrations.2 Indeed it is 45 cm−1 lower in frequency than the ν(FeO) mode reported for the [FeV(O)(bTAML)]− (bTAML = biuret TetraAmido Macrocyclic Ligand) complex,56 commensurate with the 0.07 Å longer FeNTs distance found for [FeV(NTs)(TAML)]− by EXAFS analysis.31 DFT calculations on this complex find a more acute FeN–S angle at 125°, thereby essentially uncoupling the two vibrations and allowing the actual FeN stretching frequency to be observed.
X-ray diffraction quality crystals could not be obtained for 1(IV)NTs and 2(IV)NTs, but insight into their structures could be derived from EXAFS analysis of frozen samples in acetone (Fig. 4) and supported by comparison to the corresponding amido derivatives 1(III)–NHTs and 2(III)–NHTs which will be described in the next section.
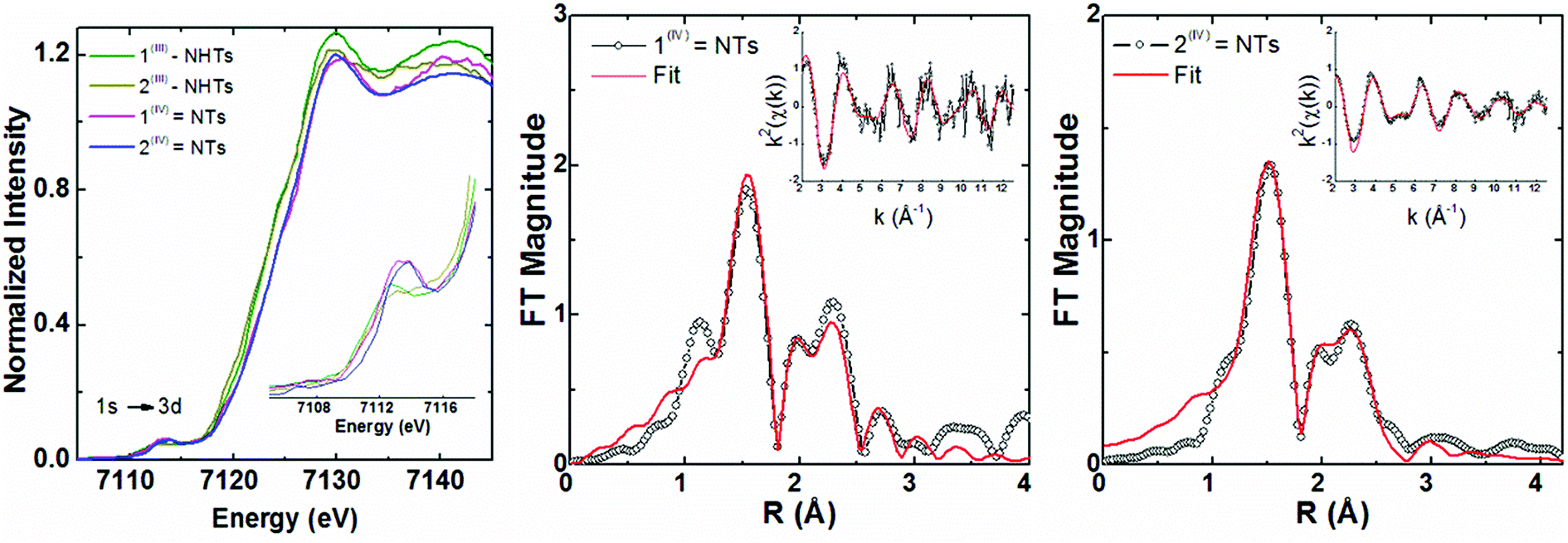 | ||
| Fig. 4 Iron K-edge XANES region (left) for 1(III)–NHTs, 2(III)–NHTs, 1(IV)NTs and 2(IV)NTs together with (right) non-phase shift corrected Fourier transforms of EXAFS data for 1(IV)NTs and 2(IV)NTs with insets showing the k-space data and fits. Experimental data are shown in black and fits in red (see fit Tables S4–S7†). | ||
Iron K-edge X-ray absorption spectroscopy (XAS) of 1(IV)NTs and 2(IV)NTs (Fig. 4) show rising edges centred at 7123.4 eV and 7123.3 eV, having pre-edge features corresponding to 1s → 3d transitions at 7113.2 eV and 7113.3 eV similar to those previously reported iron(IV) centered complexes (Table 2, ESI Table S8†).30,49,57,58 Inspection of the iron K-edge pre-edge and rising energies of the 1(III)–NHTs (1s → 3d 7112.5 eV, 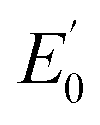 7122.5 eV) and 2(III)–NHTs (1s → 3d 7112.4 eV,
7122.5 eV) and 2(III)–NHTs (1s → 3d 7112.4 eV, 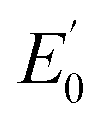 7122.3 eV) derivatives as well as the recently reported analogous 1(V)–N complex ([FeV(N)(MePy2tacn)]+2, (1s → 3d 7114.2 eV,
7122.3 eV) derivatives as well as the recently reported analogous 1(V)–N complex ([FeV(N)(MePy2tacn)]+2, (1s → 3d 7114.2 eV, 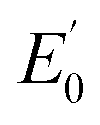 7123.8 eV))59 show a systematic shift of the XANES profile to higher energy strongly suggesting metal centred oxidation for the series.60–63 In particular, the pre-edge energy shifts in steps of about 0.9 eV across the series, consistent with a +1 change in formal oxidation state at the metal centre (Table 2 and S8†). Additionally, the pre-edge intensities increase from about 0.11 normalized units for X(III)–NHTs to 0.18 for X(IV)–NTs and 0.48 for X(V)–N due to both a change in oxidation as well as enhanced p–d mixing due to more covalent, shorter Fe–N/(NTs) bonds, causing ever increasing tetragonal distortions of the pseudo-octahedral environment.58 This is supported by EXAFS analysis (Tables S4–S7†) showing six-coordinate environments for both X(III)–NHTs and X(IV)–NTs species consisting of 5 N/O scattering atoms at ∼2.0 Å and a shorter Fe–N bond, presumably from the tosylimido moiety. For X(IV)–NTs complexes this bond, centred at 1.72 Å, is ∼0.12–0.17 Å shorter than that determined for X(III)–NTs species (∼1.84 Å–1.89 Å) and similar to that of the previously reported 3(IV)NTs (1.73 Å).30
7123.8 eV))59 show a systematic shift of the XANES profile to higher energy strongly suggesting metal centred oxidation for the series.60–63 In particular, the pre-edge energy shifts in steps of about 0.9 eV across the series, consistent with a +1 change in formal oxidation state at the metal centre (Table 2 and S8†). Additionally, the pre-edge intensities increase from about 0.11 normalized units for X(III)–NHTs to 0.18 for X(IV)–NTs and 0.48 for X(V)–N due to both a change in oxidation as well as enhanced p–d mixing due to more covalent, shorter Fe–N/(NTs) bonds, causing ever increasing tetragonal distortions of the pseudo-octahedral environment.58 This is supported by EXAFS analysis (Tables S4–S7†) showing six-coordinate environments for both X(III)–NHTs and X(IV)–NTs species consisting of 5 N/O scattering atoms at ∼2.0 Å and a shorter Fe–N bond, presumably from the tosylimido moiety. For X(IV)–NTs complexes this bond, centred at 1.72 Å, is ∼0.12–0.17 Å shorter than that determined for X(III)–NTs species (∼1.84 Å–1.89 Å) and similar to that of the previously reported 3(IV)NTs (1.73 Å).30
| Mössbauer spectroscopy | Raman v14N/15N (cm−1) | X-ray absorption | |||||
|---|---|---|---|---|---|---|---|
| δ (mm s−1) | ΔEQ (mm s−1) | 1s → 3d area (×102) | 1s → 3d (eV) |
|
Fe–N (Å) | ||
| a DFT calculated values in parenthesis. b Data from ref. 30 and 54. c Data from complex prepared in acetonitrile. | |||||||
|
1(IV)NTs
|
0.05 (0.03) | 1.09 (1.01) | 984/960 (1029) | 18.5 | 7113.2 | 7123.4 | 1.71 (1.73) |
|
2(IV)NTs
|
0.06 (0.04) | 0.73 (0.77) | 1016/994c (1063) | 18.0 | 7113.3 | 7123.3 | 1.72 (1.72) |
|
3(IV)NTs
|
0.02 | 0.98 | 998/975 | 18.0 | 7113.1 | 7123.0 | 1.73 |
To further our understanding of X(IV)–NTs complexes theoretical models of both 1(IV)NTs and 2(IV)NTs were explored with density functional theory. Geometry optimized structures at the B3LYP/6-311+g** level together with electronic energies at the B3LYP/cc-pVTZ level favor a triplet structure for X(IV)–NTs complexes with predicted Fe–NTs bond distances of 1.73 Å and 1.72 Å respectively with a second shell of 5 N scattering atoms at ∼2.03 Å, comparable to experimentally determined EXAFS metrics (Table 2). In the quintet X(IV)–NTs models on the other hand, the Fe–NTs distance is still centred around 1.72 Å, but the remaining ligands are split into two scattering shells of 3 N at 2.10 Å and 2 N at 2.20 Å, well outside the EXAFS experimental fit parameters. In addition, Mössbauer parameters calculated for the triplet structures are in good agreement with experimentally derived values (Table 2), suggesting that in addition to geometry the electronic structure is also well described by the theoretical models. It is important to highlight that similar to the iron(III) analogues (vide infra) the quadrupole splitting for 1(IV)NTs is larger than that of 2(IV)NTs, suggesting a more centrosymmetric environment for 2(IV)NTs. Finally, a description of the electronic structure in terms of a molecular orbital picture was carried out at the B3LYP/def2-TZVP level to facilitate comparison with the previously described electronic structure of 3(IV)NTs data30 (Fig. 5). Similar to 3(IV)NTs we find a highly covalent Fe–NTs interaction in the X(IV)–NTs species with spin density shared by both the iron center (∼1.11) and the N of the tosylimido ligand (∼0.7). As such the question of a iron(IV)–N imido or iron(III)–N− imido radical pair may arise, the latter situation being recently described by Neidig et al. for a pyrazolyl derived high valent iron–imido complex.38 It is important to note however, that the Mössbauer isomer shift for the pyrazolyl complex is 0.20 mm s−1 with a quadrupole splitting of 1.96 mm s−1, parameters that may indeed be attributed to an iron(III) center, and which indeed is quite similar to that of the corresponding iron(III)–amido derivative (0.26 mm s−1) and to values for X(III)–NHTs complexes reported herein. Secondly, the experimentally derived XAS rising edges for our family of complexes consistently show metal based oxidation which when correlated with their corresponding Mössbauer isomer shifts and rising edges of similar Fe–imido complexes fall in a range best attributable to formally iron(IV) centers (Fig. S15†).30,59,64 Lastly, it is important to note that spin density alone may not be a good measure for inferring oxidation state for FeE (E = O, N) type centers. Experimental and theoretical descriptions of FeO centers, that are isoelectronic to FeN, carried out by Que and Solomon show that the highly covalent nature of the Fe–O bond increases the unpaired spin-density on the O-atom.65 Systematic theoretical descriptions by Neese et al. further show that the unpaired spin-density is expected to increase with metal oxidation state and is further favoured in the π* manifold in the case of N due to the smaller effective nuclear charge of N over O resulting in a more covalent π-interaction for iron–imido complexes.66 FeO bonds on the other hand are predicted to have a stronger σ-interaction which helps rationalize the shorter FeE bonds in iron–oxo moieties (1.63 Å).30,49,66 In the FeNTs case the weaker σ-interaction results in nearly degenerate Fe dx2−y2 and Fe dz2 energies as opposed to FeO where the Fe dx2−y2 and Fe dz2 are well separated.65 Therefore the X(IV)–NTs complexes are best described as having a formally +4 oxidation spin 1 iron center similar to the previously reported 3(IV)NTs complex.30 Employing both unrestricted corresponding orbitals (Fig. S16†), which highlight doubly, singly or spin paired molecular orbitals based on the spatial overlap of spin up and down pairs, as well as quasi-restricted orbitals, which allow for the “familiar” picture of doubly and singly occupied as well as empty orbitals,67 a highly mixed and covalent Fe d-orbital manifold is evidenced. Yet, the primary contribution to the SOMO orbitals is the iron metal center yielding a π(Op)4(Fedxy)2π*(Fedxz,yz)2σ*(Fedx2−y2)0σ*(Fedz2)0 arrangement giving an effective bond order of 2 for the FeN moiety (Fig. 5).
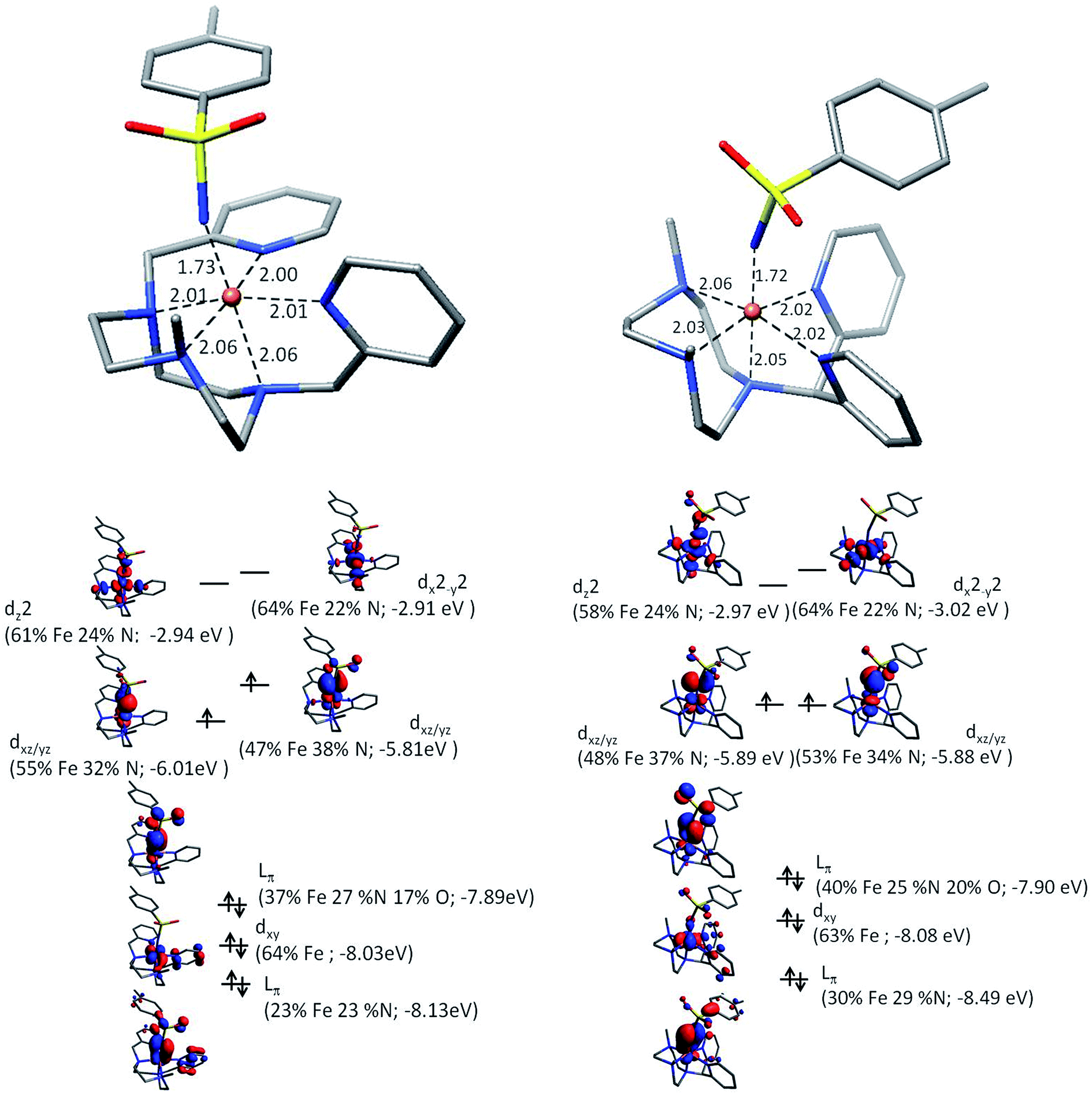 | ||
| Fig. 5 (Top) Computationally derived geometries showing first coordination shell bond distances. (Bottom) Schematic of FeNTs molecular orbitals showing the iron d-manifold using quasi-restricted orbitals (0.05 isovalue) for 1(IV)NTs (left) and 2(IV)NTs (right). | ||
To date only a few iron(IV)–imido species have been prepared and structurally characterised. X-ray diffraction examples include three41 and four coordinate iron complexes.6,23,43,45,68,69 Most of these contain a distorted tetrahedral geometry,6,23,45,68,69 but a complex with a cis-divacant octahedron geometry has also been recently described.43 Notably, all of them exhibit significantly shorter FeN bonds of 1.61–1.64 Å in comparison to octahedral systems. The aforementioned 3(IV)NTs (Fe=N 1.73 Å from EXAFS data) is the only example structurally and electronically comparable to 1(IV)NTs and 2(IV)NTs. On the other hand, examples of ferric–imido radical complexes have been described,38–40 including an example of an octahedral complex.38 FeN distances determined for 1(IV)NTs and 2(IV)NTs fall between those of high-spin three and four coordinate ferric–imido radical complexes (1.76–1.77 Å)18,40 and the computed distance for the single example of the low-spin octahedral complex (1.696 Å) recently described by Neidig and Maron.38 Therefore, the distinctive Mössbauer parameters but not the Fe–N distance allow the differentiation between the two electronic structures.
Synthesis and characterization of 1(III)–NHTs and 2(III)–NHTs
Attempts to obtain single crystals of iron(IV)–tosylimido compounds 1(IV)NTs and 2(IV)NTs by slow diethyl ether diffusion to acetonitrile or acetone solutions of the compounds failed, providing instead the corresponding ferric complexes [FeIII(NHTs)(MePy2tacn)](OTf)2 [1(III)–NHTs](OTf)2, and [FeIII(NHTs)(Me2(CHPy2)tacn)](OTf)2, [2(III)–NHTs](OTf)2 as crystalline materials. A collection of the spectroscopic data obtained for [1(III)–NHTs](OTf)2, and [2(III)–NHTs](OTf)2 is shown in Table 3.
| S | g x | δ (mm s−1) | ΔEQ (mm s−1) | Γ (mm s−1) | η | A x /gnμn | % | E o (Epre-edge) (eV) | |
|---|---|---|---|---|---|---|---|---|---|
| g y | A y /gnμn | ||||||||
| g z | A z /gnμn (T) | ||||||||
| [1(III)–NHTs](OTf)2 | 1/2 | 2.23 | 0.28 | 2.40 | 0.57 | 0 | −36.2 | 100 | 7122.5 (7112.5) |
| 2.23 | 12.8 | ||||||||
| 1.93 | 9.1 | ||||||||
| [2(III)–NHTs](OTf)2 | 1/2 | 2.24 | 0.27 | −2.03 | 0.25 | 0.84 | −23 | 100 | 7122.3 (7112.4) |
| 2.24 | 4.6 | ||||||||
| 1.95 | 3.8 |
Single crystals suitable for X-ray diffraction were obtained for [1(III)–NHTs](SbF6)2. An ORTEP diagram corresponding to its molecular structure along with selected bond distances are shown in Fig. 6. The complex contains an iron centre in a distorted octahedral geometry. Five coordination sites are occupied by nitrogen atoms from the pentadentate ligand with Fe–N distances typical for iron(III) in low-spin (1.98–2.03 Å).70–72 The sixth site is occupied by the tosylamido N atom. The two pyridine rings are perpendicular one to each other, maintaining the structure of 1(II), with one of them parallel to the Fe–N(H) axis. Fe–Py bonds are 1.990 and 1.988 Å and Fe–Nalkyl are 2.024, 2.027 and 1.986 Å. Fe–Py bonds are slightly shorter likely due to the π-acceptor character of pyridine. The Fe–N(H) distance is relatively short (1.89 Å). For comparison the Fe–N(H) bond is 0.18 Å longer than FeN in 1(IV)NTs.
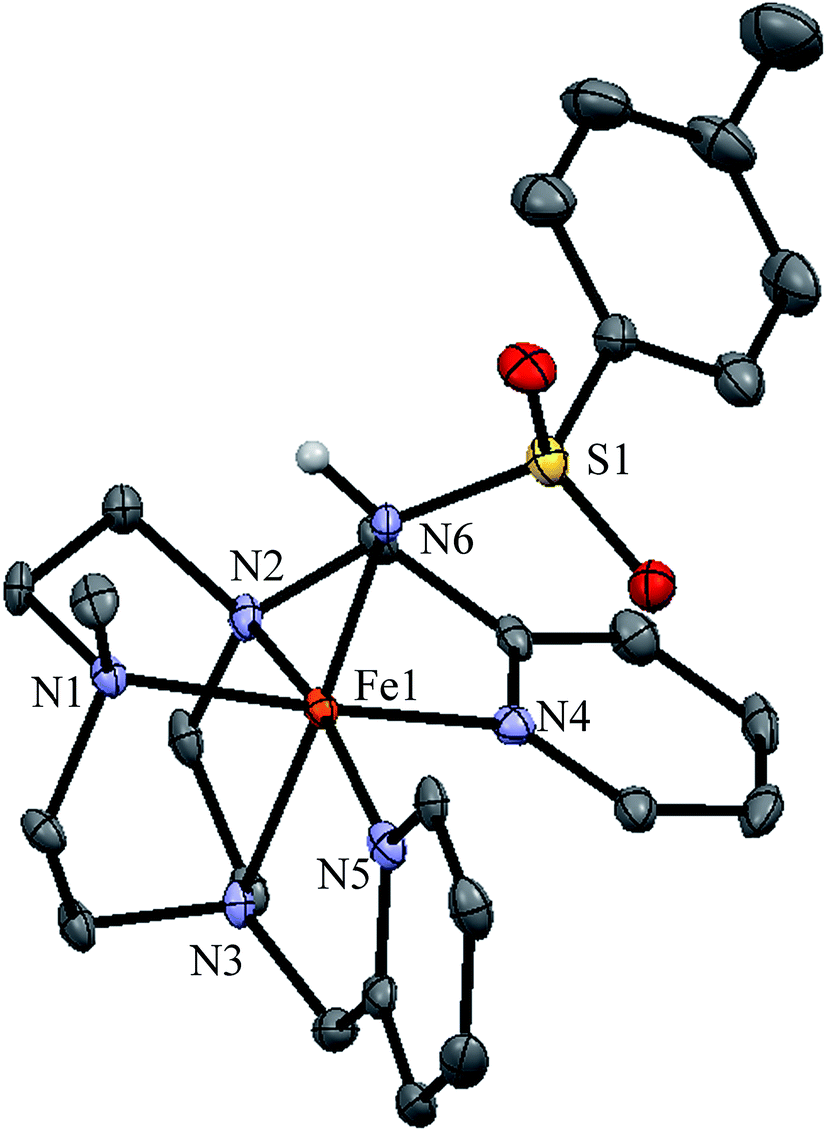 | ||
| Fig. 6 ORTEP structure of isolated ferric complex [1(III)–NHTs](SbF6)2 (50% ellipsoid probability). Counterions omitted for clarity. Selected metrical parameters; Fe1–N1 2.027(4) Å, Fe1–N2 1.986(4) Å, Fe1–N3 2.024(4) Å, Fe1–N4 1.990(4) Å, Fe1–N5 1.988(4) Å, Fe1–N6 1.894(4) Å, Fe1–N6–S1 133.4(3)°. | ||
The Fe–N(H) distance in 1(III)–NHTs is very similar to the values reported by Spasyuk et al. for octahedral complexes [FeIII(NHR)(B2Pz4Py)] (R = C6H4C(CH3)3 or adamantane, 1.869 and 1.854 Å, respectively).38 This is in line with the low-spin character of the iron center in all three complexes. By contrast, longer Fe–N(H) distances in the range 1.95–2.00 Å are noted for the high-spin five coordinate ferric amido complexes [FeIII(NHTol)H22] and [FeIII(NH–SO2C6H4R)TPA2C(O)NHtBu](OTf)2 reported respectively by Borovik73 and Chang.74
EPR studies of 1(III)–NHTs and 2(III)–NHTs at 2 K (Fig. 7) show axial spectra (geff,⊥ = 2.23 and geff,// = 1.93) and (geff,⊥ = 2.21 and geff,// = 1.95) indicative of S = 1/2 systems, characteristic of low-spin ferric centres. In both samples a minor high-spin signal is observed. Mössbauer parameters obtained are congruent with the presence of low-spin ferric centers for both complexes. At 80 K in the absence of a magnetic field, both show asymmetric doublets with δ ≈ 0.28 mm s−1, but differ only in ΔEQ = 2.40 and 2.03 mm s−1, respectively (Fig. 7). When recorded at 4.2 K under a field of 7 T applied parallel to the γ-rays, the spectra of both complexes split into a multiplet spread over a limited velocity range −3 to +3.5 mm s−1 which is consistent with an S = 1/2 center. The data could indeed be fitted under this assumption with the parameters listed in Table 3.
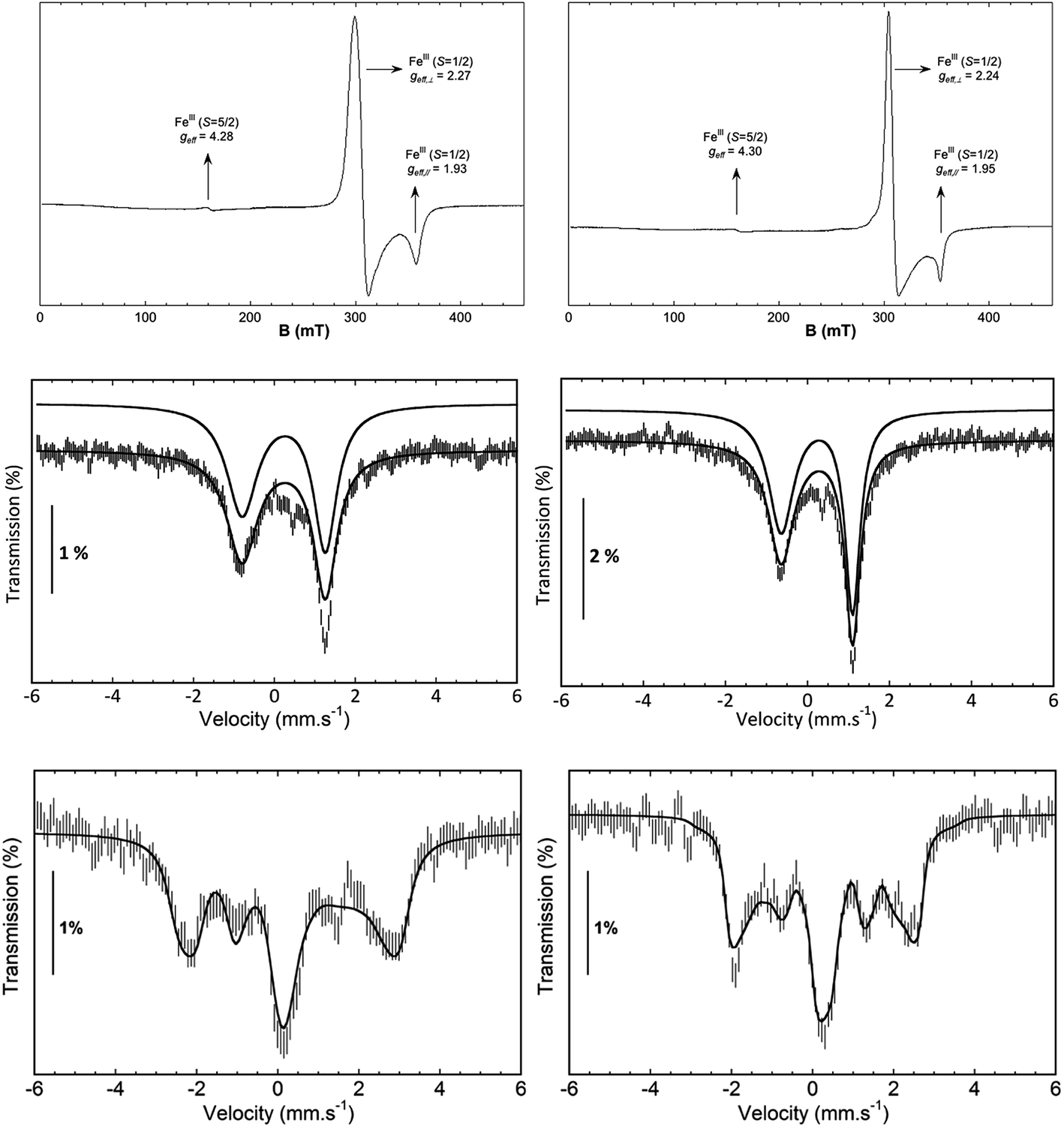 | ||
| Fig. 7 Spectroscopic data of isolated 1(III)–NHTs (left) and 2(III)–NHTs (right). (Top) EPR spectra of 1(III)–NHTs (left) and 2(III)–NHTs (right). (Middle) Mössbauer spectra of 1(III)–NHTs (left) and 2(III)–NHTs (right) recorded at 80 K in absence of field (middle) and at 4.2 K under a field of 7 T applied parallel to the γ-ray (bottom). Experimental spectrum: hatched bars, theoretical spectrum: solid line (see Table 3 for parameters). | ||
Reactivity of 1(IV)NTs and 2(IV)NTs in formal N-atom transfer reactions
1(IV)NTs and 2(IV)NTs, were freshly prepared in situ in acetonitrile at 293 K by reaction of the starting ferrous complexes with 1.2 equiv. of solid PhINTs. Reaction mixtures were filtered to remove the excess of PhINTs and rapidly reacted with thioanisole substrates. Reaction was ascertained by the bleaching of the characteristic low-energy bands in the visible spectra of the complexes (vide infra). 1H-NMR analyses at the end of the reactions show formation of sulfilimine products with yields of ∼0.5 ± 0.1 and ∼0.4 ± 0.1 equiv. (Table 4) per mol of 1(IV)NTs and 2(IV)NTs, respectively. Alternatively, when excess of PhINTs (e.g. 5 equiv.) are used and reaction mixtures were not filtered, 1(IV)NTs and 2(IV)NTs show catalytic behaviour; 1(IV)NTs reacts with MeOPhSMe and HPhSMe to provide 1.35 and 2.1 equiv. of the corresponding sulfilimine product. In analogous conditions, 2(IV)NTs provides 3.5 equiv. and 2.9 equiv. of the corresponding sulfilimine products. Blank experiments under analogous conditions but in the absence of iron complexes generate ≈0.1 equiv. of product. Thus, we conclude that 1(IV)NTs and 2(IV)NTs are competent NTs transfer agents, and that they show catalytic behaviour, although quite modest (Scheme 2).
| X | XPhS(NTs)Me yields (%) | |||
|---|---|---|---|---|
| Complex |
1(IV)NTs
|
2(IV)NTs
|
||
| Solvent | Acetonitrile | Acetone | Acetonitrile | |
| MeO | 57 | 42 | 48 | |
| Me | 46 | 43 | 44 | |
| H | 49 | 53 | 33 | |
| Cl | 44 | 43 | 29 | |
 | ||
| Scheme 2 Iron mediated reaction of thioanisole with PhINTs. | ||
Spectroscopic analysis of the reactions
NTs and 2(IV)NTs at 293 K, a reaction with two phases is apparent from the time dependent changes observed in the UV-vis spectra. In the first phase, the characteristic low energy features at 750 and 740 nm of the respective visible spectra rapidly decrease in intensity (e.g. 5–15 min for MeOPhSMe). Simultaneously a red shift was observed of the initial iron(IV)–tosylimido bands at 455 and 456 nm towards 470 and 465 nm, respectively (Fig. 8). Reaction rates for this first phase are sensitive to the concentration and nature of the sulphide, being faster as the sulphide is more electron rich, and slower as electron-poorer sulphides were employed. As stated above sulfilimine products are formed during this first phase in ca. 50% yield relative to the pre-formed X(IV)NTs. Remarkably, upon complete consumption of the initial iron(IV) at the end of this phase, as ascertained by the bleaching of the band at ∼700 nm, the spectra that result do not match those of 1(II) and 2(II), and are the same irrespective of the nature of the p-X-substituted thioanisole substrate employed. A second and slower phase (on the order of hours) follows and entails formation of 1(II) and 2(II), which show characteristic UV-vis spectra. This second process is independent of the nature and concentration of the substrate.
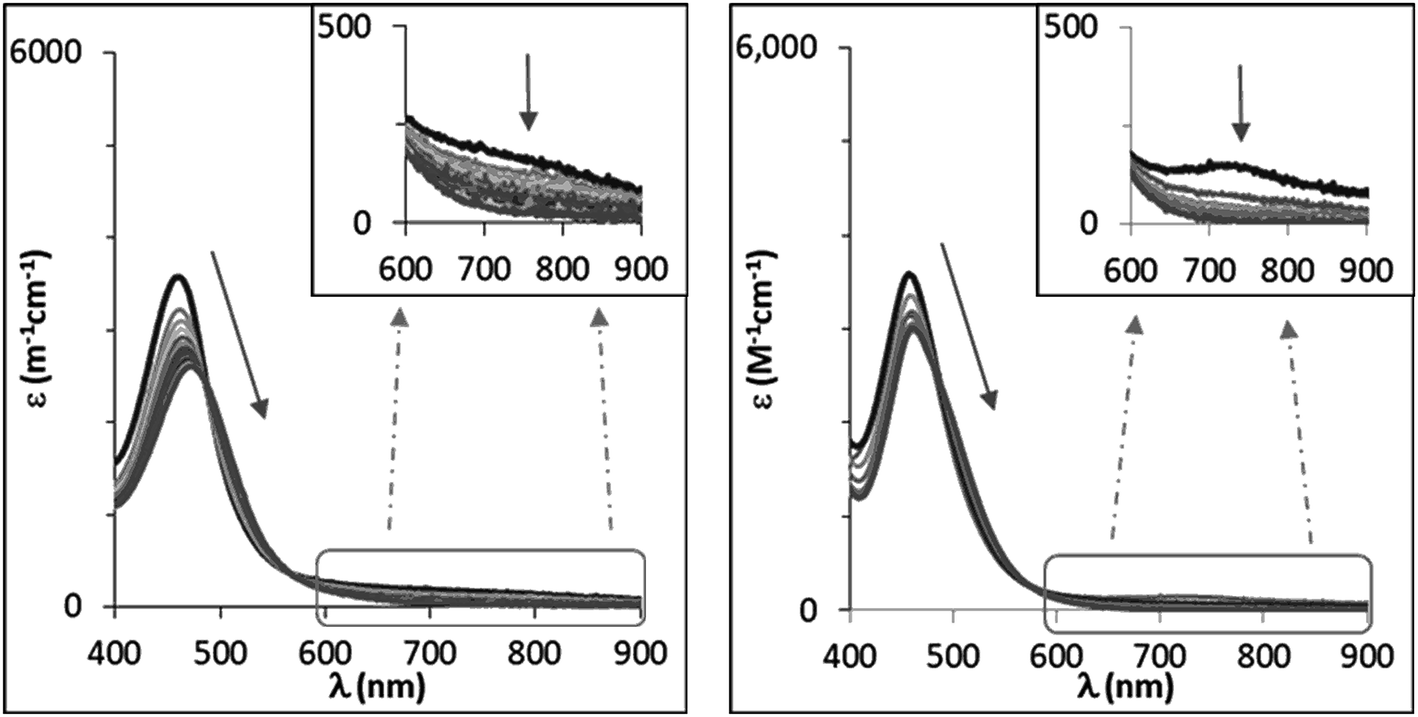 | ||
| Fig. 8 UV-vis spectra corresponding to the reaction of 1(IV)NTs (left) and 2(IV)NTs (right) upon addition of an excess of MeOPhSMe (reaction at 20 °C with 100 and 50 equiv. of substrate added respectively, [Fe] = 0.25 mM in MeCN). Spectra correspond to the first rapid phase of the reaction. | ||
Frozen samples were prepared immediately following the first rapid phase of the reaction of 1(IV)NTs and 2(IV)NTs with MeOPhSMe, and analysed by Mössbauer and EPR spectroscopy. These analyses (see ESI† for details) revealed the presence of monoiron species with an approximate relative ratio of Fe(II)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe(III) of 1:6 for the decay of 1(IV)NTs and 1:3 for 2(IV)NTs. The major ferric species formed after reaction of 1(IV)NTs and 2(IV)NTs with the sulphide can be identified as 1(III)–NHTs and 2(III)–NHTs/2(III)–OH, respectively on the basis of the agreement with the EPR and Mössbauer parameters of the independently prepared complexes. On the other hand, the parameters of the minor ferrous component observed in the Mössbauer spectra (Fig. S7 and Table S1†) are characteristic of a low-spin ferrous centre and are indicative of the formation of 1(II) and 2(II), respectively.49 Of interest, no reaction is observed when 1(IV)NTs is mixed with 1(II) or when 2(IV)NTs is mixed with 2(II). Therefore, it can be concluded that 1(III)–NHTs and 2(III)–NHTs do not originate from subsequent comproportionation reactions. The sum of the spectroscopic data and the stoichiometry of the reactions of 1(IV)NTs and 2(IV)NTs with sulphides indicates that transfer of the tosylimido moiety to the sulphide does not correspond to a 2e− N-transfer where the iron(IV) centre becomes reduced to iron(II), as it has been previously documented for 3(IV)NTs,75 but instead, a competitive and most dominant path involves formation of iron(III) species.
:Fe(III) of 1:6 for the decay of 1(IV)NTs and 1:3 for 2(IV)NTs. The major ferric species formed after reaction of 1(IV)NTs and 2(IV)NTs with the sulphide can be identified as 1(III)–NHTs and 2(III)–NHTs/2(III)–OH, respectively on the basis of the agreement with the EPR and Mössbauer parameters of the independently prepared complexes. On the other hand, the parameters of the minor ferrous component observed in the Mössbauer spectra (Fig. S7 and Table S1†) are characteristic of a low-spin ferrous centre and are indicative of the formation of 1(II) and 2(II), respectively.49 Of interest, no reaction is observed when 1(IV)NTs is mixed with 1(II) or when 2(IV)NTs is mixed with 2(II). Therefore, it can be concluded that 1(III)–NHTs and 2(III)–NHTs do not originate from subsequent comproportionation reactions. The sum of the spectroscopic data and the stoichiometry of the reactions of 1(IV)NTs and 2(IV)NTs with sulphides indicates that transfer of the tosylimido moiety to the sulphide does not correspond to a 2e− N-transfer where the iron(IV) centre becomes reduced to iron(II), as it has been previously documented for 3(IV)NTs,75 but instead, a competitive and most dominant path involves formation of iron(III) species.
NTs and 2(IV)NTs (0.25–4 mM) with sulphides were explored in acetonitrile. Reactions were monitored by following the evolution over time of the absorbance of the low-energy bands corresponding to the iron(IV) species (Fig. 8). Data was satisfactorily adjusted to first order decay processes according to eqn (1)| d[FeIV]/dt = −kobs[FeIV] | (1) |
NTs] or [2(IV)NTs].
The respective pseudo-first order rate constants (kobs) were then calculated from this fitting.
In the first place, reactions of 1(IV)NTs and 2(IV)NTs at a fixed concentration (0.25 mM) against different concentrations of thioanisole substrate, used in large excess (30–220 mM) were analysed (Fig. 9, panels A and B). Observed reaction rates (kobs) were found to be linearly dependent on sulphide concentration, indicating that they are pseudo-first order rate constants with the expression kobs = k′[sulphide].
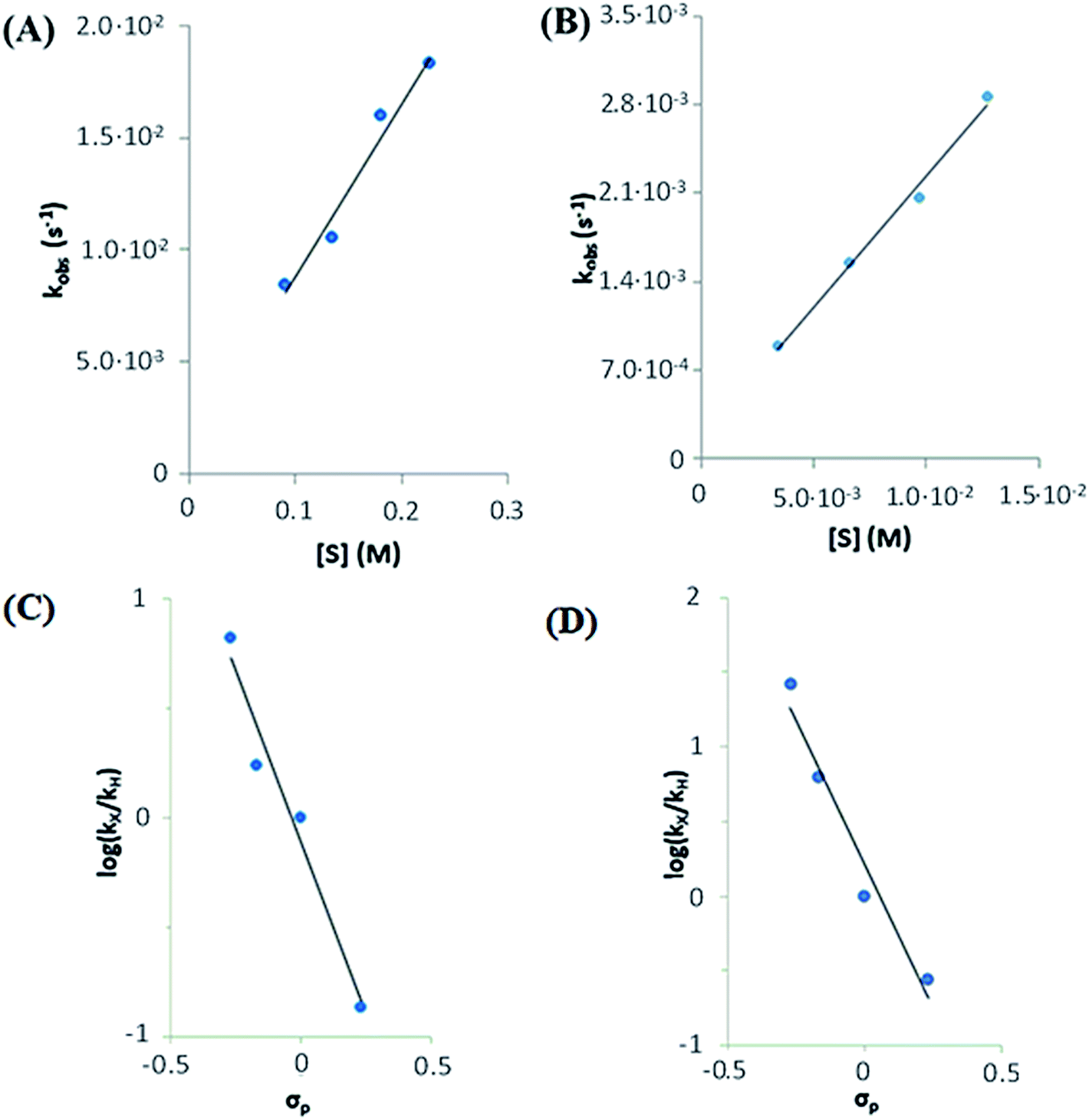 | ||
| Fig. 9 Top: rate dependence on substrate concentration of reactions of (A) 1(IV)NTs (left) with MeOPhSMe substrate (denoted as [S]) and (B) 2(IV)NTs with MeOPhSMe substrate (right) at 293 K. Bottom: Hammett plot representing log(kX/kH) against Hammett parameter (σp) for the reactions of (C) 1(IV)NTs (left) and (D) 2(IV)NTs (right) with p-X-thioanisoles in acetonitrile at 293 K. kx values correspond to the second order rate constants obtained by measuring kobs values at different p-X-thioanisole concentrations. | ||
The k′ values for 1(IV)NTs and 2(IV)NTs were determined for para-substituted thioanisole substrates (XPhSMe, X = MeO, Me, H, Cl) and are shown in Table 5. A Hammett plot was represented by plotting 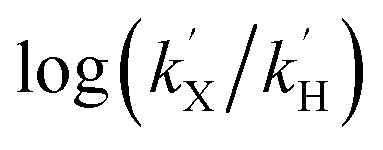 against Hammett parameters (σp). The obtained plots (Fig. 9, panels C and D) show linearity with a slope of ρ = −3.12 and ρ = −3.89 for 1(IV)NTs (kobs values were determined in reactions with a common concentration of 1(IV)NTs and 2(IV)NTs respectively). These values are relatively large in magnitude compared to those described for OAT reactivity examples with iron(IV)–oxo compounds, which range between ρ = −1 to −2,76–79 but are in good agreement with that associated with N atom transfer with 3(IV)NTs (−3.35).47 Moreover, when k′ values were plotted against the oxidation potentials of corresponding thioanisole substrates, a linear plot was obtained with slopes of −5.9 and −7.8 V−1 for 1(IV)NTs and 2(IV)NTs respectively (Fig. S8†). For comparison, for OAT with iron(IV)–oxo processes those values range between −2 and −3 V−1. The relatively large slopes exhibited by 1(IV)NTs and 2(IV)NTs in comparison with 1(IV)O and 2(IV)O indicate that the former reactions proceed with larger charge transfer in the transition states. In fact, the herein determined values are reminiscent of reactions where a single electron transfer process is the rate determining step.47,79 However, as will be discussed in the following lines, thermodynamic considerations discard this scenario.
against Hammett parameters (σp). The obtained plots (Fig. 9, panels C and D) show linearity with a slope of ρ = −3.12 and ρ = −3.89 for 1(IV)NTs (kobs values were determined in reactions with a common concentration of 1(IV)NTs and 2(IV)NTs respectively). These values are relatively large in magnitude compared to those described for OAT reactivity examples with iron(IV)–oxo compounds, which range between ρ = −1 to −2,76–79 but are in good agreement with that associated with N atom transfer with 3(IV)NTs (−3.35).47 Moreover, when k′ values were plotted against the oxidation potentials of corresponding thioanisole substrates, a linear plot was obtained with slopes of −5.9 and −7.8 V−1 for 1(IV)NTs and 2(IV)NTs respectively (Fig. S8†). For comparison, for OAT with iron(IV)–oxo processes those values range between −2 and −3 V−1. The relatively large slopes exhibited by 1(IV)NTs and 2(IV)NTs in comparison with 1(IV)O and 2(IV)O indicate that the former reactions proceed with larger charge transfer in the transition states. In fact, the herein determined values are reminiscent of reactions where a single electron transfer process is the rate determining step.47,79 However, as will be discussed in the following lines, thermodynamic considerations discard this scenario.
NTs and 2(IV)NTs with selected XPhSMe at 293 K
Comparison between reaction rates determined for 1(IV)NTs and 2(IV)NTs with respect to those previously described for 3(IV)NTs deserve some comment (Table 5). Of note, even when performed at a higher temperature (293 K vs. 273 K), reactions of 1(IV)NTs and 2(IV)NTs are nearly two orders of magnitude slower than those of 3(IV)NTs. These observations suggest that 1(IV)NTs and 2(IV)NTs are milder oxidants. Comparison with reaction rates for oxo-transfer reactions to sulphides by the corresponding oxo complexes 1(IV)O and 2(IV)O is also pertinent. Comparison between 3(IV)NTs and 3(IV)O revealed the former to react with sulphide five times faster than the latter.75 In contrast, oxo transfer reactions by 1(IV)O and 2(IV)O are systematically faster than tosylimido transfer by 1(IV)NTs and 2(IV)NTs, with reaction rates between two to twenty times larger when comparing complexes with the same pentadentate ligand.
NTs and 2(IV)NTs with p-X-ArSMe substrates is shown in Scheme 3. The large slopes in the Hammett plots led us to initially consider the possibility that the reaction was initiated by an electron transfer (ET). However, this scenario is discarded on the basis of thermochemical considerations; DFT computed E1/2(FeIV/FeIII) values for the pairs 1(IV)NTs/1(III)NTs and 2(IV)NTs/2(III)NTs are 0.87 and 0.97 V vs. SHE (see Section 5 in the ESI for details, attempts to determine the values experimentally by cyclic voltammetry and by red-ox titrations with ferrocene proved unsuccessful), while the computed potential for the single electron oxidation of thioanisole to the corresponding sulfide cation radical (p-H-ArS˙+Me) is 1.32 V vs. SHE. These values indicate that the ET reaction is highly endergonic, discarding its viability. In addition, DFT computed E1/2(FeIV/FeIII) values for the pairs 1(IV)O/1(III)O and 2(IV)O/2(III)O are 0.08 and 0.19 V vs. SHE respectively (see ESI† for details). These values indicate that they are milder single electron oxidants than the corresponding tosylimido analogues and that ET from thioanisole to these iron(IV)–oxo complexes is also highly endergonic.
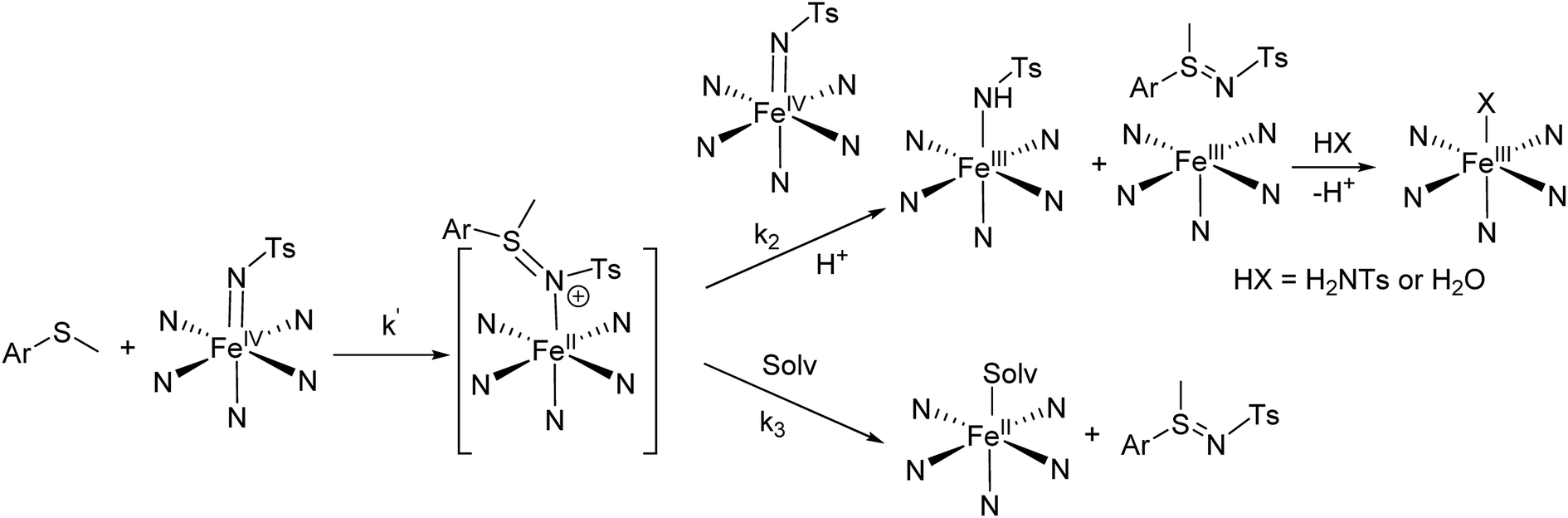 | ||
| Scheme 3 Mechanism proposal for the first phase of the reactions of iron(IV)–tosylimido complexes with thioanisole substrates. Solv. stands for solvent molecule. | ||
Alternatively, reaction of 1(IV)NTs and 2(IV)NTs with p-X-ArSMe substrates is proposed to take place via a two-step process and involves two molecules of the iron(IV)–tosylimido complex (1(IV)NTs or 2(IV)NTs) for each molecule of sulphide. In the first, rate determining step, the sulphide reacts via a nucleophilic attack on the tosylimido N atom of a first molecule of iron(IV)–tosylimido complex. This attack results in tosylimido transfer to the substrate, forming a putative [FeII(p-X-ArS(NTs)Me)(L)], 1–2(II)ArSNTs intermediate that may undergo sulfilimine dissociation and acetonitrile binding to yield [FeII(CH3CN)(L)]2+, as earlier documented for 3(IV)NTs.47 However, in the case of 1(IV)NTs and 2(IV)NTs, the latter appears to be a minor path (k3 < k2), and instead 1–2(II)ArSNTs can be oxidized by a second molecule of 1(IV)NTs or 2(IV)NTs, to form one equivalent of sulfilimine, and two equivalents of ferric species. One of the two retains a tosylimido ligand and converts to 1(III)–NHTs or 2(III)–NHTs presumably by proton transfer from adventitious water. Formation of additional amounts of 1(III)–NHTs or 2(III)–NHTs/2(III)–OH from the five coordinate ferric species [FeIII(L)] may originate from reaction with residual H2NTs and adventitious water.
Therefore, while the three iron(IV)–imido complexes 1–3(IV)NTs react with sulphides via an initial two-electron tosylimido transfer step, the reactions that follow this step are dependent on the nature of L. Structural/steric factors depending on the specific complex may account for the different reactivity, by limiting bimolecular reactions or favouring dissociation. Specifically, the N4Py ligand may provide a sterically more demanding scaffold than the tacn based ligands (MePy2tacn and Me2(CHPy2)tacn). This factor presumably favors rapid sulfilimine dissociation from 3(II)ArSNTs and disfavors its reaction with 3(IV)NTs.
Reactivity of 1(IV)NTs and 2(IV)NTs toward H-atom abstraction reactions
The potential HAT reactivity of 1(IV)NTs and 2(IV)NTs against substrates containing weak C–H bonds was studied by monitoring the bleaching of the characteristic visible bands upon addition of solutions of selected substrates (xanthene, dihydroanthracene (DHA), cyclohexadiene (CHD) and fluorene). No reaction was observed with toluene and ethylbenzene. Reactions were performed in acetone and in acetonitrile at 293 K under a N2 atmosphere. UV-vis monitoring shows that the reactions lead to the formation of 1(III)–NHTs or 2(III)–NHTs along with substrate oxidation products resulting from a formal dehydrogenation process (–H2). Ferrous complexes 1(II) or 2(II), and products resulting from tosylimido transfer were not detected. Nearly quantitative reaction yields for the formation of anthracene from DHA for 1(IV)NTs and 2(IV)NTs were obtained (0.47 and 0.49 equiv. anthracene/equiv. of iron), confirming the involvement of two iron(IV) molecules for the formation of one molecule of product. Therefore, Fe(IV)NTs complexes behave as 1e- oxidants in C–H oxidation reactions, as also occurs with their Fe(IV)O analogs.3,48,80,81
Reactions were studied kinetically. The decay of 1(IV)NTs and 2(IV)NTs exhibits an exponential behavior upon addition of different substrate concentrations. Unexpectedly, the observed rates (kobs, s−1), extracted from the exponential fits, do not vary linearly with the substrate concentrations and instead saturation profiles are observed (Fig. 10).
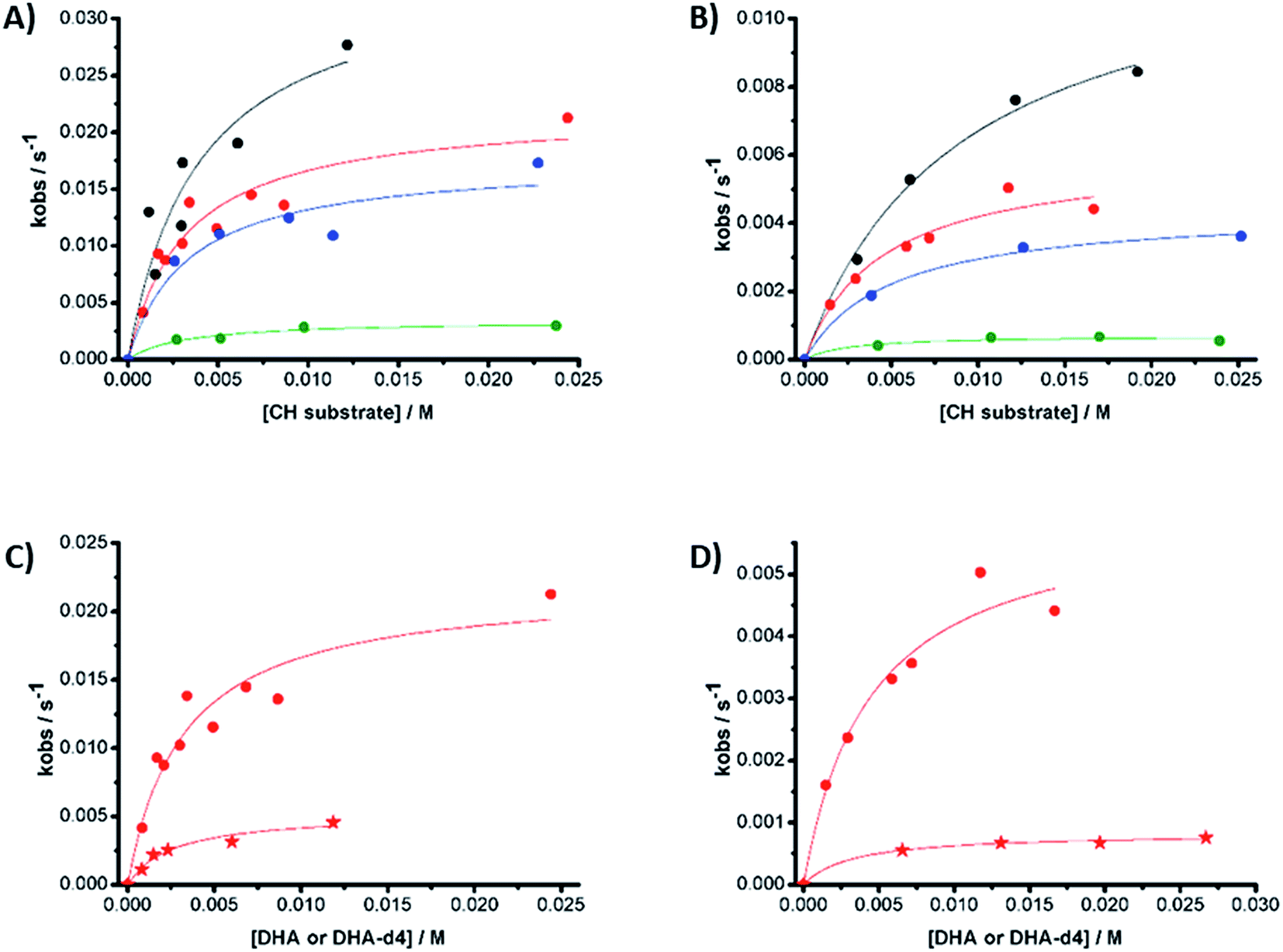 | ||
| Fig. 10 Observed reaction rates (kobs) derived from the reaction of (A) 1(IV)NTs and (B) 2(IV)NTs against different substrates in acetonitrile solution at 293 K under N2. Substrates: black = xanthene; red = DHA; blue = CHD; green = fluorene. (C) 1(IV)NTs and (D) 2(IV)NTs against DHA (circles) and DHA-d4 (stars). | ||
Conditions that lead to rate saturation profiles involve the specific mechanistic feature of presenting a fast reversible reaction step that precedes a slower irreversible step. Thus, by fitting the data with the pre-equilibrium approximation model:
| kobs = (Keq × k × [CH])/(1 + Keq × [CH]) | (2) |
The values of k (s−1) and Keq (M−1) reported in Table 6 could be derived.
NTs and 2(IV)NTs against different hydrocarbons in acetonitrile at 293 K
| BDE, kcal mol−1 | Substrate |
1(IV)NTs
|
2(IV)NTs
|
||
|---|---|---|---|---|---|
| k, s−1 | K eq, M−1 | k, s−1 | K eq, M−1 | ||
| 75.5 | Xanthene | 3.5 ± 0.7 (10−2) | 254 ± 118 | 1.3 ± 0.9 (10−3) | 112 ± 19 |
| 77 | DHA | 2.2 ± 0.2 (10−2) | 315 ± 81 | 6.0 ± 0.6 (10−3) | 227 ± 67 |
| DHA-D4 | 5.2 ± 0.6 (10−3) | 390 ± 115 | 8.3 ± 0.4 (10−4) | 314 ± 74 | |
| 78 | CHD | 1.8 ± 0.2 (10−2) | 301 ± 154 | 4.4 ± 0.2 (10−3) | 203 ± 31 |
| 80 | Fluorene | 3.4 ± 0.3 (10−3) | 329 ± 118 | 7.0 ± 0.9 (10−4) | 415 ± 301 |
The logarithms of the extrapolated rates k (s−1) values corrected by the number of CH (nH) contained in each substrate (log(k/nH) = log(k′)) exhibit a linear dependence versus the BDE of the studied substrates (Fig. S11†), with slopes of −0.21 and −0.26 kcal−1 mol for 1(IV)NTs and 2(IV)NTs complexes, respectively. Notably, these slopes are in good agreement with those determined for HAT reactions of iron(IV)–oxo complexes with the same substrates (between −0.1 and −0.4 kcal−1 mol),48,77,82,83 reinforcing the credibility of the kinetic analysis. This suggests the contribution of a H-atom abstraction event in the rate-determining step of substrates oxidation reaction.84 Consistently, reactions with deuterated DHA (DHA-D4) give rise to k′ values that reveal a primary KIE of 4 and 7 for 1(IV)NTs and 2(IV)NTs, respectively.
The values observed for Keq are all in the range, within experimental error, of 150–500 M−1 for both 1(IV)NTs and 2(IV)NTs, and they are barely related to the BDE of the substrate involved in the reaction. Furthermore, reaction with deuterated DHA (DHA-D4) produce k′ values that reveal a primary KIE of 4 and 7 for 1(IV)NTs and 2(IV)NTs, respectively, while equilibrium constants (Keq) remain basically constant (Table 6 and Fig. 10). These results strongly suggest that a hydrogen atom abstraction is not involved in the first reversible reaction step, but instead contribute to the second, irreversible, rate-determining step of the oxidation.
The reaction of 1(IV)NTs with DHA has also been studied in acetone as solvent. Again, rate constants show saturation profiles with increasing substrate concentration. The overall process is accelerated in acetone, with a kacetone/kCH3CN ratio of 5 at 20 °C, while equilibrium constants Keq are very similar, with a Keq acetone/Keq CH3CN ratio of 0.8 (Table S3 and Fig. S12†). Unfortunately, direct comparison with the reaction rates exhibited by 3(IV)NTs and oxo–iron complexes 1(IV)O and 2(IV)O, is not possible because of the different rate laws.
NTs and 2(IV)NTs with hydrocarbons is best described as a stepwise mechanism in which the HAT event (which represents the rate-determining step of the process) is preceded by a pre-equilibrium step (Scheme 4). Most conclusive is the observation that, unlike k′, the equilibrium constants (Keq) do not relate with the BDE of the substrates and do not present a KIE effect. Furthermore, Keq constants appear to be, within error, only slightly sensitive to the nature of the substrate. Finally, a reaction order for 1(IV)NTs was determined to be 1, both at low and high substrate loadings, excluding the generation of dimers as active species.
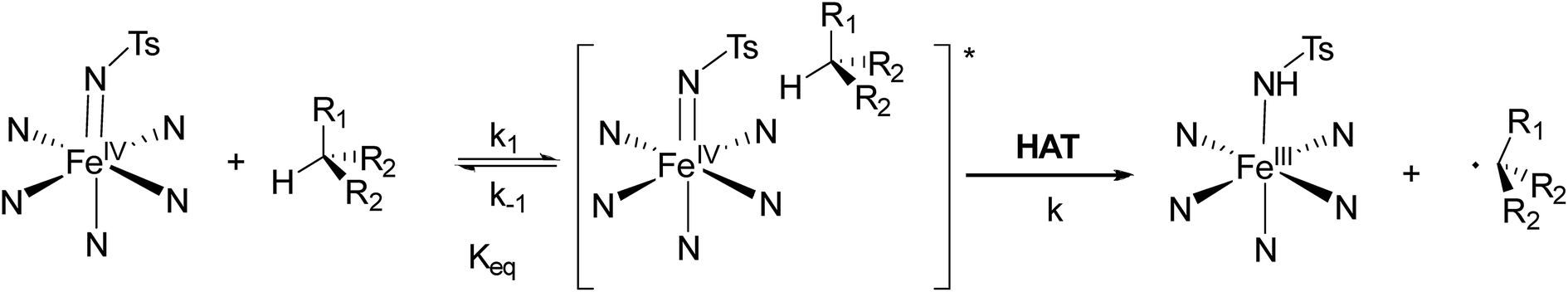 | ||
| Scheme 4 Mechanism proposal for the reactions of iron(IV)–tosylimido complexes 1(IV)NTs and 2(IV)NTs with hydrocarbons. | ||
The exact nature of this pre-equilibrium step cannot be determined, because the intermediate species that form do not show distinctive spectroscopic features. However, kinetic evidence for the formation of association complexes preceding hydrogen atom transfer by metal oxo species85–87 and free radicals88,89 have precedents in the literature. With few exceptions,85 these reactions involve substrates with polarized hydrogen atoms that can engage in hydrogen donor interactions. In the current reactions, reversible hydrogen bond interaction of the substrates with the FeIVNTs can be reasonably discarded because of the apolar nature of the hydrocarbons, and because acetonitrile and acetone are polar solvents that should limit weak hydrogen-bond interactions. Instead, we suggest that the substrate may engage in a series of weak interactions with the electron poor aromatic rings of the tosylimido and ligand pyridine moieties.
Notably, the saturation profiles observed for the reaction of 1(IV)NTs and 2(IV)NTs with hydrocarbons are in contrast with the kinetic behavior of the analogous reactions performed by the related Fe(IV)O species,48,81 by 3(IV)NTs (ref. 75) and by ferric–imido/imidyl radicals,11,38,90,91 for which rate constants vary linearly with substrates concentration. With the single exception of 3(IV)NTs, these reactions are kinetically described as single step HAT processes. In the case of 3(IV)NTs, reversible ET is proposed to precede HAT,47 formally resulting in a hydride transfer reaction. A thermochemical analysis indicates that an analogous ET can not be proposed to account for the pre-equilibrium observed in the reactions of 1(IV)NTs and 2(IV)NTs; the DFT computed single electron oxidation of cyclohexadiene to the cyclohexadienyl radical is 1.61 V vs. SHE, rendering outer sphere ET oxidation by 1(IV)NTs and 2(IV)NTs highly endergonic.
Important differences arise also when comparing KIE's; the values determined for 1(IV)NTs and 2(IV)NTs are much smaller than the remarkably large value (kH/kD = 567) obtained for the octahedral ferric imidyl radical complex described by Spasyuk et al.38 but instead is closer to that of 3(IV)NTs,47 (kH/kD = 7) and to Betley's low-coordinate ferric-amidyl radical complex (kH/kD = 13).11
Rationalization of the HAT reactivity of 1(IV)NTs and 2(IV)NTs may be done by estimating the BDE of the N–H bond in the resulting 1(III)–NHTs and 2(III)–NHTs. As earlier indicated, the pKa and E1/2(FeIV/FeIII) values necessary for determining these BDE's can not be obtained experimentally, and were then derived from DFT methods (B3LYP, see ESI† for details). From this analysis, the deduced BDE's for N–H bonds in 1(III)–NHTs and 2(III)–NHTs are 91.6 and 94.0 kcal mol−1, which rationalizes the HAT ability of 1(IV)NTs and 2(IV)NTs. A cautionary note is that the analysis predicts that 2(IV)NTs should be much more reactive than 1(IV)NTs, but this is not observed. This suggests that steric factors may also contribute in a significant extent to modulate the relative reactivities of the two complexes.
Conclusion
Iron(IV)–tosylimido species 1(IV)NTs and 2(IV)NTs bearing pentadentate aminopyridine ligands, based on the tacn platform are described. They constitute rare examples of octahedral iron(IV)–imido complexes. These metastable high valent iron species have been extensively characterised by means of UV-vis, HR-MS, 1H-NMR, resonance Raman, Mössbauer and X-ray absorption spectra (XANES and EXAFS) as well as DFT calculations. Their reactivity in formal N-transfer and HAT reactions has been investigated. The reactivity of 1(IV)NTs and 2(IV)NTs towards thioanisole substrates is shown to proceed via an initial NTs transfer, followed by reaction with a second molecule of FeIVNTs, overall forming ferric complexes. This behaviour contrasts with that found for iron(IV)–oxo species with the same substrates, which unexceptionally proceed via single step 2e− oxo-transfer processes, producing one equivalent of ferrous complex for each molecule of sulfoxide. Complexes 1(IV)NTs and 2(IV)NTs are HAT agents and react with hydrocarbons containing weak C–H bonds via a two-step process entailing an initial substrate binding equilibria followed by a rate determining HAT reaction. Thus, reactions proceed via an unusually gated HAT mechanism. Again, this reactivity differs from the one exhibited by the analogous iron(IV) oxo complexes, which undergo classical single-step bimolecular HAT processes. Thus, iron(IV)–imido complexes exhibit a singular and rich reactivity that departs from those observed for the isoelectronic oxo analogues, extending the reactivity of high-valent iron species.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge beamline Spline at the European Synchrotron Radiation Facility (ESRF), Grenoble, France, the SAMBA beamline at SOLEIL synchrotron, St. Aubin, France and the XAFS beamline at Elettra Synchrotron, Trieste, Italy. This work has been partially supported by Labex ARCANE and CBH-EUR-GS (ANR-17-EURE-0003). Financial support from the Spanish Ministry of Science CTQ2015-70795-P (M. C.), CTQ2016-80038-R (J. L. F.) and Generalitat de Catalunya (ICREA Academia Award to M. C. and 2017 SGR 01378). STR of UdG are acknowledged for experimental support. Work on nonheme high-valent iron complexes at the University of Minnesota is supported by the US National Science Foundation (CHE-161665391, L. Q.).References
- J. Hohenberger, K. Ray and K. Meyer, Nat. Commun., 2012, 3, 720 CrossRef PubMed.
- A. R. McDonald and L. Que Jr, Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS.
- K.-B. Cho, H. Hirao, S. Shaik and W. Nam, Chem. Soc. Rev., 2016, 45, 1197–1210 RSC.
- W. Nam, Acc. Chem. Res., 2015, 48, 2169–2494 CrossRef PubMed.
- X. Lu, X.-X. Li, M. S. Seo, Y.-M. Lee, M. Clémancey, P. Maldivi, J.-M. Latour, R. Sarangi, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2019, 141, 80–83 CrossRef CAS PubMed.
- M. P. Mehn and J. C. Peters, J. Inorg. Biochem., 2006, 100, 634–643 CrossRef CAS PubMed.
- P. Liu, E. L. M. Wong, A. W. H. Yuen and C. M. Che, Org. Lett., 2008, 10, 3275–3278 CrossRef CAS PubMed.
- J. F. Berry, Comments Inorg. Chem., 2009, 30, 28–66 CrossRef CAS.
- C.-M. Che, C.-Y. Zhou and E. L.-M. Wong, in Iron Catalysis: Fundamentals and Applications, ed. B. Plietker, 2011, vol. 33, pp. 111–138 Search PubMed.
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591–595 CrossRef CAS PubMed.
- M. J. T. Wilding, D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2017, 139, 12043–12049 CrossRef CAS PubMed.
- H. Y. Wang, Y. X. Li, Z. M. Wang, J. Lou, Y. L. Xiao, G. F. Qiu, X. M. Hu, H. J. Altenbach and P. Liu, RSC Adv., 2014, 4, 25287–25290 RSC.
- R. Singh, M. Bordeaux and R. Fasan, ACS Catal., 2014, 4, 546–552 CrossRef CAS PubMed.
- Y. Liu, X. Guan, E. L.-M. Wong, P. Liu, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2013, 135, 7194–7204 CrossRef CAS PubMed.
- S. W. Liang and M. P. Jensen, Organometallics, 2012, 31, 8055–8058 CrossRef CAS.
- T. Hatanaka, R. Miyake, Y. Ishida and H. Kawaguchi, J. Organomet. Chem., 2011, 696, 4046–4050 CrossRef CAS.
- K. L. Klotz, L. M. Slominski, M. E. Riemer, J. A. Phillips and J. A. Halfen, Inorg. Chem., 2009, 48, 801–803 CrossRef CAS PubMed.
- D. A. Iovan, M. J. T. Wilding, Y. Baek, E. T. Hennessy and T. A. Betley, Angew. Chem., Int. Ed., 2017, 56, 15599–15602 CrossRef CAS PubMed.
- S. Hong, X. Y. Lu, Y. M. Lee, M. S. Seo, T. Ohta, T. Ogura, M. Clemancey, P. Maldivi, J. M. Latour, R. Sarangi and W. Nam, J. Am. Chem. Soc., 2017, 139, 14372–14375 CrossRef CAS PubMed.
- B. Pandey, M. Jaccob and G. Rajaraman, Chem. Commun., 2017, 53, 3193–3196 RSC.
- R. Patra and P. Maldivi, J. Mol. Model., 2016, 22, 278 CrossRef PubMed.
- P. Leeladee, G. N. L. Jameson, M. A. Siegler, D. Kumar, S. P. de Visser and D. P. Goldberg, Inorg. Chem., 2013, 52, 4668–4682 CrossRef CAS PubMed.
- I. Nieto, F. Ding, R. P. Bontchev, H. B. Wang and J. M. Smith, J. Am. Chem. Soc., 2008, 130, 2716–2717 CrossRef CAS PubMed.
- S. A. Cramer and D. M. Jenkins, J. Am. Chem. Soc., 2011, 133, 19342–19345 CrossRef CAS PubMed.
- S. A. Cramer, R. H. Sanchez, D. F. Brakhage and D. M. Jenkins, Chem. Commun., 2014, 50, 13967–13970 RSC.
- P. P. Chandrachud, H. M. Bass and D. M. Jenkins, Organometallics, 2016, 35, 1652–1657 CrossRef CAS.
- F. Avenier, E. Goure, P. Dubourdeaux, O. Seneque, J. L. Oddou, J. Pecaut, S. Chardon-Noblat, A. Deronzier and J. M. Latour, Angew. Chem., Int. Ed., 2008, 47, 715–717 CrossRef CAS PubMed.
- E. Goure, F. Avenier, P. Dubourdeaux, O. Seneque, F. Albrieux, C. Lebrun, M. Clemancey, P. Maldivi and J. M. Latour, Angew. Chem., Int. Ed., 2014, 53, 1580–1584 CrossRef CAS PubMed.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- E. J. Klinker, T. A. Jackson, M. P. Jensen, A. Stubna, G. Juhasz, E. L. Bominaar, E. Munck and L. Que Jr, Angew. Chem., Int. Ed., 2006, 45, 7394–7397 CrossRef CAS PubMed.
- S. Hong, K. D. Sutherlin, A. K. Vardhaman, J. J. Yan, S. Park, Y. M. Lee, S. Jang, X. Y. Lu, T. Ohta, T. Ogura, E. I. Solomon and W. Nam, J. Am. Chem. Soc., 2017, 139, 8800–8803 CrossRef CAS PubMed.
- L. Bucinsky, M. Breza, W. T. Lee, A. K. Hickey, D. A. Dickie, I. Nieto, J. A. DeGayner, T. D. Harris, K. Meyer, J. Krzystek, A. Ozarowski, J. Nehrkorn, A. Schnegg, K. Holldack, R. H. Herber, J. Telser and J. M. Smith, Inorg. Chem., 2017, 56, 4751–4768 CrossRef PubMed.
- R. Patra, G. Coin, L. Castro, P. Dubourdeaux, M. Clemancey, J. Pecaut, C. Lebrun, P. Maldivi and J. M. Latour, Catal. Sci. Technol., 2017, 7, 4388–4400 RSC.
- E. Goure, D. Senthilnathan, G. Coin, F. Albrieux, F. Avenier, P. Dubourdeaux, C. Lebrun, P. Maldivi and J. M. Latour, Angew. Chem., Int. Ed., 2017, 56, 4305–4309 CrossRef CAS PubMed.
- M. F. Shehata, S. K. Ayer and J. L. Roizen, J. Org. Chem., 2018, 83, 5072–5081 CrossRef CAS PubMed.
- I. T. Alt, G. Claudia and P. Bernd, Angew. Chem., Int. Ed., 2017, 56, 10582–10586 CrossRef CAS PubMed.
- V. Bagchi, A. Kalra, P. Das, P. Paraskevopoulou, S. Gorla, L. Ai, Q. Wang, S. Mohapatra, A. Choudhury, Z. Sun, T. R. Cundari and P. Stavropoulos, ACS Catal., 2018, 8, 9183–9206 CrossRef CAS.
- D. M. Spasyuk, S. H. Carpenter, C. E. Kefalidis, W. E. Piers, M. L. Neidig and L. Maron, Chem. Sci., 2016, 7, 5939–5944 RSC.
- M. J. T. Wilding, D. A. Iovan, A. T. Wrobel, J. T. Lukens, S. N. MacMillan, K. M. Lancaster and T. A. Betley, J. Am. Chem. Soc., 2017, 139, 14757–14766 CrossRef CAS PubMed.
- D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2016, 138, 1983–1993 CrossRef CAS PubMed.
- L. Wang, L. R. Hu, H. Z. Zhang, H. Chen and L. Deng, J. Am. Chem. Soc., 2015, 137, 14196–14207 CrossRef CAS PubMed.
- H. Z. Zhang, Z. W. Ouyang, Y. S. Liu, Q. Zhang, L. Wang and L. Deng, Angew. Chem., Int. Ed., 2014, 53, 8432–8436 CrossRef CAS PubMed.
- K. Searles, S. Fortier, M. M. Khusniyarov, P. J. Carroll, J. Sutter, K. Meyer, D. J. Mindiola and K. G. Caulton, Angew. Chem., Int. Ed., 2014, 53, 14139–14143 CrossRef CAS PubMed.
- M. E. Moret and J. C. Peters, Angew. Chem., Int. Ed., 2011, 50, 2063–2067 CrossRef CAS PubMed.
- C. M. Thomas, N. P. Mankad and J. C. Peters, J. Am. Chem. Soc., 2006, 128, 4956–4957 CrossRef CAS PubMed.
- A. K. Vardhaman, Y. M. Lee, J. Jung, K. Ohkubo, W. Nam and S. Fukuzumi, Angew. Chem., Int. Ed., 2016, 55, 3709–3713 CrossRef CAS PubMed.
- S. Kumar, A. S. Faponle, P. Barman, A. K. Vardhaman, C. V. Sastri, D. Kumar and S. P. de Visser, J. Am. Chem. Soc., 2014, 136, 17102–17115 CrossRef CAS PubMed.
- D. Wang, K. Ray, M. J. Collins, E. R. Farquhar, J. R. Frisch, L. Gomez, T. A. Jackson, M. Kerscher, A. Waleska, P. Comba, M. Costas and L. Que Jr, Chem. Sci., 2013, 4, 282–291 RSC.
- A. Company, G. Sabenya, M. Gonzalez-Bejar, L. Gomez, M. Clemancey, G. Blondin, A. J. Jasniewski, M. Puri, W. R. Browne, J. M. Latour, L. Que Jr, M. Costas, J. Perez-Prieto and J. Lloret-Fillol, J. Am. Chem. Soc., 2014, 136, 4624–4633 CrossRef CAS PubMed.
- X. P. Shan and L. Que Jr, J. Inorg. Biochem., 2006, 100, 421–433 CrossRef CAS PubMed.
- E. J. Klinker, J. Kaizer, W. W. Brennessel, N. L. Woodrum, C. J. Cramer and L. Que Jr, Angew. Chem., Int. Ed., 2005, 44, 3690–3694 CrossRef CAS PubMed.
- W. F. R. Rasheed, C. S. Abelson, P. O. Peterson, W.-M. Ching, Y. Guo and L. Que Jr, J. Biol. Inorg Chem., 2019, 24, 533–545 CrossRef CAS PubMed.
- N. Ortega-Villar, V. M. Ugalde-Saldívar, M. C. Muñoz, L. A. Ortiz-Frade, J. G. Alvarado-Rodríguez, J. A. Real and R. Moreno-Esparza, Inorg. Chem., 2007, 46, 7285–7293 CrossRef CAS PubMed.
- E. J. Klinker, PhD, University of Minnesota, 2005.
- M. P. Mehn, S. D. Brown, D. M. Jenkins, J. C. Peters and L. Que Jr, Inorg. Chem., 2006, 45, 7417–7427 CrossRef CAS PubMed.
- S. Pattanayak, A. J. Jasniewski, A. Rana, A. Draksharapu, K. K. Singh, A. Weitz, M. Hendrich, L. Que Jr, A. Dey and S. Sen Gupta, Inorg. Chem., 2017, 56, 6352–6361 CrossRef CAS PubMed.
- J.-U. Rohde, S. Torelli, X. Shan, M. H. Lim, E. J. Klinker, J. Kaizer, K. Chen, W. Nam and L. Que Jr, J. Am. Chem. Soc., 2004, 126, 16750–16761 CrossRef CAS PubMed.
- P. Chandrasekaran, S. C. E. Stieber, T. J. Collins, L. Que Jr, F. Neese and S. DeBeer, Dalton Trans., 2011, 40, 11070–11079 RSC.
- G. Sabenya, L. Lázaro, I. Gamba, V. Martin-Diaconescu, E. Andris, T. Weyhermüller, F. Neese, J. Roithova, E. Bill, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2017, 139, 9168–9177 CrossRef CAS PubMed.
- R. Sarangi, S. DeBeer George, D. J. Rudd, R. K. Szilagyi, X. Ribas, C. Rovira, M. Almeida, K. O. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 2316–2326 CrossRef CAS PubMed.
- C. C. Lu, S. D. George, T. Weyhermuller, E. Bill, E. Bothe and K. Wieghardt, Angew. Chem., Int. Ed., 2008, 47, 6384–6387 CrossRef CAS PubMed.
- M. Kawai, T. Yamaguchi, S. Masaoka, F. Tani, T. Kohzuma, L. Chiang, T. Storr, K. Mieda, T. Ogura, R. K. Szilagyi and Y. Shimazaki, Inorg. Chem., 2014, 53, 10195–10202 CrossRef CAS PubMed.
- L. K. Blusch, K. E. Craigo, V. Martin-Diaconescu, A. B. McQuarters, E. Bill, S. Dechert, S. DeBeer, N. Lehnert and F. Meyer, J. Am. Chem. Soc., 2013, 135, 13892–13899 CrossRef CAS PubMed.
- N. Aliaga-Alcalde, S. D. George, B. Mienert, E. Bill, K. Wieghardt and F. Neese, Angew. Chem., Int. Ed., 2005, 44, 2908–2912 CrossRef CAS PubMed.
- A. Decker, J.-U. Rohde, L. Que Jr and E. I. Solomon, J. Am. Chem. Soc., 2004, 126, 5378–5379 CrossRef CAS PubMed.
- J. F. Berry, S. D. George and F. Neese, Phys. Chem. Chem. Phys., 2008, 10, 4361–4374 RSC.
- S. Ye, C.-Y. Geng, S. Shaik and F. Neese, Phys. Chem. Chem. Phys., 2013, 15, 8017–8030 RSC.
- A. K. Verma, T. N. Nazif, C. Achim and S. C. Lee, J. Am. Chem. Soc., 2000, 122, 11013–11014 CrossRef CAS.
- C. T. Saouma and J. C. Peters, Coord. Chem. Rev., 2011, 255, 920–937 CrossRef CAS PubMed.
- Y. Zang, J. Kim, Y. Dong, E. C. Wilkinson, E. H. Appelman and L. Que Jr, J. Am. Chem. Soc., 1997, 119, 4197–4205 CrossRef CAS.
- Y. Nishida, K. Kino and S. Kida, J. Chem. Soc., Dalton Trans., 1987, 1157–1161 RSC.
- J. C. Noveron, R. Herradora, M. M. Olmstead and P. K. Mascharak, Inorg. Chim. Acta, 1999, 285, 269–276 CrossRef CAS.
- R. L. Lucas, D. R. Powell and A. S. Borovik, J. Am. Chem. Soc., 2005, 127, 11596–11597 CrossRef CAS PubMed.
- H. S. Soo, M. T. Sougrati, F. Grandjean, G. J. Long and C. J. Chang, Inorg. Chim. Acta, 2011, 369, 82–91 CrossRef CAS.
- A. K. Vardhaman, P. Barman, S. Kumar, C. V. Sastri, D. Kumar and S. P. de Visser, Angew. Chem., Int. Ed., 2013, 52, 12288–12292 CrossRef CAS PubMed.
- O. Planas, M. Clemancey, J.-M. Latour, A. Company and M. Costas, Chem. Commun., 2014, 50, 10887–10890 RSC.
- A. Company, I. Prat, J. R. Frisch, D. R. Mas-Ballesté, M. Güell, G. Juhász, X. Ribas, D. E. Münck, J. M. Luis, L. Que Jr and M. Costas, Chem.–Eur. J., 2011, 17, 1622–1634 CrossRef CAS PubMed.
- C. V. Sastri, M. S. Seo, M. J. Park, K. M. Kim and W. Nam, Chem. Commun., 2005, 1405–1407 RSC.
- Y. Goto, T. Matsui, S.-i. Ozaki, Y. Watanabe and S. Fukuzumi, J. Am. Chem. Soc., 1999, 121, 9497–9502 CrossRef CAS.
- K.-B. Cho, X. Wu, Y.-M. Lee, Y. H. Kwon, S. Shaik and W. Nam, J. Am. Chem. Soc., 2012, 134, 20222–20225 CrossRef CAS PubMed.
- E. Andris, R. Navrátil, J. Jašík, T. Terencio, M. Srnec, M. Costas and J. Roithová, J. Am. Chem. Soc., 2017, 139, 2757–2765 CrossRef CAS PubMed.
- J. Kaizer, E. J. Klinker, N. Y. Oh, J.-U. Rohde, W. J. Song, A. Stubna, J. Kim, E. Munck, W. Nam and L. Que Jr, J. Am. Chem. Soc., 2004, 126, 472–473 CrossRef CAS PubMed.
- T. K. Paine, M. Costas, J. Kaizer and L. Que Jr, J. Biol. Inorg Chem., 2006, 11, 272–276 CrossRef CAS PubMed.
- X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS PubMed.
- I. Garcia-Bosch, A. Company, C. W. Cady, S. Styring, W. R. Browne, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2011, 50, 5648–5653 CrossRef CAS PubMed.
- G. B. Wijeratne, B. Corzine, V. W. Day and T. A. Jackson, Inorg. Chem., 2014, 53, 7622–7634 CrossRef CAS PubMed.
- T. Ishizuka, S. Ohzu, H. Kotani, Y. Shiota, K. Yoshizawa and T. Kojima, Chem. Sci., 2014, 5, 1429–1436 RSC.
- M. Salamone, G. A. DiLabio and M. Bietti, J. Org. Chem., 2012, 77, 10479–10487 CrossRef CAS PubMed.
- M. Salamone, G. A. DiLabio and M. Bietti, J. Am. Chem. Soc., 2011, 133, 16625–16634 CrossRef CAS PubMed.
- R. E. Cowley, N. A. Eckert, S. Vaddadi, T. M. Figg, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2011, 133, 9796–9811 CrossRef CAS PubMed.
- R. E. Cowley and P. L. Holland, Inorg. Chem., 2012, 51, 8352–8361 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1918211. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02526j |
| This journal is © The Royal Society of Chemistry 2019 |