
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-valent homobimetallic Rh complexes: influence of ligands on the structure and the intramolecular reactivity of Rh–H intermediates†
Pascal
Jurt
a,
Oleg G.
Salnikov
bc,
Thomas L.
Gianetti
 *ad,
Nikita V.
Chukanov
bc,
Matthew G.
Baker
a,
Grégoire
Le Corre
a,
Jaap E.
Borger
a,
Rene
Verel
a,
Sébastien
Gauthier
e,
Olaf
Fuhr
g,
Kirill V.
Kovtunov
bc,
Alexey
Fedorov
af,
Dieter
Fenske
g,
Igor V.
Koptyug
bc and
Hansjörg
Grützmacher
*a
*ad,
Nikita V.
Chukanov
bc,
Matthew G.
Baker
a,
Grégoire
Le Corre
a,
Jaap E.
Borger
a,
Rene
Verel
a,
Sébastien
Gauthier
e,
Olaf
Fuhr
g,
Kirill V.
Kovtunov
bc,
Alexey
Fedorov
af,
Dieter
Fenske
g,
Igor V.
Koptyug
bc and
Hansjörg
Grützmacher
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zürich, Switzerland. E-mail: hgruetzmacher@ethz.ch
bInternational Tomography Center, SB RAS, Novosibirsk, Russia
cNovosibirsk State University, Novosibirsk, Russia
dDepartment of Chemistry and Biochemistry, University of Arizona, Tucson, USA. E-mail: tgianetti@email.arizona.edu
eUniversité de Rennes, CNRS, ISCR-UMR 6226, F-35000 Rennes, France
fDepartment of Mechanical and Process Engineering, ETH Zürich, Switzerland
gInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Eggenstein-Leopoldshafen, Germany
First published on 8th July 2019
Abstract
Supporting two metal binding sites by a tailored polydentate trop-based (trop = 5H-dibenzo[a,d]cyclohepten-5-yl) ligand yields highly unsymmetric homobimetallic rhodium(I) complexes. Their reaction with hydrogen rapidly forms Rh hydrides that undergo an intramolecular semihydrogenation of two C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds of the trop ligand. This reaction is chemoselective and converts CC bonds to a bridging carbene and an olefinic ligand in the first and the second semihydrogenation steps, respectively. Stabilization by a bridging diphosphine ligand allows characterization of a Rh hydride species by advanced NMR techniques and may provide insight into possible elementary steps of H2 activation by interfacial sites of heterogeneous Rh/C catalysts.
C bonds of the trop ligand. This reaction is chemoselective and converts CC bonds to a bridging carbene and an olefinic ligand in the first and the second semihydrogenation steps, respectively. Stabilization by a bridging diphosphine ligand allows characterization of a Rh hydride species by advanced NMR techniques and may provide insight into possible elementary steps of H2 activation by interfacial sites of heterogeneous Rh/C catalysts.
Introduction
Bimetallic transition metal complexes have been investigated since the 1970s,1 however despite numerous reports, only a few complexes are widely used in catalytic applications. The most prominent is a rhodium(II) acetate dimer and its derivatives exploited for carbene transfer reactions.2 These and more recent bimetallic systems typically outperform monometallic alternatives in terms of activity and selectivity, functional group tolerance, and catalyst loading.3–5 The interest in homobimetallic catalysts has recently seen a renaissance.6 Selected catalytic applications include diazo-free cyclopropanation,7 small molecule activation,8,9 hydrogenation,10 hydroformylation11 and C–C coupling reactions.12 Notably, heterobimetallic complexes serve as models of the transmetallation step for the latter process.13–17 Most of these bimetallic species fall into two groups: (i) heterobimetallic early-late transition metal complexes, or (ii) symmetric bimetallic complexes with a core consisting of two mid-to-late transition metals.18 Group (i) typically relies on ligands that combine in close proximity both soft and hard moieties, and feature a highly polar dative interaction from the late to the early transition metal.19 Group (ii) relies on symmetric bridging ligands, often leading to an apolar bimetallic interaction.20 Complexes of the group (ii) have been of particular interest for the understanding of reactivity of small clusters.21–23Rhodium nanoparticles on support materials are widely used in heterogeneous catalysis and various industrial processes especially for hydrogenation–dehydrogenation reactions.24 But the mode of interaction between the supported rhodium sites and H2 is not fully understood.25 A recent DFT study reported that hydrogen activation by small rhodium clusters on a carbon support features a bridging hydride species and a hydride ligand (Scheme 1a) that could be transfered to the carbon support.26 However, such intermediates have not been observed experimentally to date. Low valent homobimetallic rhodium complexes serve as molecular models for supported Rh sites towards understanding the hydrogen activation on such materials.19 While representation of supported heterogeneous catalysts by a bimetallic model significantly reduces their complexity, this approach allows for a reliable identification of reaction products and is therefore insightful, despite the apparent oversimplification of the intrinsic complexity of heterogeneous catalysts. That said, examples of well-defined bimetallic Rh(I)–Rh(I) systems capable of activating H2 are scarce.27Scheme 1b presents a rare dirhodium(I) complex that, according to DFT calculations, activates dihydrogen leading to one bridging and one terminal hydride ligand, in a similar fashion as mentioned above for the supported rhodium centers. This mode of the hydrogen activation dissymmetrizes the dirhodium complex while simultaneously forming a stabilizing Rh–Rh bond.28 Again, experimental evidence for such a dirhodium dihydride species is still lacking, although dissymmetric complexes with a dirhodium(I) core containing chloride ligands in place of the hydrides were reported.29 A symmetric dirhodium dihydride complex, which forms an intermetallic bond, was characterized by NMR and IR (Scheme 1c).30
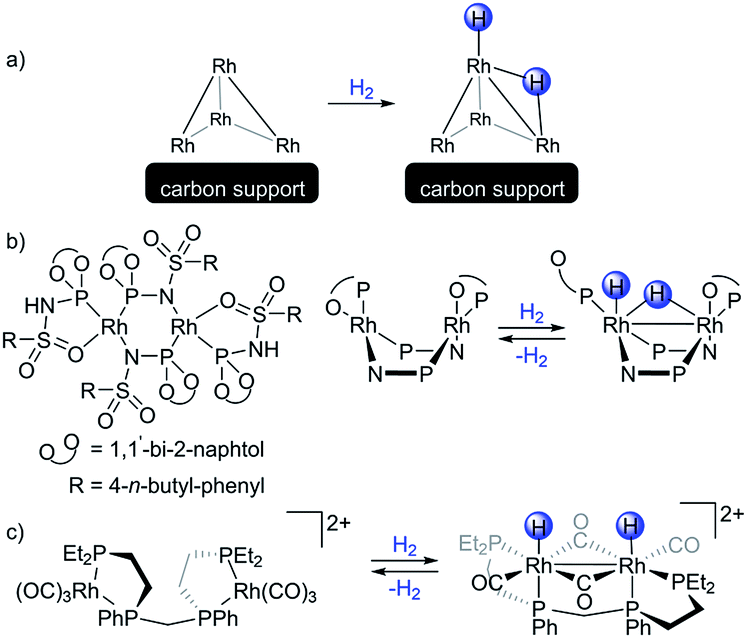 | ||
| Scheme 1 (a) Hydrogen activation as calculated by DFT for small rhodium clusters on a carbon support. (b) A bimetallic Rh hydrogenation catalyst (left) and its proposed hydrogen activation mode, according to DFT (right). (c) A rare dirhodium(II) dihydride characterized by NMR. | ||
In contrast to the exploitation of complexes with symmetric ligands, the advantages of dissymmetric ligands to control the reactivity of two adjacent Rh centers are underutilized.31 The dissymmetry of the electronic environment imposed by the ligand could not only enable otherwise inaccessible reactivity manifolds,19,32 but also allows modelling the environment and complexity of heterogeneous catalysts (metal-support interface, surface defects, etc.). In particular, a non-innocent ligand with multiple unsaturated C–C bonds could provide insights in the reactivity of Rh/C interfacial sites of metallic Rh nanoclusters or nanoparticles on carbon-based supports.
Previous work by the Grützmacher group showed that the bidentate concavely shaped tropPPh2 (trop = 5H-dibenzo[a,d]cyclohepten-5-yl) ligand featuring both a σ-donor (Ph2P group) and a π-accepting binding site (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ctrop) enables a strong binding33 to several transition metal centers including Pd,34 Rh35,36 and Ir.36,37 In order to synthesize a homobimetallic low valent dirhodium complex, we developed a tailored {(TMS)CC}2tropPPh2 ligand with a second binding site created by two alkyne moieties (vide infra). This framework supports a Rh2(I) complex with labile triflate ligands cis to the polarized intermetallic Rh–Rh bond and demonstrates cooperativity of two metal sites in the hydrogen activation. However, resulting hydrides react intra-molecularly by adding hydrogen to the alkyne units of the {(TMS)CC}2tropPPh2 ligand. We performed in depth NMR studies, including the use of the parahydrogen-induced polarization (PHIP) technique,38–42 to elucidate the structure and transformations of these Rh hydride intermediates that convert the trop ligand into a carbene-like motif via semihydrogenation of the first CC bond. Remarkably, the semihydrogenation of the second CC bond proceeds with a different chemoselectivity forming a cis-olefinic ligand. Tuning the Rh coordination sphere by exchanging one triflate for a bis(diphenylphosphino)methane (dppm) ligand inhibits the semihydrogenation steps and allows to characterize the intermediate rhodium hydride species by NMR. Overall, reactivity of Rh2 complexes in a carbon-rich ligand environment offers mechanistic insight on the net H2 activation across a Rh–Rh bond and interaction of Rh–H species with a carbon support in Rh/C heterogeneous catalysts.
Ctrop) enables a strong binding33 to several transition metal centers including Pd,34 Rh35,36 and Ir.36,37 In order to synthesize a homobimetallic low valent dirhodium complex, we developed a tailored {(TMS)CC}2tropPPh2 ligand with a second binding site created by two alkyne moieties (vide infra). This framework supports a Rh2(I) complex with labile triflate ligands cis to the polarized intermetallic Rh–Rh bond and demonstrates cooperativity of two metal sites in the hydrogen activation. However, resulting hydrides react intra-molecularly by adding hydrogen to the alkyne units of the {(TMS)CC}2tropPPh2 ligand. We performed in depth NMR studies, including the use of the parahydrogen-induced polarization (PHIP) technique,38–42 to elucidate the structure and transformations of these Rh hydride intermediates that convert the trop ligand into a carbene-like motif via semihydrogenation of the first CC bond. Remarkably, the semihydrogenation of the second CC bond proceeds with a different chemoselectivity forming a cis-olefinic ligand. Tuning the Rh coordination sphere by exchanging one triflate for a bis(diphenylphosphino)methane (dppm) ligand inhibits the semihydrogenation steps and allows to characterize the intermediate rhodium hydride species by NMR. Overall, reactivity of Rh2 complexes in a carbon-rich ligand environment offers mechanistic insight on the net H2 activation across a Rh–Rh bond and interaction of Rh–H species with a carbon support in Rh/C heterogeneous catalysts.
Results and discussion
Synthesis and characterization of 10,11-di-(trimethylsilyl)acetylene-5H-dibenzo[a,d]cyclohepten-5-diphenylphosphine and its Rh2 complexes
The tropketone 143 was converted to the polydentate trop ligand 5 in four steps with an overall yield of 34%, utilizing a Sonogashira protocol and conventional functional group transformation reactions (Fig. 1, top panel). Mixing 5 with one equivalent of [(C2H4)2RhCl]2 leads to the chloro bridged dimer 6 having two adjacent rhodium centers per trop ligand (83% yield). The abstraction of the chloride ligands in 6 with silver triflate gives the monomeric homobimetallic complex 7 in 82% yield. According to single crystal X-ray diffraction of 7, two triflate ligands bridge between the two rhodium centers. The addition of one equivalent of diphenylphosphinomethane (dppm) displaces one triflate ligand to form the dppm adduct 8 (83% yield, Fig. 1, bottom panel).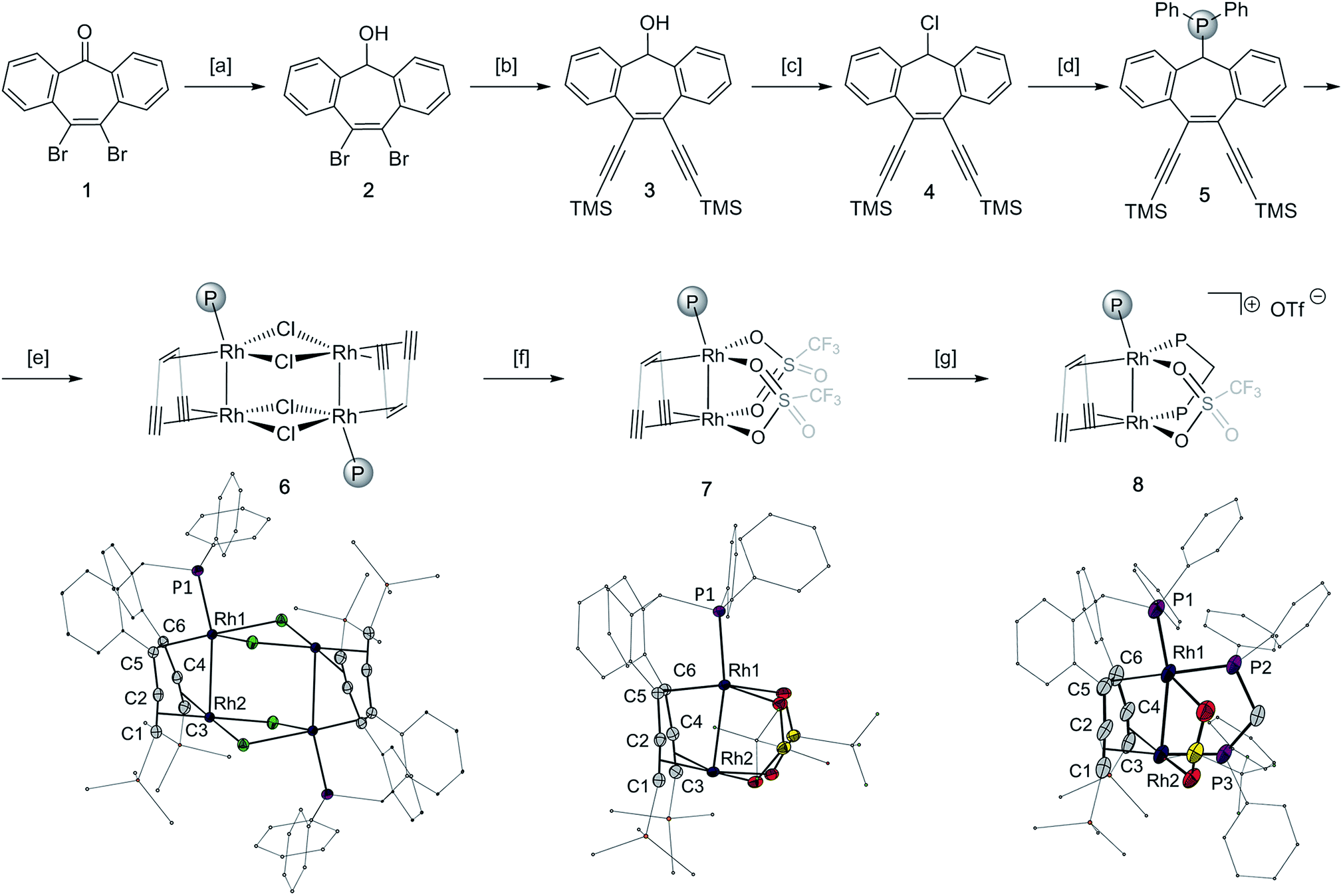 | ||
| Fig. 1 [a] NaBH4, MeOH, 98%. [b] TMSCCH, 5 mol% [Pd(PPh3)4], CuI, toluene, Et3N, 76%. [c] SOCl2, DCM, 0 °C, 80%. [d] LiPPh2, toluene, 63%. [e] [(C2H4)RhCl]2, benzene, 82%. [f] AgOTf, DCM, 83%. [g] Ph2PCH2PPh2, DCM/hexane, 80%. The representation of ligand 5 is simplified in structures 6–8 for clarity. The crystal structures of bimetallic complexes 6–8 are given below the respective chemical structures and have the solvent molecules and non-coordinated triflate anions omitted for clarity. | ||
Complexes 6–8 were characterized by single crystal X-ray diffraction methods. All complexes possess a distorted square planar geometry around Rh1 and a nearly ideal square planar environment around Rh2 (τRh1 = 0.29, 0.46 and 0.37, and τRh2 = 0.06, 0.01 and 0.03 for 6, 7 and 8 respectively).44 The Rh–Rh contact in monomeric 7 is 2.6297(2) Å, that is 0.21 Å shorter than in dimeric 6 (2.8464(3) Å, Table 1). This shortening of the Rh–Rh distances is accompanied by an elongation of the P–Rh bond from 2.1829(3) Å in the chloro bridged dimer 6 to 2.2115(5) Å in the monomeric triflate complex 7, indicating that the Rh–Rh interaction in 7 is stronger. However, the bond lengths of the coordinated C–C multiple bonds as well as the respective C–Rh distances do not differ much between 6 and 7 (Table 1). Dppm adduct 8 has a Rh–Rh bond of 2.7691(7) Å, an intermediate value between those of 6 and 7. Additional significant changes are observed in the alkene/alkyne bonds trans to P2 and P3, which are elongated in 8 (Rh1-ct(C5–C6) 1.981(7) Å and Rh2-ct(C1–C2) 2.232(7) Å in 8 compared to Rh1-ct(C5–C6) 1.913(2) Å and Rh2-ct(C1–C2) 2.062(2) Å in 7), explained by the higher trans influence of the phosphine ligand.45
| Bond length or angle | 6 | 7 | 8 |
|---|---|---|---|
| Rh1–Rh2 | 2.8464(3) | 2.6297(2) | 2.7691(7) |
| Rh1–P1 | 2.1829(3) | 2.2115(5) | 2.2385(17) |
| C1–C2 | 1.242(3) | 1.246(3) | 1.235(10) |
| C3–C4 | 1.247(4) | 1.248(3) | 1.230(9) |
| C5–C6 | 1.499(3) | 1.495(3) | 1.475(10) |
| ct(C1–C2)–Rh2 | 2.077(2) | 2.062(2) | 2.232(7) |
| ct(C3–C4)–Rh2 | 2.054(3) | 2.057(2) | 2.051(6) |
| ct(C5–C6)–Rh1 | 1.962(2) | 1.913(2) | 1.981(7) |
| P1–Rh1–Rh2 | 162.708(19) | 168.488(16) | 164.92(5) |
| Rh1–Rh2-ct(C1–C2) | 78.83(7) | 87.48(6) | 80.82(19) |
| Rh1–Rh2-ct(C3–C4) | 81.01(7) | 83.07(6) | 80.93(19) |
| ct(C1–C2)–Rh2-ct(C3–C4) | 89.67(10) | 82.19(6) | 85.9(2) |
| P1–Rh1-ct(C5–C6) | 91.43(7) | 90.56(8) | 91.4(2) |
The 13C NMR olefinic chemical shifts for the chloro bridged dimer 6 and the monomeric triflate complex 7 are similar (δ13C = 37.9 vs. 37.0 ppm for 6 and 7, respectively). However, a strong shielding is observed for both triple bonds in the triflate complex (δ13C = 99.4 to 86.6 ppm for TMS–CC and δ13C = 74.0 to 65.3 for TMS–CC for 6 and 7, respectively) suggesting that Rh2 site in 7 is more electron-rich than in 6. Likely, the Rh–Rh bond can be best described as a dative bond where electron donation from Rh2 into the antibonding orbital of the Rh1–P bond occurs, similar to the bonding in early-late bimetallic transition metal complexes.19 For complex 8, this is supported by calculations, as the HOMO−2 and HOMO−3 orbitals show a clear overlap between the two metal centers, with a larger orbital contribution of Rh2 (Fig. S1†).
Reactivity of [Rh2{(TMS)C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C}2tropPPh2] complexes with H2
C}2tropPPh2] complexes with H2
Adding hydrogen (1–2 bar) to the headspace of a J. Young NMR tube with monomeric triflate complex 7 dissolved in CD3CN and following the reaction progress by NMR allows observing a stepwise hydrogenation of the two triple bonds of the supporting {(TMS)CC}2tropPPh2 ligand. The first alkyne semihydrogenation step proceeds quantitatively within 15 min and, strikingly, yields carbene species 9 (Scheme 2). Such chemoselective hydrogenation is uncommon and was only previously observed for alkynes bound to d8 metal centers with a Pt(II)–(μ-H)3–Pt(II) core.46 The semihydrogenation of the remaining triple bond leads to a coordinated alkene 10 (Scheme 2) and proceeds with a slower rate requiring ca. 20 h at 2 bar of H2 for quantitative conversion. The carbene complex 9 and the carbene alkene complex 10 display characteristic signals in 13C NMR spectra assigned to the bridging carbene at δ13C = 171.1 and 166.3 ppm and the methylene carbon at δ13C = 35.5 and 36.7 ppm, respectively. The methylene group formed is identified by the two diastereotopic proton signals in the 1H NMR spectrum at δ1H = 2.38, 3.18 and 1.99, 3.03 ppm for 9 and 10, respectively, showing a geminal 2JHH coupling of 12.1 and 13.1 Hz. 2D NMR experiments (COSY, HSQC and HMBC) support the structural assignment of complexes 9 and 10; this data is presented in the ESI (Fig. S2–S5†). The bridging nature of the carbene ligand is further characterized by two rather different JCRh coupling constants for 9 (1JCRh = 33.9, 11.1 Hz) and for 10 (1JCRh = 35.7, 11.2 Hz), which suggests that the Rh2 center has a closer contact to the carbene carbon than Rh1 (Scheme 2). Results of a single crystal X-ray analysis of 10 are consistent with the proposed structure, although the quality of data for 10 is rather poor (Fig. S6†). The observed 1JCRh coupling constants of 33.9 and 35.7 Hz are similar to the earlier reported values for dinuclear rhodium(I) complexes with bridging carbenes which likewise show 1JCRh in the range of 30–32 Hz.6 The assignment of cis-semihydrogenation in 10 is supported by two 1H NMR signals at δ1H = 2.58 and 5.07 ppm coupled to each other with 3JHH = 11.3 Hz.
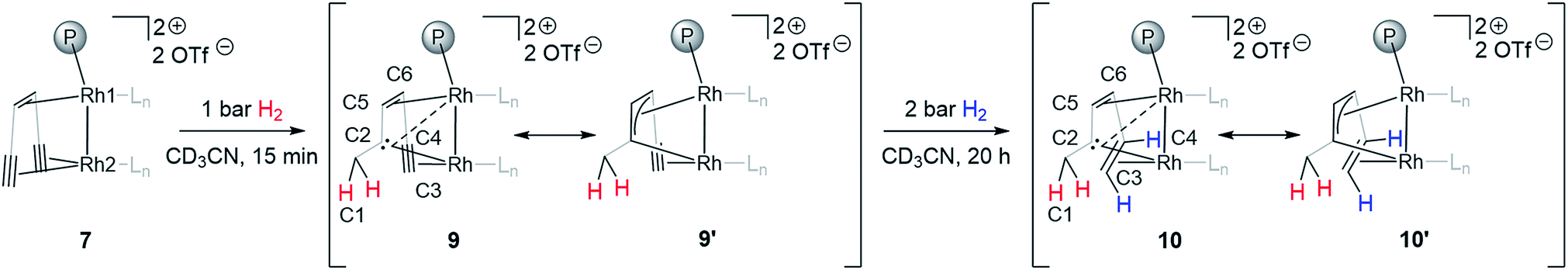 | ||
| Scheme 2 Stepwise semihydrogenation of 7 in CD3CN at room temperature followed by in situ NMR spectroscopy. Ln is CD3CN. The representation of ligand 5 (Scheme 1) was simplified for clarity. | ||
The two olefinic carbons of the central trop double bond in 6–8 (C5 and C6 in Fig. 1) are strongly shielded, most likely due to the anisotropic effects of the neighboring alkyne group (the chemical shifts range from δ13C = 37.0–48.7 ppm). Upon hydrogenation, these carbons show a remarkable difference in the chemical shift (9: δ13C = 43.3 and 98.8 ppm, 10: δ13C = 62.9 and 100.0 ppm for C6 and C5 respectively, see Scheme 2). The chemical shift change in C6 is consistent with hydrogenation of the alkyne to alkene, lowering its anisotropic effect. However, the strong deshielding of C5 in both complexes indicates an overlap between the C2 carbene p orbital and the trop double bond, leading to an allyl-like bonding around Rh1.47 This inference is further confirmed by the JCRh coupling constants, which are much smaller for the C5 carbon than for the C6 carbon (Table 2, numbering in the trop ligand is according to Scheme 2). A similar bonding motif was reported with a dinickel(I) core in the solid state.48 To summarize, complexes 9 and 10 contain a carbene carbon in conjugation with the central double bond of the trop ligand and this allyl-type ensemble is bridging to the Rh(I)–Rh(I) fragment. The ligand sphere of Rh2 in 9 and 10 in solution is likely stabilized by CD3CN.
| Nucleus | Position | 7 | 9 | 10 | |||
|---|---|---|---|---|---|---|---|
| δ (ppm) | J (Hz) | δ (ppm) | J (Hz) | δ (ppm) | J (Hz) | ||
| a No proton showed a significant coupling to Rh2 in the 1H–103Rh HMQC spectrum. | |||||||
| 13C | C1 | 75.4 | J CRh = 11.9 | 35.5 | 36.7 | ||
| C2 | 97.0 | J CRh = 7.7 | 171.1 | J CRh = 33.9, 11.1 | 166.3 | J CRh = 35.7, 11.2 | |
| C3 | 75.4 | J CRh = 11.9 | 76.9 | J CRh = 11.6 | 57.3 | J CRh = 15.6 | |
| C4 | 97.0 | J CRh = 7.7 | 101.7 | J CRh = 8.1 | 81.5 | J CRh = 12.4, 2.7 | |
| C5 | 38.1 | J CRh = 14.9 | 98.8 | J CRh = 7.6 | 100.0 | J CRh = 7.1 | |
| C6 | 38.1 | J CRh = 14.9 | 43.3 | J CRh = 11.0 | 62.9 | J CRh = 10.8 | |
| 1H | C1H2 | 2.38, 3.18 | 2 J HH = 12.1 | 1.99, 3.03 | 2 J HH = 13.1 | ||
| 31P | 104.6 | 1 J PRh1 = 185.6 | 59.7 | 1 J PRh1 = 127.2 | 67.0 | 1 J PRh1 = 136.0 | |
| 2 J PRh2 = 4.6 | 2 J PRh2 = 18.4 | 2 J PRh2 = 7.1 | |||||
| 103Rh | P–Rh1–Rh2 | −6852 | 1 J Rh1P = 186 | −7184 | 1 J Rh1P = 126 | −7163 | 1 J Rh1P = 136 |
| P–Rh1–Rh2 | —a | −6733 | −7066 | ||||
Furthermore, a characteristic shielding is observed in the 31P NMR spectra when comparing the trop phosphorus signal at δ31P = 104.6 ppm in the monomeric triflate complex 7 with the respective signals at δ31P = 59.7 and 67.0 ppm in the carbene complex 9 and the carbene alkene complex 10. This change is accompanied by a shift to a smaller 1JRh1P coupling constant in 9 and 10 (1JRh1P = 127.2 and 136.0 Hz) as compared to that in 7 (1JRh1P = 185.6 Hz), indicating a higher trans influence of the Rh2 center in 9 and 10 compared to 7. This suggests weakening of the P–Rh bond that is offset by strengthening of the Rh–Rh bond, attributed to the interaction with the bridging carbene ligand.49 The latter could also be viewed as a distorted dirhodacyclopropane.50 Analysis of the 103Rh NMR data reveals that Rh1 is significantly shifted upfield when comparing 7 with 9 and 10 (δ103Rh = −6852, −7184, and −7163 ppm for 7, 9 and 10, respectively) further supporting a more electron rich Rh core. Comparing 9 and 10 using the 103Rh NMR shift of Rh2 suggests that the Rh2 site in 10 is more electron rich than in 9 (δ103Rh = −6733, −7066 ppm for 9 and 10, respectively),51 which is consistent with a change in the ligand sphere from an alkyne to a weaker π accepting alkene. These results indicate that the bimetallic core becomes more electron rich with each hydrogenation step.
We have also performed the semihydrogenation of the ligand in a stepwise manner, where the carbene complex 9 was formed first under H2, followed by deuteration to the carbene alkene complex 10 under a D2 atmosphere. In this case, deuterium is only incorporated at the double bond (blue hydrogens atoms in Scheme 2) indicating irreversible ligand hydrogenation. This observation is consistent with the results of an experiment where 10 was formed in situ under an atmosphere of H2 and subsequently placed under an atmosphere of D2. In this case no deuterium incorporation is observed within 14 h, indicating that both hydrogenation steps are irreversible (Fig. S7 and S8†).
To gain further insight on how the H2 molecule is activated at the Rh2 fragment, parahydrogen (p-H2)52,53 was used instead of H2 for the hydrogenation of monomeric triflate complex 7, resulting in no detectable hyperpolarized Rh hydride intermediates. Interestingly, the only PHIP hyperpolarized signals observed in the 1H NMR spectra are assigned to the methylene CH2 protons of carbene complex 9 (Fig. S10 and S11†), which establishes a pairwise hydrogen addition to the triple bond of the trop ligand when forming the bridging carbene complex 9.
Next, we added 1–2 bar of H2 to the solution of dppm adduct 8 in CD3CN in a J. Young NMR tube and observed no formation of the bridging carbene-like moiety. Instead, a mixture of 8 and a Rh dihydride complex 11 (Fig. 2a) is formed that is stable for several days at room temperature. Two characteristic hydride signals are observed at approximately δ1H = −16.2 and −20.5 ppm. Dihydride 11 was further characterized by low temperature NMR experiments as well as using the PHIP technique. The removal of the H2 (or the D2) atmosphere from a J. Young NMR tube containing a mixture of the dppm adduct 8 and the dihydride 11 cleanly reforms 8, indicating a fully reversible hydrogen activation (Fig. S12 and S13†). This is further supported by EXSY spectroscopy, variable temperature NMR and a partially negative line-shape (PNL) of the orthohydrogen peak in PHIP NMR (Fig. S14–S17†).38,54,55 Since PNL does also occur in PHIP experiments with 7, the same mode of hydrogen activation is likely occurring with both species, 7 and 8. Upon formation of cis-dihydride 11, the Rh1 NMR resonance δ103Rh1 = −7758 ppm is significantly shifted to lower frequencies. This low-frequency shift exceeds even the ones observed for 9 (δ103Rh1 = −7184 ppm) and 10 (δ103Rh1 = −7163 ppm). The Rh2 nucleus (δ103Rh = −7630 ppm) shows likewise a strong shift to lower frequencies relative to the 103Rh2 nuclei in 9 and 10 (δ103Rh2 = −6733 and −7066 ppm, respectively), which is in agreement with the influence of strongly σ donating hydrides on both metal centers (Fig. 2b). 1H{31P} J-resolved 2D NMR spectroscopy reveals the JHH and JHRh coupling constants (Fig. 2c). The two hydrides remain coupled in complex 11, revealed by the 2JHH = 15.4 Hz splitting in the indirect dimension. The hydride at δ1H = −16.2 ppm shows an additional 4JHH = 5.8 Hz coupling to another proton, assigned by COSY to one of the methylene protons of the dppm ligand (red in Fig. 2a). The NMR characterization of 11 was also completed by 1H, 13C, 19F, 29Si and 31P spectra (Fig. S21–S27†). Altogether, this data confirmed that dppm adduct 8 activates hydrogen reversibly and is in equilibrium with the dihydride species 11. No semihydrogenation of the triple bonds is observed in this case.
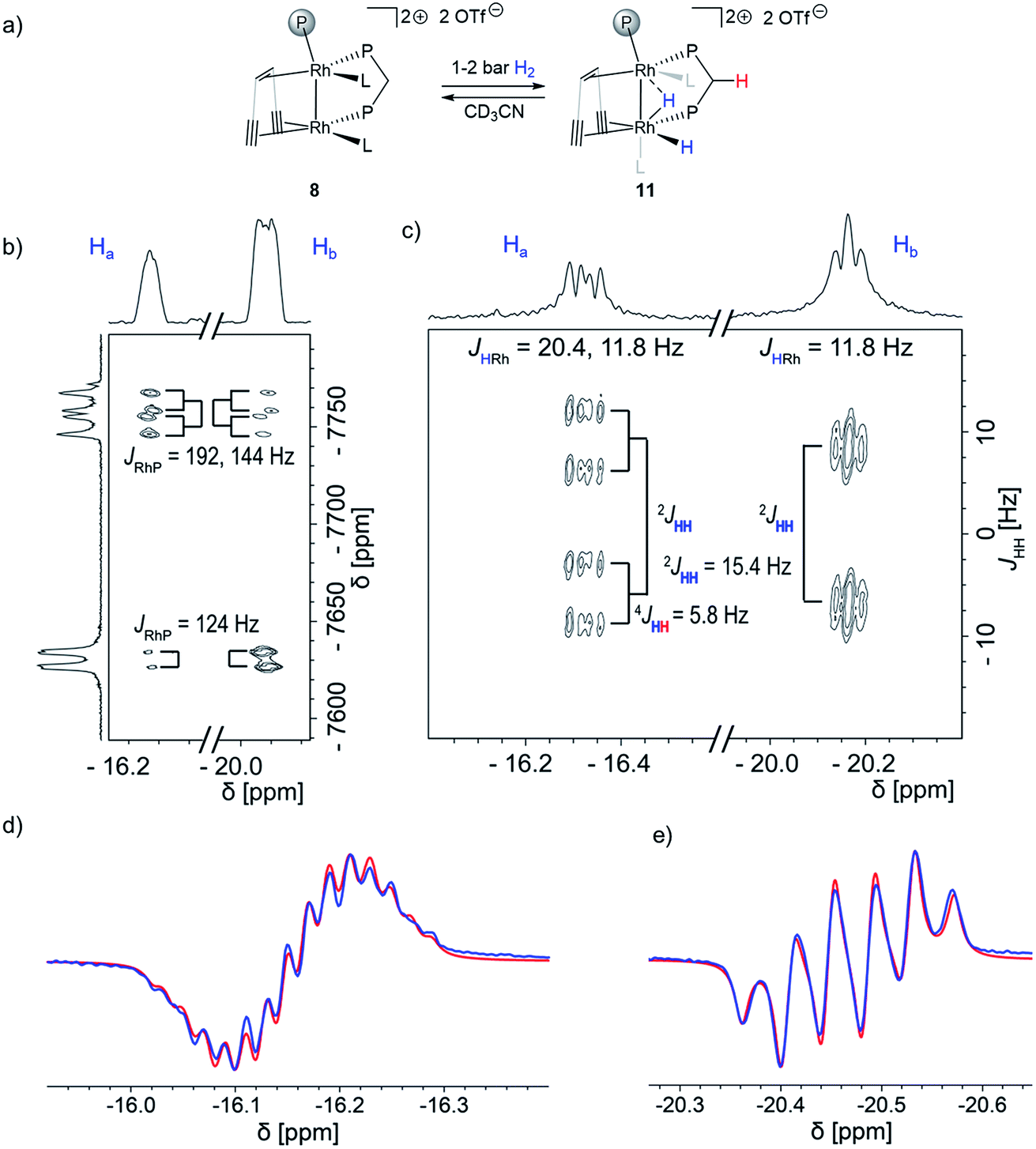 | ||
| Fig. 2 Proposed activation of hydrogen by complex 8 (L is one acetonitrile ligand) and relevant spectroscopic data: (a) reaction of 8 with hydrogen, as proposed on the basis of NMR experiments, (b) J-resolved 1H{31P} spectrum, (c) 1H–103Rh HMQC spectrum, (d) PHIP 1H spectrum (blue) with simulation (red), of hydride Ha, (e) PHIP 1H{31P} spectrum (blue) with simulation (red) of hydride Hb. The representation of ligand 5 was simplified in structures 8, 11 and 12 for clarity. | ||
Recording the J-resolved spectrum with 31P decoupling allows extracting the JHRh coupling constants (Fig. 2c). While the hydride at δ1H = −16.3 ppm appears as a doublet of doublets (JHRh = 20.4, 11.8 Hz), the hydride at δ1H = −20.2 ppm appears as a pseudo-triplet (JHRh = 11.8 Hz). The observed 2JHH coupling of 15.4 Hz is larger than typically found in traditional cis hydrides formed via oxidative addition (2JHH = 7.2–9.5 Hz),56 suggesting another geometry since higher coupling constants indicate larger angles between the substituents. Overall, these results are consistent with a bridging geometry for the dihydride 11 (Fig. 2a). Assignment of Hacis to the intermetallic bond is based on the observation of a remarkable long-range 4JHH coupling of Ha to a CH2 proton (4JHH = 5.8 Hz, highlighted red in Fig. 2a and b). Unfortunately, recording a J-resolved spectrum with 103Rh decoupling was not successful due to the large difference in the chemical shift, and it was not possible to eliminate the JHRh couplings from both metal centers at once. Therefore, the JHP coupling constant could not be accessed with this approach. However, the 1H–31P coupling constants could be extracted by fitting the observed PHIP signals (Fig. 2d and e). The PHIP spectrum in Fig. 2e was recorded with a 31P decoupling, which selectively eliminates only 31P couplings arising from the dppm moiety. This allows distinguishing coupling constants between the hydrides and dppm/trop phosphorous centers. In addition, PHIP experiments allowed to determine the sign of the 2JHH coupling constant between the two hydrides which is negative 2JHH = −15.4 Hz.
The proton Ha shows an 2JHP coupling to the dppm 31P center of 2JHP = 13 and 6 Hz, indicating a cis2JHP coupling.56 The second hydrogen atom Hb is likely close to the cis position of the trop phosphine center, as suggested by the couplings of 2JHP = 24 and 18 Hz. The coupling to the second dppm phosphorous center is substantially smaller (JHP = 4 Hz). However, the large JHH coupling constant as well as the similar coupling constants to both Rh centers (resulting in the pseudo-triplet in Fig. 2b) are consistent with an interaction with the second Rh center. This assignment allows reporting a cis coupling constant of 2JHRh = 11.8 Hz. This indicates that the close Rh–Rh contact is preserved after the addition of dihydrogen. Having assigned Ha as the terminal hydride, while Hb interacts with the two Rh centers unsymmetrically, the Rh–Rh core can be described either as a Rh(II)–Rh(II) center or a Rh(I) → Rh(III) polarized bond, due to the bridging nature of Hb. Both descriptions imply a close Rh–Rh contact. We tested if this hydride reactivity could also be observed with other ligands and subjected tricyclohexylphosphine, diphenylphosphine oxide and triazabicyclodecene ligands to conditions of the in situ PHIP experiments demonstrating that only the dppm ligand leads to the hydride species. However, all PHIP experiments with the added ligand except triazabicyclodecene showed the hyperpolarized signals of methylene CH2 protons with very similar chemical shifts and coupling constants as observed for 9. In addition, a partially negative lineshape signal for the orthohydrogen was observed for all ligands, suggesting that hydrogen activation is reversible (Fig. S31–S33†).
DFT calculations (ωB97X-D/def2-SVP) support the proposed reversible hydrogen activation of the monomeric triflate complex 8 leading to the dihydride 11 (Scheme 3). From the computed 8-SM intermediate, two H2 adducts can be formed: with the H2 molecule coordinated cis (8-H2) or trans (8a-H2) to Rh1 (Scheme 3). Both complexes can undergo oxidative addition steps via activated complexes which are located at energetically low lying transition states ([TS1]‡: ΔG = 11.4 kcal mol−1; [TS2]‡: ΔG = 7.1 kcal mol−1), leading to dihydrides 8a-HHox and 11-HHox, respectively. With the exception of 11-MeCN, all intermediates are relatively close in energy and are expected to be accessible from 8-SM at room temperature. Since 11-HHox has an open coordination site trans to Rh1, binding of one acetonitrile ligand occurs giving a more stable species 11-MeCN (ΔG = −19.8 kcal mol−1) with a distorted octahedral coordination environment around Rh2, as expected for Rh(III) d6 complexes. The energy of the coordinatively saturated 11-MeCN is only slightly higher than that of 8-MeCN (ΔG = −12.3 and −16.8 kcal mol−1, respectively). These two complexes are therefore expected to slowly interconvert and be observable in solution, which is indeed supported by NMR spectroscopy. We note that ΔΔG values from DFT calculations and variable temperature NMR measurements obtained from a Van't Hoff plot (Fig. S16†) are in reasonable agreement (ΔΔG = 4.5 and 1.1 kcal mol−1, respectively). It is of note that since the geometrical reorganization in going from [TS2]‡ to 11-HHox is only minimal, the barrier for this step is very low and we attribute the small positive energy difference between 11-HHox and [TS2]‡ to the numerical inaccuracy of the applied DFT method. In addition, we note that the bimetallic hydrogen activation pathway involving a four-membered M2H2 transition state was not considered because this reaction is symmetry forbidden.57
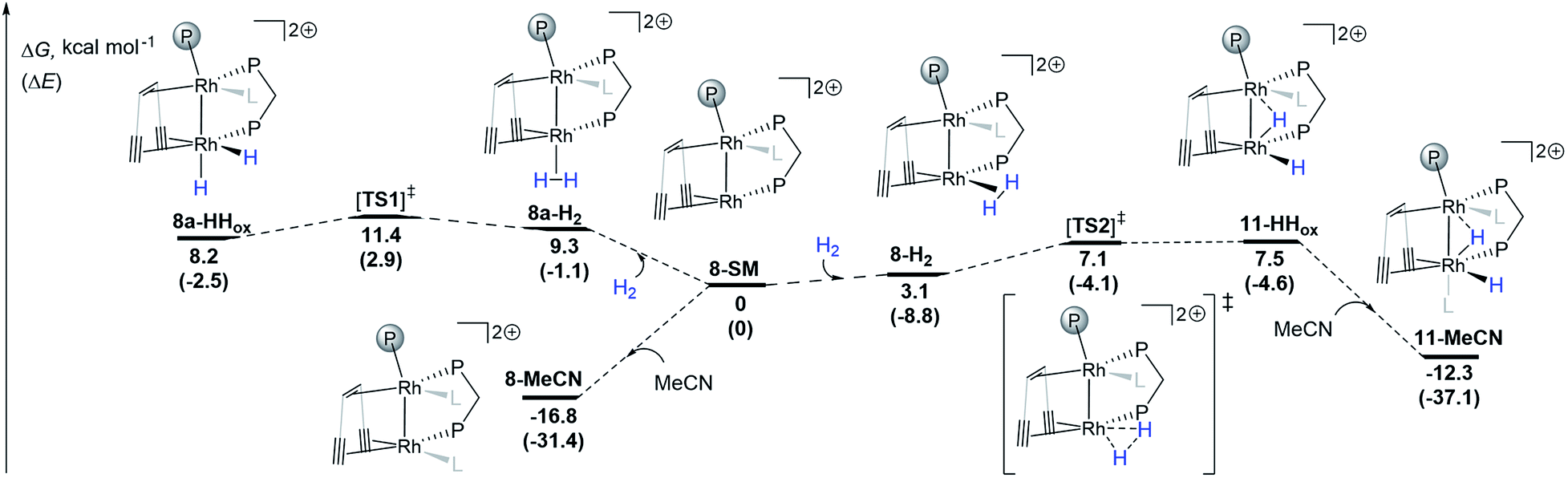 | ||
| Scheme 3 DFT calculations (Gaussian09, ωB97X-D/def2-SVP) for the hydrogen activation pathways from 8-SM to 8a-HHox and 11-HHox. L is one acetonitrile ligand. The transition states were confirmed to connect the two respective energy minima by IRC calculations (see ESI†). | ||
Interestingly, a bridging hydride resembling that in 11 has been proposed and calculated in both rhodium-based homogeneous bimetallic28 and heterogeneous systems.26 This observation further underlines the utility of low valent homobimetallic complexes with ligands containing alkene and alkyne binding sites in understanding elementary steps in heterogeneous catalysts which are deposited on carbon support materials.
Bimetallic dirhodium complexes 7–9 and the monometallic catalyst 12 were then tested as catalysts for the semihydrogenation of phenylacetylene (5% catalyst loading, 25 °C, Fig. 3, for details see ESI†). No activation period was observed (Fig. S41†). The dppm adduct 8 showed highest performance with a selectivity for styrene of 96% at 78% conversion and kobs around 3–5 times higher than those for monomeric triflate complex 7 and carbene complex 9 (Table 3, entries 1–3). The similar rates kobs observed with 7 and 9 indicate that the in situ hydrogenation of 7 to 9 takes place under the catalytic conditions and leads to the same active species, consistent with the presence of the hyperpolarized CH2 signals characteristic for 9 in these catalytic reactions according to in situ PHIP NMR (Fig. S42†). Importantly, PHIP results also show that the dihydride species 11 form from 8 under catalytic conditions (Fig. S43†).
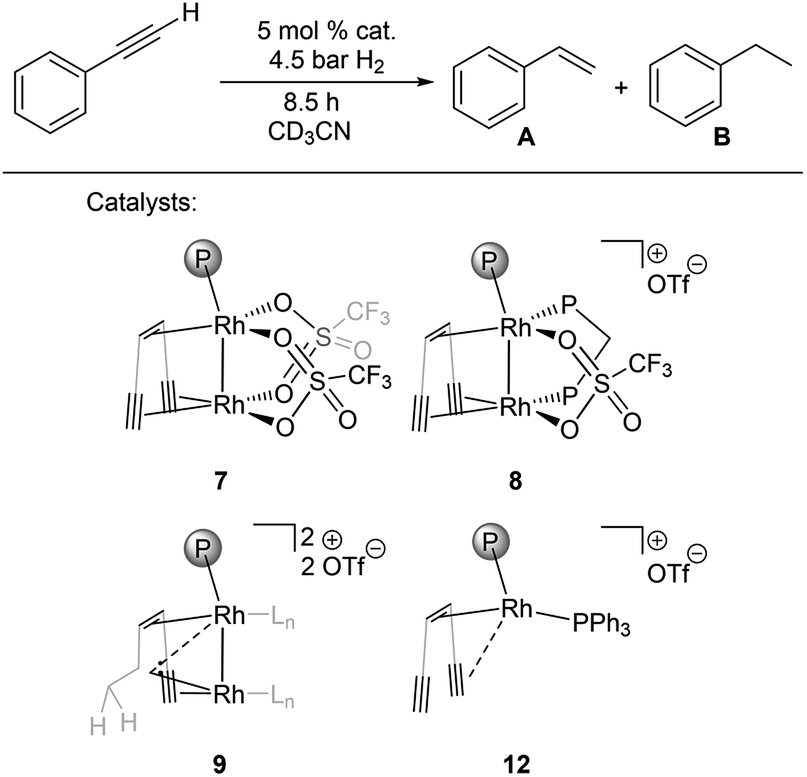 | ||
| Fig. 3 Catalytic semihydrogenation of phenylacetylene by bimetallic catalysts 7–9 and the benchmark monometallic catalyst 12 (Ln is CD3CN, {(TMS)CC}2tropPPh2 ligand is simplified for clarity). | ||
| Entry | Catalyst | Conv. (%) | Sel. A (%) | Sel. B (%) | k obs (10−2 h−1) |
|---|---|---|---|---|---|
| 1 | 7 | 39 | 90 | 10 | 5.9 |
| 2 | 8 | 79 | 97 | 3 | 23 |
| 3 | 9 | 39 | 97 | 3 | 6.7 |
| 4 | 12 | 16 | >99 | — | 0.95 |
Remarkably, the monometallic catalyst 12 converts phenylacetylene significantly slower than all tested bimetallic complexes, with a rate of kobs = 9.5 × 10−3 h−1 (Table 3, entry 4). We speculate that the second metal serves as an electron reservoir which helps to avoid the formation of an inert trop-Rh(III) d6 species58 formed by oxidative addition of dihydrogen. PHIP experiment performed under catalytic conditions with the ddpm adduct 8 indicates a pairwise hydrogen transfer to phenylacetylene forming styrene, i.e. hydrogen is activated by 8 to give dihydride 11, followed by the transfer of the two hydrides to the same substrate molecule (Fig. S43†).
Conclusions
The {(TMS)CC}2tropPPh2 molecule with one phosphane, one alkene, and two alkyne donor sites was designed to allow the synthesis of a new family of low-valent homobimetallic Rh(I)–Rh(I) complexes. The reactivity of these complexes provides insight into possible metal–metal cooperation in hydrogen activation reactions. The unique ligand environment of the Rh2 site in {(TMS)CC}2tropPPh2 complexes makes this ligand an interesting model for the interface between metal clusters and particles and a carbon support material as found in Rh/C heterogeneous catalysts. Two distinct modes of intramolecular hydrogenation of the ligand were identified leading to a bridging carbene or an olefin ligand. When stabilized by a dppm ligand, an intermediate hydride species could be characterized by advanced NMR methods which show that this species has a structure with a bridging and a terminal hydride ligand.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We gratefully acknowledge financial support of Swiss National Science Foundation. I. V. K. acknowledges the grant from the Russian Science Foundation (19-13-00172) for the support of hydrogenation experiments with parahydrogen. K. V. K thanks SB RAS integrated research program (# 0333-2018-0006/II.1.13) for parahydrogen activation studies and Ministry of Science and Higher Education of the RF (AAAA-A16-116121510087-5) for access to NMR equipment. A. F. thanks the Holcim Stiftung for the Habilitation fellowship.Notes and references
- H. Vahrenkamp, Angew. Chem., Int. Ed., 1978, 17, 379–392 CrossRef.
- M. P. Doyle, Chem. Rev., 1986, 86, 919–939 CrossRef CAS.
- H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC.
- H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS.
- J. Adams and D. M. Spero, Tetrahedron, 1991, 47, 1765–1808 CrossRef CAS.
- J. F. Berry and C. M. Thomas, Dalton Trans., 2017, 46, 5472–5473 RSC.
- Y.-Y. Zhou and C. Uyeda, Angew. Chem., Int. Ed., 2016, 55, 3171–3175 CrossRef CAS.
- J. Ye, R. C. Cammarota, J. Xie, M. V. Vollmer, D. G. Truhlar, C. J. Cramer, C. C. Lu and L. Gagliardi, ACS Catal., 2018, 8, 4955–4968 CrossRef CAS.
- R. B. Siedschlag, V. Bernales, K. D. Vogiatzis, N. Planas, L. J. Clouston, E. Bill, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2015, 137, 4638–4641 CrossRef CAS.
- M. K. Karunananda and N. P. Mankad, ACS Catal., 2017, 7, 6110–6119 CrossRef CAS.
- M. E. Broussard, B. Juma, S. G. Train, W.-J. Peng, S. A. Laneman and G. G. Stanley, Science, 1993, 260, 1784–1788 CrossRef CAS.
- D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705–1718 Search PubMed.
- M.-E. Moret, in Higher Oxidation State Organopalladium and Platinum Chemistry, ed. A. J. Canty, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 157–184 Search PubMed.
- R. J. Oeschger, D. H. Ringger and P. Chen, Organometallics, 2015, 34, 3888–3892 CrossRef CAS.
- D. Serra, M.-E. Moret and P. Chen, J. Am. Chem. Soc., 2011, 133, 8914–8926 CrossRef CAS.
- R. J. Oeschger and P. Chen, J. Am. Chem. Soc., 2017, 139, 1069–1072 CrossRef CAS.
- R. J. Oeschger and P. Chen, Organometallics, 2017, 36, 1465–1468 CrossRef CAS.
- J. P. Krogman and C. M. Thomas, Chem. Commun., 2014, 50, 5115–5127 RSC.
- R. C. Cammarota, L. J. Clouston and C. C. Lu, Coord. Chem. Rev., 2017, 334, 100–111 CrossRef CAS.
- D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840–850 CrossRef CAS.
- P. M. Maitlis, H. C. Long, R. Quyoum, M. L. Turner and Z.-Q. Wang, Chem. Commun., 1996, 1–8 RSC.
- P. M. Maitlis, J. Organomet. Chem., 2004, 689, 4366–4374 CrossRef CAS.
- P. M. Maitlis and V. Zanotti, Catal. Lett., 2008, 122, 80–83 CrossRef CAS.
- C. H. Bartholomew and R. J. Farrauto, Hydrogenation and Dehydrogenation of Organic Compounds, John Wiley and Sons, Hoboken, NJ, 2nd edn, 2005 Search PubMed.
- P. Serna and B. C. Gates, Acc. Chem. Res., 2014, 47, 2612–2620 CrossRef CAS.
- A. Dutta and P. Mondal, J. Phys. Chem. C, 2018, 122, 16925–16939 CrossRef CAS.
- A. S. Weller and J. S. McIndoe, Eur. J. Inorg. Chem., 2007, 4411–4423 CrossRef CAS.
- F. W. Patureau, S. de Boer, M. Kuil, J. Meeuwissen, P.-A. R. Breuil, M. A. Siegler, A. L. Spek, A. J. Sandee, B. de Bruin and J. N. H. Reek, J. Am. Chem. Soc., 2009, 131, 6683–6685 CrossRef CAS.
- A. J. Esswein, A. S. Veige and D. G. Nocera, J. Am. Chem. Soc., 2005, 127, 16641–16651 CrossRef CAS.
- R. C. Matthews, D. K. Howell, W.-J. Peng, S. G. Train, W. D. Treleaven and G. G. Stanley, Angew. Chem., Int. Ed. Engl., 1996, 35, 2253–2256 CrossRef CAS.
- J. F. Berry and C. C. Lu, Inorg. Chem., 2017, 56, 7577–7581 CrossRef CAS.
- I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef CAS.
- J. Thomaier, S. Boulmaâz, H. Schönberg, H. Rüegger, A. Currao, H. Grützmacher, H. Hillebrecht and H. Pritzkow, New J. Chem., 1998, 22, 947–958 RSC.
- L. Bettucci, C. Bianchini, W. Oberhauser, M. Vogt and H. Grützmacher, Dalton Trans., 2010, 39, 6509–6517 RSC.
- H. Schönberg, S. Boulmaâz, M. Wörle, L. Liesum, A. Schweiger and H. Grützmacher, Angew. Chem., Int. Ed., 1998, 37, 1423–1426 CrossRef.
- F. Breher, H. Rüegger, M. Mlakar, M. Rudolph, S. Deblon, H. Schönberg, S. Boulmaâz, J. Thomaier and H. Grützmacher, Chem.–Eur. J., 2004, 10, 641–653 CrossRef CAS PubMed.
- S. Boulmaâz, S. Loss, H. Schönberg, S. Deblon, M. Wörle, R. Nesper, H. Grützmacher and M. Mlakar, Chem. Commun., 1998, 2623–2624 RSC.
- A. S. Kiryutin, G. Sauer, A. V. Yurkovskaya, H.-H. Limbach, K. L. Ivanov and G. Buntkowsky, J. Phys. Chem. C, 2017, 121, 9879–9888 CrossRef CAS.
- R. A. Green, R. W. Adams, S. B. Duckett, R. E. Mewis, D. C. Williamson and G. G. R. Green, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 67, 1–48 CrossRef CAS.
- T. C. Eisenschmid, R. U. Kirss, P. P. Deutsch, S. I. Hommeltoft, R. Eisenberg, J. Bargon, R. G. Lawler and A. L. Balch, J. Am. Chem. Soc., 1987, 109, 8089–8091 CrossRef CAS.
- C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541–5542 CrossRef CAS.
- S. B. Duckett and N. J. Wood, Coord. Chem. Rev., 2008, 252, 2278–2291 CrossRef CAS.
- B. Le Bourdonnec and R. E. Dolle, US Pat., 20080275131, 2008.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- P. K. Sajith and C. H. Suresh, Dalton Trans., 2010, 39, 815–822 RSC.
- G. Banditelli and A. L. Bandini, Organometallics, 2006, 25, 1578–1582 CrossRef CAS.
- W. A. Herrmann, Angew. Chem., Int. Ed., 1978, 17, 800–812 CrossRef.
- C. A. Laskowski and G. L. Hillhouse, Chem. Sci., 2011, 2, 321–325 RSC.
- A. Pidcock, R. E. Richards and L. M. Venanzi, J. Chem. Soc. A, 1966, 1707–1710 RSC.
- W. A. Herrmann, J. Plank, D. Riedel, M. L. Ziegler, K. Weidenhammer, E. Guggolz and B. Balbach, J. Am. Chem. Soc., 1981, 103, 63–75 CrossRef CAS.
- P. B. Graham, M. D. Rausch, K. Taeschler and W. Von Philipsborn, Organometallics, 1991, 10, 3049–3052 CrossRef CAS.
- O. G. Salnikov, H.-J. Liu, A. Fedorov, D. B. Burueva, K. V. Kovtunov, C. Copéret and I. V. Koptyug, Chem. Sci., 2017, 8, 2426–2430 RSC.
- K. V. Kovtunov, D. A. Barskiy, R. V. Shchepin, A. M. Coffey, K. W. Waddell, I. V. Koptyug and E. Y. Chekmenev, Anal. Chem., 2014, 86, 6192–6196 CrossRef CAS.
- V. V. Zhivonitko, V.-V. Telkki, K. Chernichenko, T. Repo, M. Leskelä, V. Sumerin and I. V. Koptyug, J. Am. Chem. Soc., 2014, 136, 598–601 CrossRef CAS.
- D. A. Barskiy, K. V. Kovtunov, I. V. Koptyug, P. He, K. A. Groome, Q. A. Best, F. Shi, B. M. Goodson, R. V. Shchepin, A. M. Coffey, K. W. Waddell and E. Y. Chekmenev, J. Am. Chem. Soc., 2014, 136, 3322–3325 CrossRef CAS.
- A. Koch and J. Bargon, Inorg. Chem., 2001, 40, 533–539 CrossRef CAS.
- G. Trinquier and R. Hoffmann, Organometallics, 1984, 3, 370–380 CrossRef CAS.
- F. F. Puschmann, H. Grützmacher and B. d. Bruin, J. Am. Chem. Soc., 2010, 132, 73–75 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental spectra and modelling details. CCDC 1919533, 1919534, 1919542, 1919543 and 1919546. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02683e |
| This journal is © The Royal Society of Chemistry 2019 |