
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalized DNA nanostructures as scaffolds for guided mineralization†
Francesca
Kim
a,
Tong
Chen
a,
Trevor
Burgess
a,
Prakash
Rasie
a,
Tim Luca
Selinger
a,
Andrea
Greschner
b,
Georgios
Rizis
a and
Karina
Carneiro
 *a
*a
aFaculty of Dentistry, University of Toronto, Toronto, ON M5G 1G6, Canada. E-mail: karina.carneiro@utoronto.ca
bInstitut National de la Recherche Scientifique (INRS), EMT Research Center, Varennes, QC J3X 1S2, Canada
First published on 27th September 2019
Abstract
The field of DNA nanotechnology uses synthetic DNA strands as building blocks for designing complex shapes in one-, two- and three-dimensions. Here, we investigate whether DNA nanostructures are feasible platforms for the precise organization of polyaspartic acid (pAsp), a known mineral carrier, with a goal towards biomimetic mineralization for enamel regeneration. We describe the preparation of DNA–pAsp conjugates and their subsequent assembly into ordered nanostructures. Covalent attachment of pAsp to DNA was noted to hinder DNA nanostructure formation past a certain threshold (50% pAsp) when tested on a previously published DNA system. However, a simplified double stranded DNA system (3sDH system) was more robust and efficient in its pAsp incorporation. In addition, the 3sDH system was successful in organizing mineral inducing groups in one dimension at repeating intervals of 28.7 ± 4.0 nm, as determined by atomic force microscopy. Our results demonstrate that DNA nanostructures can be functionalized with pAsp and act as a platform to investigate guided mineralization.
Introduction
DNA nanotechnology is a field where synthetic DNA strands are programmed to self-assemble into designed architectures.1 A plethora of self-assembled materials in one-, two-, and three-dimensions have been constructed, and are increasingly being exploited as highly organized scaffolds that can direct specific molecular arrangements.2 DNA is addressable at the nanometer-level due to its base-pairing properties, thus DNA assemblies have been successfully used as scaffolds for the precise positioning of a variety of groups, ranging from proteins3 and enzymes,4 to oligomers,5 polymers6 and preformed nanoparticles. The potential of DNA to modulate the morphology of inorganic materials7 and the ability of DNA nanostructures to serve as templates8 and moulds9 have been previously demonstrated. Moreover, recent findings elegantly demonstrate the replication of complex geometric information by DNA hybrid materials.9,10 In particular, extended DNA assemblies such as two-dimensional networks and nanotubes are ideal for guiding biomineralization on length scales ranging from nanometers to millimeters, as is necessary for the regeneration of mineralized tissues such as tooth and bone.1a,11,12The presence of calcium, phosphate and/or carbonate ions is essential to the process of biomimetic mineralization; thus, proper assembly and stability in calcium-containing solutions that mimic in vivo conditions are key requirements during in vitro scaffold validation. Conditions used for DNA self-assembly into nanostructures typically comprise magnesium-containing buffers that shield the negative charges of the phosphate backbone. Crystallographic studies have shown that the nature of the cation affects the local helical structures of DNA duplexes,13 which in turn may affect the overall assembly of the DNA nanostructure. Few previous studies have investigated how the nature and presence of the cation, and the amount of curvature within the DNA nanostructure affect the 3D conformation of the final construct.14 A more detailed investigation of the behaviour of DNA nanostructures in calcium-containing solutions is necessary in order to design hybrid organic–inorganic nanostructures as suitable materials for guiding mineralization.
In addition to the scaffolding properties of self-assembled DNA nanostructures, under the appropriate conditions double-stranded DNA (dsDNA) has shown a role in guiding mineralization through the polymer-induced liquid precursor (PILP) method.15 To initiate the PILP process, it is hypothesized that anionic polymers with a high linear charge density sequester both calcium counter ions and excess phosphate ions in solution, resulting in the formation of ion-rich fluid domains via liquid–liquid phase separation. The fluid domains then coalesce and aggregate to form amorphous calcium phosphate that adsorbs onto heterogeneous surfaces and acts as a transient precursor to crystallization. It is proposed that a biological equivalent of this process likely plays an important role in biomineralization, with non-collagenous proteins acting as mineral promoters.9In vitro studies have shown that other negatively charged polymers such as polyaspartic acid (pAsp) also participate in the PILP process, and can achieve guided mineralization when deposited on a scaffold such as intrafibrillar collagen in dentin.16 This system has also been used to successfully recreate the nanostructure of bone.17 These studies have focused on the passive incorporation of unbound pAsp to scaffolds.18 Here we describe the synthesis of DNA–pAsp conjugates and their incorporation into a DNA nanostructure; we investigate whether bound pAsp can mediate calcium phosphate (CaP) mineral formation for their potential use as scaffolds for guided mineralization. In the intended design, the DNA component of our model system determines the scaffold dimensions and morphology, which are ultimately transferred to the mineral species. The pAsp moiety, in turn, mediates the PILP process, which promotes mineral formation. In comparing DNA assembly platforms, our study has revealed early design rules for DNA scaffold robustness under PILP conditions. Our results indicate that the precise organization of pAsp along DNA nanostructures, through covalent or supramolecular interactions, alters the course of the PILP process, and influences both the organization of amorphous calcium phosphate, and the morphology of the growing mineral across multiple length scales.
Results and discussion
The one strand DNA nanotube (1sNT)19 was the initial DNA scaffold of choice due to its easy and reliable self-assembly in one-dimension, and its relative design simplicity. This previously published DNA structure consists of a single strand of DNA that is self-complementary and designed to fold into a DNA sheet, which then rolls into a nanotube (Fig. 1A). To test the feasibility of this scaffold system under conditions relevant to biomineralization, the assembly fidelity of the 1sNT nanostructure in calcium-containing buffers (thermal anneal, 90 → 4 °C) was assessed by gel electrophoresis, circular dichroism and atomic force microscopy (AFM) (Fig. 1B, S1 and S2†). In a previous study, DNA which normally assembled into a two-dimensional grid in the presence of Mg2+ adopted a nanotube conformation when Ca2+ was used.14b The origins of this phenomenon were not discussed, however it can be presumed that the difference in ionic radius – Ca2+ is ∼27% larger than Mg2+ – may result in different inter-duplex spacing in tightly packed DNA structures. As observed in the DNA tile-based system employed by Yan et al.,20 small local changes in spacing can induce curvature over a large length scale and promote the formation of nanotubes. The successful assembly of a DNA origami nanostructure has also been demonstrated in magnesium-free buffers.21 In the case of the 1sNT DNA used herein, where the native structure is already curved, the use of Ca2+ in place of Mg2+ did not change the resulting morphology based on width and height measurements as determined by AFM (Table 1). When these sample solutions were exposed to phosphate ions, minerals were observed, but they were disorganized and randomly deposited on the surface as detected by AFM (Fig. S5†). | ||
| Fig. 1 Self-assembly of the one strand nanotube (1sNT)19 in magnesium- and calcium-containing buffer solutions. (A) Scheme of the self-assembly of the 1sNT through a thermal anneal (90 to 4 °C); (B) AFM images of the 1sNT in buffers containing magnesium (left) and calcium (right) ions. | ||
| Sample | Width (nm) | Height (nm) |
|---|---|---|
| 1sNT in TAMg | 66.4 ± 8.2 | 4.8 ± 0.2 |
| 1sNT in TACa | 66.7 ± 7.4 | 4.0 ± 0.2 |
In an effort to achieve guided mineralization, we incorporated a known mineral inducer, polyaspartic acid (pAsp), into our system. pAsp was chosen due to its well-studied mineralization properties, and for its ability to induce guided mineralization in collagen matrices.22 pAsp was incorporated into 1sNT assemblies either through passive addition, or through covalent coupling to a DNA strand. For passive addition, 1sNT DNA and pAsp were assembled into nanotubes and nanospheres, respectively, as previously described (Fig. S4†).16a,19,22
Varying concentrations of pAsp (2.5, 5, 7.5 and 10% w/w), pre-assembled into nanospheres, were incubated with the 1sNT DNA nanotubes in the presence of Ca2+ to investigate any possible interactions between the DNA and the polymers. We hypothesized that pAsp nanospheres would interact with DNA nanotubes through electrostatic interactions, following a similar deposition mechanism as that observed in the PILP method.9 We expected to observe pAsp polymer aggregates adsorbing on the DNA nanotubes and acting as precursors to calcium phosphate crystallization. At lower concentrations of pAsp, AFM characterization displayed adsorbed nanospheres along the nanotube surface (Fig. S3†). These nanospheres were attributed to pAsp as they presented similar dimensions as the pAsp nanosphere controls (Fig. S4A†). As the concentration of pAsp increased, the deposition process appeared to be non-selective, as the nanospheres were randomly dispersed both on the 1sNT DNA nanotubes and the mica surface. This lack of selectivity behaviour was also characterized by liquid AFM, at 10% pAsp (Fig. S4D†). A limitation of passively adding pAsp spheres to DNA nanotubes is that it led to random distribution, suggesting that the growth of minerals nucleated within these pAsp accumulations would also be random as more pAsp is incorporated onto the template.
Towards improved control over the linear distribution of mineral growth sites on the DNA scaffold, we designed hybrid materials by covalently attaching pAsp to the DNA strand that folds into the 1sNT. Briefly, an amino-terminal modification yielding a thiol group on the pAsp molecule was performed by acylation with chloroacetic anhydride, followed by nucleophilic displacement with thioacetic acid. De-protection under alkaline conditions yielded a free terminal thiol (pAsp-SH), which was reacted with a DNA strand modified with a 5′-terminal maleimide. The reaction conditions followed a slight modification on a previously published thiol–maleimide conjugate addition reaction (Fig. 2A and S6†).23 The reaction yield was optimized from ∼10% to ∼97%, as calculated by ImageJ analysis of the crude reaction mixture in denaturing polyacrylamide gels (Fig. S7 and S8†). The conjugates were purified by denaturing gel excision followed by electroelution. The DNA–pAsp conjugate purity was ascertained by reduced electrophoretic mobility on a denaturing gel using silver staining methods (Fig. S9†). The use of the silver stain instead of our standard stain (GelRed®) was necessary in order to clearly visualize the purified the DNA–pAsp conjugates. We hypothesize that since GelRed® binds to DNA exclusively via intercalation,24 DNA–pAsp self-assembled into nanospheres over the purification process, which could sterically hinder the insertion of the stain between DNA base pairs. In addition, the DNA–pAsp conjugate had a slower elution time than the naked DNA by anion exchange chromatography (Fig. S10†). To our knowledge, the conjugation reaction between DNA and pAsp, which are mutually repulsive under most conditions due to the high anionic charge density of both macromolecular species, has not been previously described.
In order to test whether DNA–pAsp could properly be incorporated into the 1sNT system, samples with varying amounts of conjugate in addition to unmodified 1sNT DNA strands were subjected to a thermal annealing cycle (90 → 4 °C) in a calcium-containing buffer to promote DNA nanotube assembly as previously described for the unmodified nanotubes. Interestingly, the nanotube dimensions and morphology were affected by increasing the proportion of DNA–pAsp conjugate relative to unmodified DNA present in the system; increasing amounts of DNA–pAsp showed an inhibitory effect on nanotube formation, as judged by the presence of smaller, improperly assembled nanotubes observed during AFM analysis. Co-assembly of DNA–pAsp with 10 equivalents of unmodified DNA yielded nanotubes that were much shorter and kinked compared to the unmodified nanotubes (Fig. 2B). At an increased DNA–pAsp proportion of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, kinked nanotube bundles were observed alongside amorphous aggregates and misassembled structures; AFM images of the resulting structures are shown in Fig. 2C. DNA nanotubes did not properly assemble as the pAsp content was further increased, which suggests that 1:2 incorporation is close to a morphological boundary. This disrupted assembly behaviour may be due to steric constraints caused by the polymer during the assembly of the single stranded DNA into nanotubes. Alternatively, electrostatic complexation between polymer chains/DNA and calcium ions in solution may favour smaller assemblies over the desired linear scaffold. It is worth noting, however, that the system showed no clear selectivity for unmodified DNA during the structure formation process, as proper nanotubes could, in principle, have formed by assembly of DNA chains alone without incorporation of DNA–pAsp. These results suggest that, locally, DNA chains functionalized with pAsp preserve their binding ability through base pairing, while on a larger length scale the attached pAsp chains have a marked morphological effect on the resulting assemblies.
:2, kinked nanotube bundles were observed alongside amorphous aggregates and misassembled structures; AFM images of the resulting structures are shown in Fig. 2C. DNA nanotubes did not properly assemble as the pAsp content was further increased, which suggests that 1:2 incorporation is close to a morphological boundary. This disrupted assembly behaviour may be due to steric constraints caused by the polymer during the assembly of the single stranded DNA into nanotubes. Alternatively, electrostatic complexation between polymer chains/DNA and calcium ions in solution may favour smaller assemblies over the desired linear scaffold. It is worth noting, however, that the system showed no clear selectivity for unmodified DNA during the structure formation process, as proper nanotubes could, in principle, have formed by assembly of DNA chains alone without incorporation of DNA–pAsp. These results suggest that, locally, DNA chains functionalized with pAsp preserve their binding ability through base pairing, while on a larger length scale the attached pAsp chains have a marked morphological effect on the resulting assemblies.
 | ||
| Fig. 2 Synthesis and self-assembly of 1sNT–pAsp conjugates. (A) Synthetic scheme of the coupling of pAsp to DNA. Self-assembly of 1sNT in the presence of (B) 1:10 DNA-pAsp:DNA and (C) 1:2 DNA-pAsp:DNA in calcium-containing buffers. | ||
To address the steric constraints imposed by the pAsp and the resulting morphological effects seen during co-assembly, we designed a simplified linear system which, unlike the 1sNT, does not need to curve or otherwise fold to create a stable 1D structure. This system (three strands double helix, 3sDH) consists of three DNA strands (S1, S2, S3), with sticky ends that come together to form a one-dimensional scaffold (Fig. 3A). The strand bearing the pAsp moiety was designed such that upon assembly the polymer is always facing the same side of the DNA duplex (i.e.: every 84 bases, or 8 helical turns); the remaining two strands were designed to form a linear structure only in the presence of the pAsp–conjugated DNA component. Only pAsp-modified S3 strands were used during assembly, which corresponded to pAsp incorporation levels above those achieved by the 1sNT. Non-denaturing gel experiments support the formation of the intended assembly product in the 3sDH system, as judged from the lack of bands related to the building blocks and the presence of a lower-mobility product ascribed to the 1D assembly (Fig. S1†).
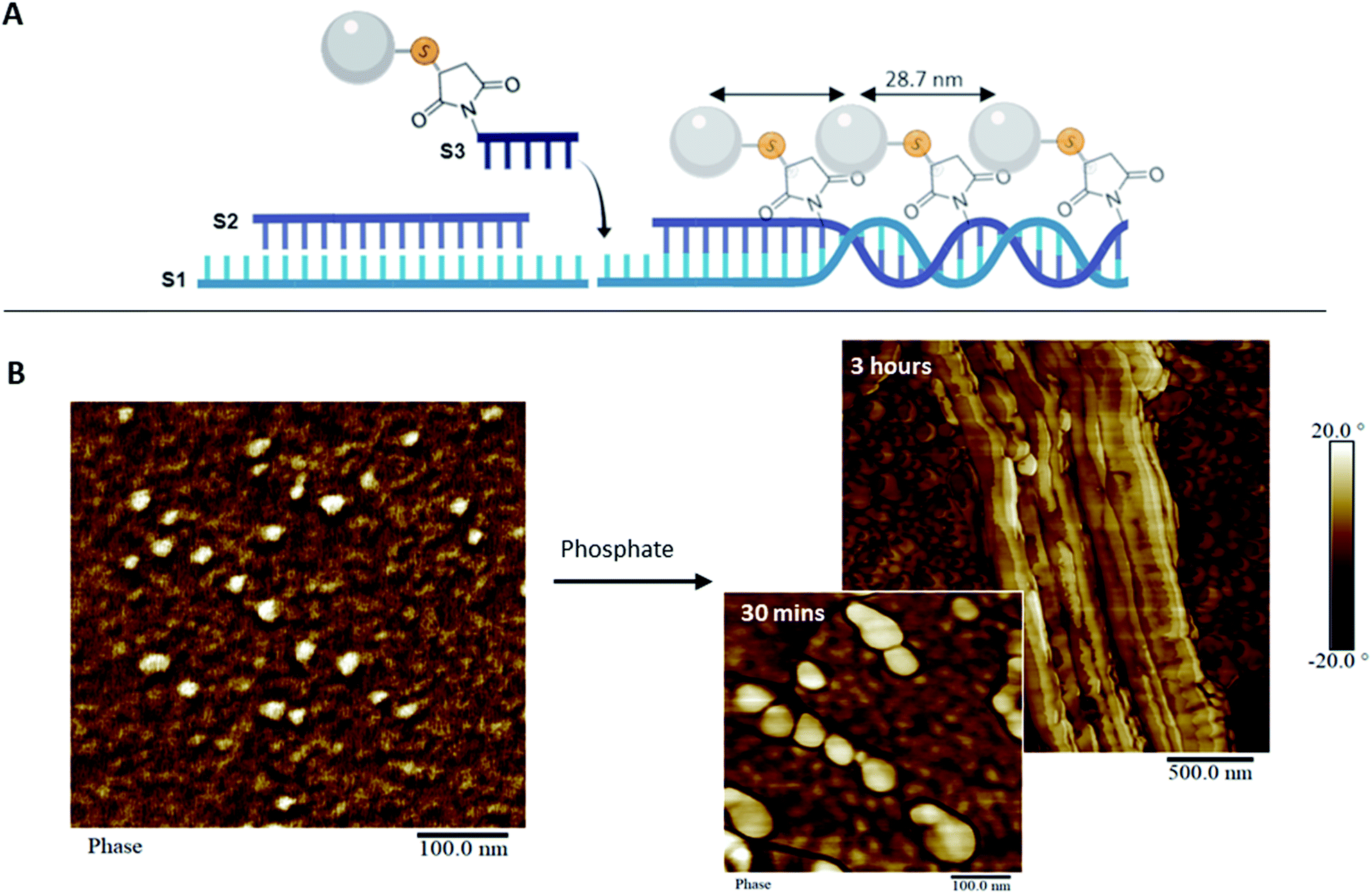 | ||
| Fig. 3 pAsp organization in 1D. (A) Scheme of the assembly of the 3sDH system composed of three DNA strands (S1, S2, and S3-pAsp) that elongate in 1D. (B) AFM image of the 3sDH-pAsp system in calcium buffer (left), and upon addition of phosphate and incubation for 30 minutes (right, bottom) and 3 hours (right, top). | ||
Linear scaffold structures were also characterized by AFM (Fig. 3B and S12†), where the pAsp nanospheres successfully incorporated into the scaffold. It should be noted that pAsp assembles into nanospheres in the DNA–pAsp concentration range of our co-assembly experiments, and that the sizes observed for pAsp and DNA–pAsp structures are comparable; DNA base pairing, therefore, did not interfere with nanosphere formation. Given the lack of bundling, or of gel network structures, nanosphere formation is presumed to have involved some amount of free pAsp, not bound to or reversibly associated with the DNA scaffold. Where observed, however, the pAsp nanospheres were predominantly aligned and had a centre-to-centre distance of 28.7 ± 5.2 nm, which corresponds to the spacing between each adjacent pAsp conjugation site in the 3sDH design (theoretical distance of 28.6 nm). To explain these findings, we hypothesize that each scaffold-bound spherical structure, formed of multiple DNA–pAsp chains, forms DNA base pairs predominantly through the S3 sequence of a single DNA strand of the nanosphere. Further, at the stoichiometric amounts used here, each unbound DNA–pAsp molecule would leave excess S1 and S2 DNA elsewhere in the system. Despite the presence of these, the data showed a preference for adjacent DNA–pAsp binding sites to be occupied: the observed periodicity suggests that some 3sDH assemblies bind fully to DNA–pAsp and are decorated with nanospheres at every available binding site. We believe these co-exist, for the most part, with S1 and S2 strands that remain partially hybridized but do not elongate. Further studies are underway to determine the origins and boundary conditions of this all-or-nothing binding mechanism; it is worth mentioning that this behaviour has been previously reported in other dynamic self-assembled systems.6a,25 This simple system, which contained periodically spaced binding sites for the pAsp–DNA conjugates, was successful in patterning pAsp groups in 1D on the nanoscale. Furthermore, in this design, the spacing between pAsp sites was dictated by the inherent periodicity of the binding sites on the 1D scaffold, which is tunable through DNA sequence selection. This high level of spatial precision and control are necessary in order to investigate guided mineralization at the molecular level and determine the underlying structure–property relationships of biomaterials involved in this process.
In comparing the co-assembly behaviour of the 1sNT and 3sDH scaffolds in the presence of pAsp, the relative simplicity of the latter design offered a clear improvement in pAsp incorporation density. In turn, this limited the amount of smaller, discrete assemblies and of free pAsp nanospheres in solution. As the curvature of a sheet-like DNA structural intermediate is central to nanotube assembly in the 1sNT system, it is likely that macromolecule functionalization on one side of the sheet, which promotes further curvature beyond the optimum, led to the progressive loss of long-range order at higher incorporation levels. Structure formation in the 3sDH system is more closely tied to DNA base pairing alone than local curvature phenomena; as such, only the 3sDH was deemed sufficiently robust for use as a long-range mineralization scaffold. To explore its feasibility in guided mineralization, the pAsp-functionalized 3sDH assemblies were exposed to phosphate ions at a 1.67:1 (calcium:phosphate) mole ratio consistent with the ratio necessary for hydroxyapatite formation, in order to induce mineralization. The reaction was incubated for up to 3 hours prior to surface deposition and characterization by AFM and transmission electron microscopy (TEM, Fig. 3B and S14†). After 30 minutes, swollen nanospheres formed of DNA–pAsp, presumably carrying amorphous calcium phosphate, were observed juxtaposed along the length of the 1D scaffold. After 3 hours incubation, the DNA nanostructures had an increased height measurement, ranging from 12.5 to 63.3 nm (unmodified DNA duplex has an average height of 1 to 2 nm by AFM).26 Unmodified DNA provides limited contrast under TEM without staining or mineral nucleation. Therefore, the visibility of organized nanospheres in our samples may indicate the presence of mineral aggregates along the DNA, which provide greater electron density (and hence contrast under TEM) than organic materials such as DNA or polypeptides. These results demonstrate that mineralization can be guided and successfully controlled through DNA scaffolds. Though the degree of crystallinity in our system was not determined, the minerals observed are likely to be mostly amorphous. As such, these organized amorphous minerals may be useful intermediates that remain stable in vitro, and could potentially form crystals over time in vivo. Moving forward, this system could be used to organize mineral-inducing groups in a variety of orientations, which will allow for the examination of the spatial regulation inherent to guided mineralization. In addition, this synthetic system provides a platform to further investigate the process of biomimetic mineralization.
Conclusion
We designed DNA–pAsp conjugates and investigated their assembly behaviour. These conjugates were able to position themselves into DNA nanostructures; however, the resilience to the incorporation of pAsp–DNA conjugates varied with each system. The previously published 1sNT system was unable to form nanotubes when conjugate ratios were greater than 1:2. The 3sDH was more robust and efficient in its pAsp incorporation. Subsequently, the 3sDH system was able to successfully organize the mineral-inducing group in one-dimension. Current efforts are directed towards the development of a dynamic DNA scaffold for guided mineralization of tissues such as tooth and bone. Fundamentally, the ability to vary the density and the pattern of macromolecules on the DNA scaffold through self-assembly will enable scientists to probe for collective or long-range effects during the mineralization process; individual crystal growth sites may be close enough to interact, and the sum of these mutual interactions are likely to influence the overall mineralization process. The conditions described herein aim to more closely mimic hydroxyapatite formation in vivo, which does not occur at isolated sites.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to thank NSERC, MITACS, Colgate and the Bertha Rosenstadt Endowment for funding, the Peter Backx Foundation for the vibration-free AFM table, and Michael J. Greschner for the preparation of Blender images. We acknowledge BioRender for partial preparation of Fig. 1–3.Notes and references
- (a) K. M. Carneiro, N. Avakyan and H. F. Sleiman, in Long-range assembly of DNA into nanofibers and highly ordered networks, 2013 Search PubMed; (b) N. C. Seeman, J. Theor. Biol., 1982, 99, 237 CrossRef CAS; (c) N. R. Kallenbach, R.-I. Ma and N. C. Seeman, Nature, 1983, 305, 829 CrossRef CAS.
- (a) G. Chan and D. J. Mooney, Trends Biotechnol., 2008, 26, 382 CrossRef CAS PubMed; (b) N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068 CrossRef; (c) J. W. de Vries, F. Zhang and A. Herrmann, J. Controlled Release, 2013, 172, 467 CrossRef CAS PubMed.
- (a) O. I. Wilner and I. Willner, Chem. Rev., 2012, 112, 2528 CrossRef CAS PubMed; (b) S. I. Liang, J. M. McFarland, D. Rabuka and Z. J. Gartner, J. Am. Chem. Soc., 2014, 136, 10850 CrossRef CAS PubMed; (c) A. A. Greschner, X. Ropagnol, M. Kort, N. Zuberi, J. Perreault, L. Razzari, T. Ozaki and M. A. Gauthier, J. Am. Chem. Soc., 2019, 141, 3456 CrossRef CAS PubMed.
- J. Fu, Y. R. Yang, A. Johnson-Buck, M. Liu, Y. Liu, N. G. Walter, N. W. Woodbury and H. Yan, Nat. Nanotechnol., 2014, 9, 531 CrossRef CAS PubMed.
- (a) T. Trinh, P. Chidchob, H. S. Bazzi and H. F. Sleiman, Chem. Commun., 2016, 52, 10914 RSC; (b) P. Chidchob, T. G. W. Edwardson, C. J. Serpell and H. F. Sleiman, J. Am. Chem. Soc., 2016, 138, 4416 CrossRef CAS PubMed; (c) K. M. M. Carneiro, F. A. Aldaye and H. F. Sleiman, J. Am. Chem. Soc., 2010, 132, 679 CrossRef CAS PubMed; (d) T. G. W. Edwardson, K. M. M. Carneiro, C. K. McLaughlin, C. J. Serpell and H. F. Sleiman, Nat. Chem., 2013, 5, 868 CrossRef CAS PubMed; (e) C. J. Serpell, T. G. W. Edwardson, P. Chidchob, K. M. M. Carneiro and H. F. Sleiman, J. Am. Chem. Soc., 2014, 136, 15767 CrossRef CAS PubMed; (f) K. E. Bujold, J. Fakhoury, T. G. W. Edwardson, K. M. M. Carneiro, J. N. Briard, A. G. Godin, L. Amrein, G. D. Hamblin, L. C. Panasci, P. W. Wiseman and H. F. Sleiman, Chem. Sci., 2014, 5, 2449 RSC.
- (a) K. M. M. Carneiro, G. D. Hamblin, K. D. Hanni, J. Fakhoury, M. K. Nayak, G. Rizis, C. K. McLaughlin, H. S. Bazzi and H. F. Sleiman, Chem. Sci., 2012, 3, 1980 RSC; (b) C. K. McLaughlin, G. D. Hamblin, K. D. Hänni, J. W. Conway, M. K. Nayak, K. M. M. Carneiro, H. S. Bazzi and H. F. Sleiman, J. Am. Chem. Soc., 2012, 134, 4280 CrossRef CAS PubMed; (c) M. Kwak and A. Herrmann, Chem. Soc. Rev., 2011, 40, 5745 RSC; (d) T. R. Wilks, J. Bath, J. W. de Vries, J. E. Raymond, A. Herrmann, A. J. Turberfield and R. K. O'Reilly, ACS Nano, 2013, 7, 8561 CrossRef CAS PubMed; (e) D. de Rochambeau, M. Barlog, T. G. W. Edwardson, J. J. Fakhoury, R. S. Stein, H. S. Bazzi and H. F. Sleiman, Polym. Chem., 2016, 7, 4998 RSC; (f) M.-P. Chien, A. M. Rush, M. P. Thompson and N. C. Gianneschi, Angew. Chem., Int. Ed., 2010, 49, 5076 CrossRef CAS PubMed.
- (a) M. Numata, K. Sugiyasu, T. Hasegawa and S. Shinkai, Angew. Chem., Int. Ed., 2004, 43, 3279 CrossRef CAS PubMed; (b) C. Jin, H. Qiu, L. Han, M. Shu and S. Che, Chem. Commun., 2009, 23, 3407 RSC.
- (a) S. P. Surwade, F. Zhou, B. Wei, W. Sun, A. Powell, C. O'Donnell, P. Yin and H. Liu, J. Am. Chem. Soc., 2013, 135, 6778 CrossRef CAS; (b) C. T. Diagne, C. Brun, D. Gasparutto, X. Baillin and R. Tiron, ACS Nano, 2016, 10, 6458 CrossRef CAS PubMed.
- W. Sun, E. Boulais, Y. Hakobyan, W. L. Wang, A. Guan, M. Bathe and P. Yin, Science, 2014, 346, 1258361 CrossRef PubMed.
- (a) X. Liu, F. Zhang, X. Jing, M. Pan, P. Liu, W. Li, B. Zhu, J. Li, H. Chen, L. Wang, J. Lin, Y. Liu, D. Zhao, H. Yan and C. Fan, Nature, 2018, 559, 593 CrossRef CAS; (b) E. C. Samano, M. Pilo-Pais, S. Goldberg, B. N. Vogen, G. Finkelstein and T. H. LaBean, Soft Matter, 2011, 7, 3240 RSC.
- M. Tintoré, R. Eritja and C. Fábrega, ChemBioChem, 2014, 15, 1374 CrossRef PubMed.
- A. Matsumoto, H. Kajiya, N. Yamamoto-M, T. Yanagi, A. Imamura, K. Okabe, T. Fukushima, H. Kido and J. Ohno, J. Biomed. Mater. Res., Part B, 2019, 107, 122 CrossRef CAS PubMed.
- (a) G. Minasov, V. Tereshko and M. Egli, J. Mol. Biol., 1999, 291, 83 CrossRef CAS PubMed; (b) J. H. Thorpe, B. C. Gale, S. C. M. Teixeira and C. J. Cardin, J. Mol. Biol., 2003, 327, 97 CrossRef CAS PubMed.
- (a) P. S. Lukeman, M. L. Stevenson and N. C. Seeman, Cryst. Growth Des., 2008, 8, 1200 CrossRef CAS PubMed; (b) Y. He, Y. Tian, Y. Chen, T. Ye and C. Mao, Macromol. Biosci., 2007, 7, 1060 CrossRef CAS; (c) S. C. Ngourn, H. A. Butts, A. R. Petty, J. E. Anderson and A. E. Gerdon, Langmuir, 2012, 28, 1215 CrossRef.
- (a) N. A. J. M. Sommerdijk, E. N. M. v. Leeuwen, M. R. J. Vos and J. A. Jansen, CrystEngComm, 2007, 9, 1209 RSC; (b) Y. Xu, K. C. H. Tijssen, P. H. H. Bomans, A. Akiva, H. Friedrich, A. P. M. Kentgens and N. A. J. M. Sommerdijk, Nat. Commun., 2018, 9, 2582 CrossRef PubMed.
- (a) H. Nurrohman, K. Saeki, K. M. M. Carneiro, Y.-C. Chien, S. Djomehri, S. P. Ho, C. Qin, L. B. Gower, S. J. Marshall, G. W. Marshall and S. Habelitz, J. Mater. Res., 2016, 31, 321 CrossRef CAS PubMed; (b) H. Nurrohman, K. M. M. Carneiro, J. Hellgeth, K. Saeki, S. J. Marshall, G. W. Marshall and S. Habelitz, PLoS One, 2017, 12, e0188277 CrossRef PubMed.
- (a) T. T. Thula, D. E. Rodriguez, M. H. Lee, L. Pendi, J. Podschun and L. B. Gower, Acta Biomater., 2011, 7, 3158 CrossRef CAS PubMed; (b) S. S. Jee, T. T. Thula and L. B. Gower, Acta Biomater., 2010, 6, 3676 CrossRef CAS PubMed.
- (a) B. D. Quan and E. D. Sone, J. R. Soc., Interface, 2018, 15, 20180269 CrossRef PubMed; (b) M. J. Olszta, X. Cheng, S. S. Jee, R. Kumar, Y.-Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., R, 2007, 58, 77 CrossRef; (c) J. Mahamid, A. Sharir, L. Addadi and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12748 CrossRef CAS PubMed; (d) D. V. Krogstad, D. Wang and S. Lin-Gibson, Biomacromolecules, 2015, 16, 1550 CrossRef CAS PubMed.
- H. Liu, Y. Chen, Y. He, A. E. Ribbe and C. Mao, Angew. Chem., Int. Ed., 2006, 45, 1942 CrossRef CAS.
- H. Yan, S. H. Park, G. Finkelstein, J. H. Reif and T. H. LaBean, Science, 2003, 301, 1882 CrossRef CAS.
- T. G. Martin and H. Dietz, Nat. Commun., 2012, 3, 1103 CrossRef PubMed.
- L. B. Gower, Chem. Rev., 2008, 108, 4551 CrossRef CAS.
- (a) X. Elduque, A. Sánchez, K. Sharma, E. Pedroso and A. Grandas, Bioconjugate Chem., 2013, 24, 832 CrossRef CAS PubMed; (b) B. H. Northrop, S. H. Frayne and U. Choudhary, Polym. Chem., 2015, 6, 3415 RSC.
- F. A. P. Crisafuli, E. B. Ramos and M. S. Rocha, Eur. Biophys. J., 2015, 44, 1 CrossRef CAS.
- R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808 CrossRef CAS PubMed.
- Y. Lyubchenko, L. Shlyakhtenko, R. Harrington, P. Oden and S. Lindsay, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 2137 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02811k |
| This journal is © The Royal Society of Chemistry 2019 |