
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkynylation of radicals: spotlight on the “Third Way” to transfer triple bonds
Franck
Le Vaillant
 and
Jérôme
Waser
*
and
Jérôme
Waser
*
Laboratory of Catalysis and Organic Synthesis, Ecole Polytechnique Fédérale de Lausanne, EPFL SB ISIC LCSO, BCH 4306, 1015 Lausanne, Switzerland. E-mail: jerome.waser@epfl.ch
First published on 13th August 2019
Abstract
The alkynylation of radical intermediates has been known since a long time, but had not been broadly applied in synthetic chemistry, in contrast to the alkynylation of either electrophiles or nucleophiles. In the last decade however, it has been intensively investigated leading to new disconnections to introduce versatile triple bonds into organic compounds. Nowadays, such processes are important alternatives to classical nucleophilic and electrophilic alkynylations. Efficient alkyne transfer reagents, in particular arylsulfones and hypervalent iodine reagents were introduced. Direct alkynylation, as well as cascade reactions, were subsequently developed. If relatively harsh conditions were required in the past, a new era began with progress in photoredox and transition metal catalysis. Starting from various radical precursors, alkynylations under very mild reaction conditions were rapidly discovered. This review covers the evolution of radical alkynylation, from its emergence to its current intensive stage of development. It will focus in particular on improvements for the generation of radicals and on the extension of the scope of radical precursors and alkyne sources.
 Franck Le Vaillant | Franck Le Vaillant was born in Nantes, France. He received his Master degree at the Ecole Nationale Supérieure de Chimie de Montpellier (ENSCM, France) in 2013, before joining the Trost group at Stanford University (USA) for one year as a visiting student researcher. From 2014 to 2018, he carried out his PhD studies at EPFL (Lausanne, Switzerland) under the supervision of Prof. Jérôme Waser. His research focused on photoredox-catalyzed functionalizations of radicals using hypervalent iodine reagents. He is now a SNF-postdoctoral fellow in the group of Dr Josep Cornella at the Max-Planck-Institute für Kohlenforschung in Mülheim an der Ruhr (Germany). |
 Jérôme Waser | Jérôme Waser was born in Sierre, Valais, Switzerland. He studied chemistry at ETH Zurich, where he obtained his PhD degree in 2006 with Prof. Erick M. Carreira. In 2006, he joined Prof. Barry M. Trost at Stanford University as a SNF postdoctoral fellow. Since October 2007 he has been professor of organic chemistry at the Ecole Polytechnique Fédérale de Lausanne (EPFL), where he was promoted full professor in 2019. He is a recipient of the ERC starting grant 2013 and consolidator grant 2017, the Werner prize of the Swiss Chemical Society 2014 and the Springer Heterocyclic Chemistry Award 2016. |
1. Introduction: context, background and emergence of the alkynylation of radicals
Aliphatic alkynes are not often encountered in nature, with few notable exceptions.1 Due to their rare occurrence and exceptional reactivity, aliphatic terminal alkynes have been widely used as tags in selective bioconjugation.2 In addition, alkynes are introduced in drugs to provide specific properties such as rigidity and lipophilicity.3 For example, ethynylestradiol (1) for estrogen medication, efavirenz (2) for HIV antiviral treatment, the nonsteroidal anti-inflammatory parsalmide (3) and the antihypertensive pargyline (4) exhibit high bioactivity (Fig. 1).4 Alkynes are also widely used in material sciences.3 Due to the unique features of the C–C triple bond, alkynes play an important role in synthetic chemistry.5 | ||
| Fig. 1 Drugs containing aliphatic alkynes. | ||
Applications of alkynes are dependent on efficient synthetic methods to access them. Transformations involving transfer of the triple bond can be subdivided into three classes: (1) nucleophilic, (2) electrophilic, (3) SOMOphilic alkyne transfer (Scheme 1). Historically, chemists used the intrinsic acidity of the Csp–H bond (pKa around 24–26) to prepare internal alkynes in presence of a base and an electrophilic partner (typically aldehydes, ketones, imines and alkyl halides) (Scheme 1A).6 Terminal alkynes have been also broadly used in transition metal catalyzed cross-couplings, such as the Cadiot–Chodkiewicz and the Sonogashira reactions.7
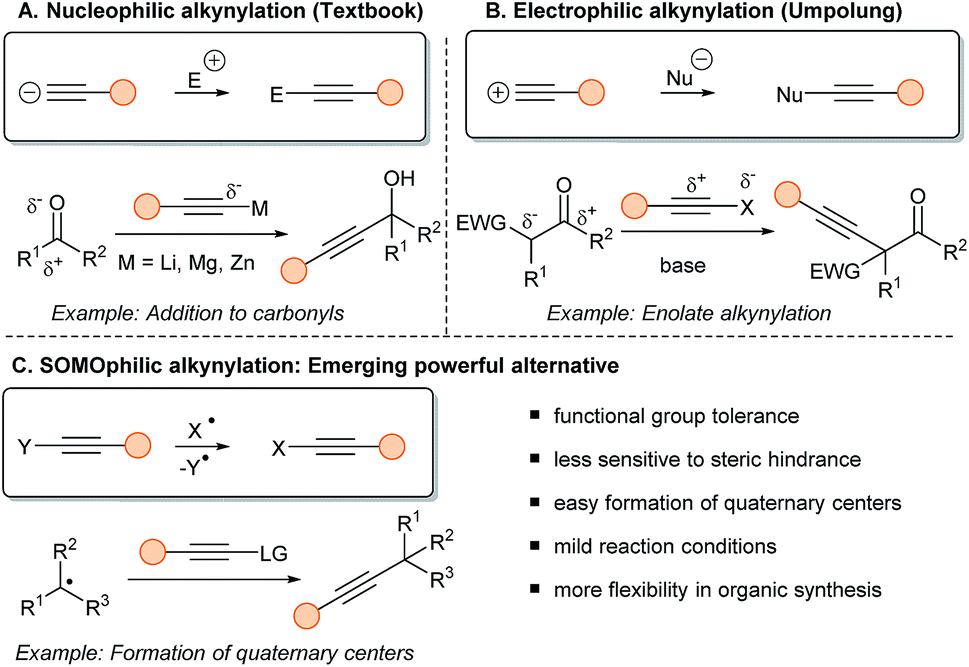 | ||
| Scheme 1 The three possible types of alkyne transfer: (A) alkynes reacting as nucleophile; (B) reacting as electrophile; (C) reacting in radical-mediated processes. | ||
After Umpolung of the reactivity,8 the alkynyl moiety can be turned into an electrophile to react with nucleophiles, such as stabilized enolates (Scheme 1B).9 In that regard, several electrophilic reagents have been developed, such as halogenated alkynes (Br, I) and hypervalent iodine reagents.10 Most electrophilic alkynylations involve heteroatomic nucleophiles and arenes in presence of transition metal catalysts. The alkynylation of Csp3 centers is still very challenging using this approach.
In fact, this specific issue can be well-addressed using radical chemistry (Scheme 1C). This approach is often characterized by a high functional group tolerance, being also less sensitive to steric hindrance, resulting in easier formation of quaternary centers. Finally, the mild and often neutral reaction conditions allow more flexibility in the synthesis of alkynes. Nevertheless, SOMOphilic alkynylation faces many challenges. First, developing and harnessing new radical precursors is crucial to allow new disconnections. Then, developing milder reactions conditions is an important goal, as UV light or tin mediated transformations originally developed are not attractive for applications. Finally, to increase the efficiency of SOMOphilic alkynylations, MCRs, asymmetric transformations and more atom–economic reactions are current hot topics of investigation.
The alkynylation of heteroatom-centered radicals has been limited to the sole alkynylation of sulfonyl and phosphoranyl radicals. Therefore, the present review will cover mostly radicals located on a carbon atom (Fig. 2). Csp centered radicals are rare due to their very high energy, even if they would enable general alkynylation methods and are regularly proposed as intermediates. Due to the strong Csp–H bond (130 kcal mol−1), alkynyl radicals react fast with C–H bonds. In addition, low selectivity is observed in the reaction with aromatic rings.11 More stable aryl and vinyl radicals have been regularly used in alkynylation. Nevertheless, functionalization of these Csp2 centered radicals remains a challenge as they are still reactive species, which can lead for example to H-abstraction from solvents. Finally, radicals located at a Csp3 center are the privileged SOMOphilic species for alkynylation and can be divided in different classes depending of their substitution patterns (primary, secondary, tertiary and stabilized by heteroatoms or conjugation).
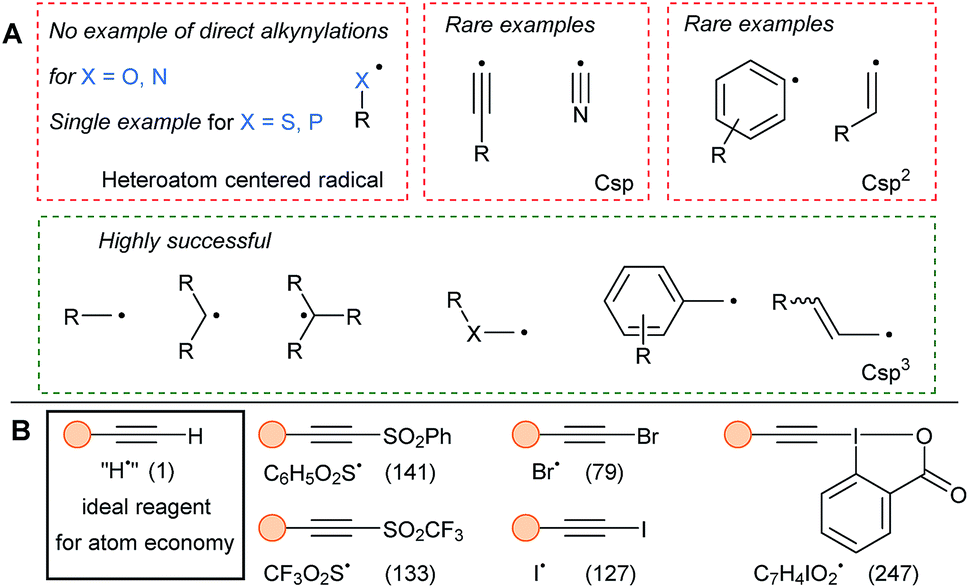 | ||
| Fig. 2 Commonly used radicals (A) and reagents (B) in SOMOphilic alkynylation. Molecular weight of the radical leaving group is written in parenthesis in g mol−1. | ||
The most used alkynylating reagents in radical chemistry are listed in Fig. 2B. Halogenoalkynes and aryl- and trifluoromethyl-sulfones were the first and are still broadly used reagents. In addition, hypervalent iodine reagents, especially ethynylbenziodoxolones (EBX), exhibit high reactivity in SOMOphilic alkynylation. All the previous reagents possess a good leaving group as substituent, that can be either easily quenched, or on the contrary, be able to carry a possible propagation chain. Finally, to avoid multi-step syntheses and considering atom-economy criteria, the ideal reagent would be the terminal alkynes.
In this review, methods for SOMOphilic alkynylation which appeared until May 1, 2019 will be presented, divided into five parts: (1) classical methods and reagents; (2) the photoredox catalysis revolution; (3) the use of transition metal catalysis; (4) asymmetric transformations; (5) the use of internal migration for remote alkynylation.
2. Classical methods for the alkynylation of radicals
Alkynylsulfones were the first successful reagents for the alkynylation of carbon radicals. The reaction is assumed to proceed via an α-addition/β-elimination sequence (Scheme 2). | ||
| Scheme 2 Putative mechanism using ethynylsulfone reagents. | ||
The first radical alkynylation was developed by Russel and Ngoviwatchai using alkylmercury halides as substrates and iodoalkynes or alkynyl sulfones as reagents (Scheme 3, eqn (1)).12 Then, Fuchs and coworkers introduced alkynyltriflones as new alkynylating reagents and developed a metal free radical alkynylation of heterocycles, ethers, sulfides and hydrocarbons (eqn (2)).13 Hydrogen Atom Transfer (HAT) process and propagation by releasing SO2 and CF3 (or perfluoroalkyl) radicals as radical chain carriers have been proposed for the reaction mechanism.14 The reaction can be initiated either by traces of peroxides/oxygen, by AIBN or by UV irradiation. This transformation could be also used for the functionalization of carbonyl C–H bonds15 and alkyl iodides (eqn (3)),16 which proved to be highly useful for the total synthesis of (+) and (−) nigellamine A2 (7),17 and mandelalide A.18
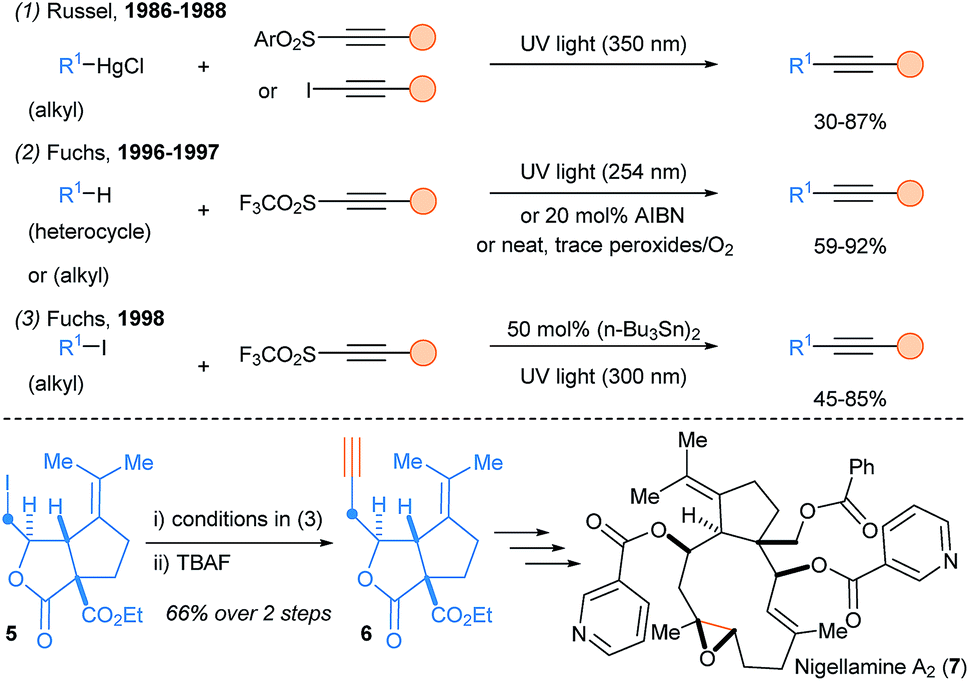 | ||
| Scheme 3 Pioneering reports on SOMOphilic alkynylation of alkyl radicals using alkynylsulfone reagents and application to the total synthesis of nigellamine A2 (7). | ||
In 2006, Renaud and coworkers developed an alkynylation of alkylcatecholboranes with alkynyl sulfones. Starting with hydroboration, a practical one-pot hydroalkynylation of olefins was realized (Scheme 4, eqn (1)).19 Finally, Landais reported the carboalkynylation of alkenes using xantates as electrophilic radical source (eqn (2)).20
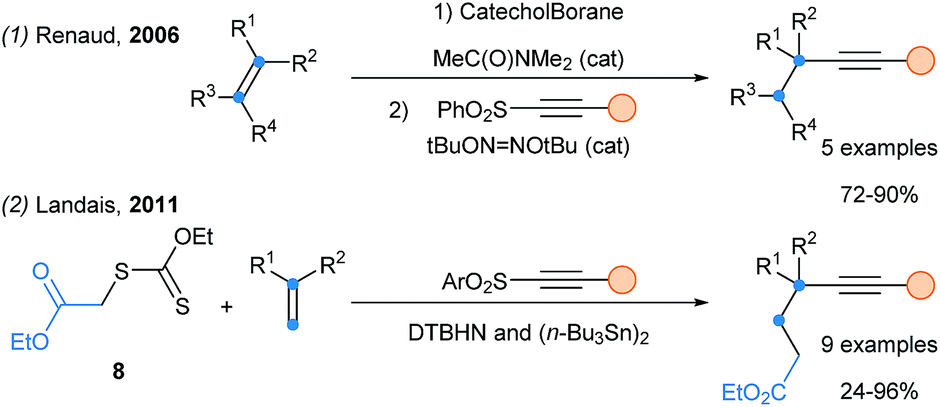 | ||
| Scheme 4 Alkynylation of olefins using alkynylsulfone reagents. | ||
In 2013, various reactive C–H bonds (Cα of alcohols, ethers, amines, amides and ureas) were alkynylated by Inoue and coworkers using a stoichiometric amount of diarylketone photosensitizer upon light irradiation.21 This transformation was an important step in the total synthesis of (+)-lactacystin (11) (Scheme 5).22 In 2017, Paul and Guin achieved the catalytic version of this C–H bond alkynylation with only 20 mol% of a diarylketone derivative under neat conditions.23
 | ||
| Scheme 5 Radical alkynylation as a key step for the synthesis of (+)-lactacystin (11) by Inoue. | ||
Alkynylsulfones have also been used in multicomponent reactions. The trifluoromethylalkynylation of unactivated alkenes was first reported by Fuchs in 1996 (ref. 13) and further developed by Li in 2017 (Scheme 6, eqn (1)).24 After a SET event, a CF3 radical is generated from Togni reagent I (12). Due to its electrophilic character, it reacts preferentially with the electron rich double bond, followed by alkynylation. This transformation was further developed by using only catalytic amount of Togni reagent II 13 with acetylenic triflones, allowing regeneration of the CF3 radical.25 Finally, Studer and coworkers used AIBN as radical initiator and allylsulfone as N-activating/protecting group, to generate a nitrogen-centered radical I (eqn (2)).26 This electrophilic radical favors a 1,5-HAT releasing a nucleophilic C-centered radical II, which is alkynylated. They then reported the desulfonylative alkynylation of alkylallylsulfones (eqn (3)). Primary, secondary and tertiary positions can be alkynylated, albeit in lower yields for primary radicals.27
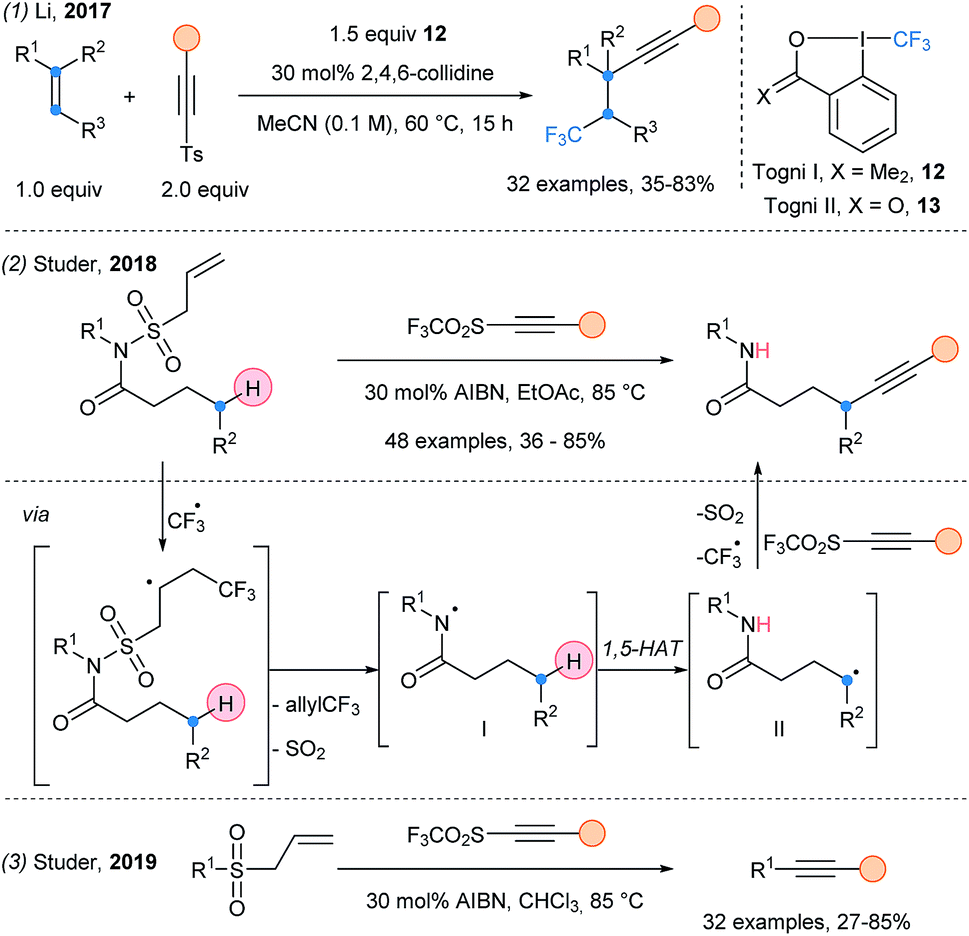 | ||
| Scheme 6 Cascade alkynylation processes using alkynylsulfone reagents. | ||
In 2002, Oshima and coworkers developed the alkynylation of α-iodocarbonyl compounds using alkynylgallium reagents and triethylborane under oxygen (Scheme 7).28 The alkyne scope is broad, but the substrate scope is somewhat limited, as secondary radicals led to low yields. In 2005, Oshima and coworkers discovered that alkynylindium reagents have similar reactivity than alkynylgallium reagents.29 Since Oshima's discovery, new applications of alkynylindium and alkynylgalliums have appeared.30
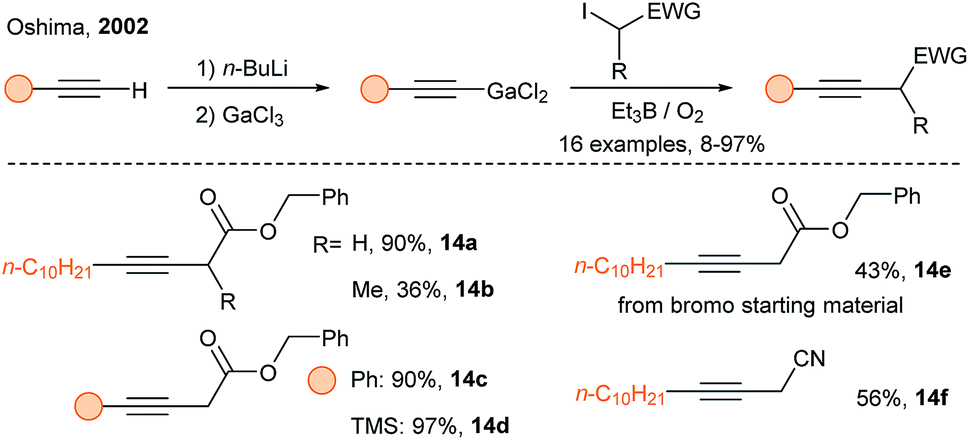 | ||
| Scheme 7 SOMOphilic alkynylation using alkynylgallium reagents. | ||
In 2015, a SOMOphilic alkynylation using terminal alkynes in aqueous media was reported (Scheme 8).31 UV light irradiation allowed generation of alkyl radicals via homolytic cleavage of alkyliodides, which upon trapping by terminal alkynes formed vinyl iodides, after an overall ATRA (Atom-Transfer Radical Addition) process. The latter readily eliminates in presence of base to release the alkyne products.
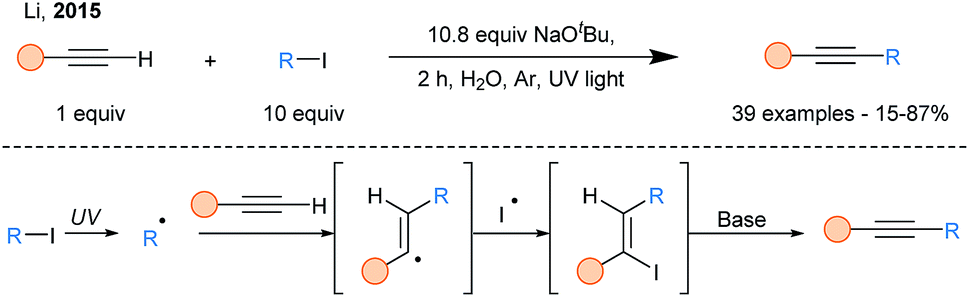 | ||
| Scheme 8 ATRA-elimination cascade for the alkylation of terminal alkynes. | ||
A direct cross-coupling of aryl alkynyliodines with arylsulfinic acids leading to alkynyl sulfones was reported by Wang and coworkers (Scheme 9).32 The proposed mechanism involved thermal homolytic cleavage of the C(sp)–I bond, leading to radicals, which can then abstract the labile hydrogen of the sulfinic acid. The resulting S-centered radical can then react with the alkynyliodide to form the alkynylsulfone along with regeneration of the iodine radical. A similar strategy was used by Li for the transition-metal free alkynylation of 2-oxindoles.33
 | ||
| Scheme 9 Iodine radical mediated HAT process for the alkynylation of arylsulfinic acids. | ||
In 2012, a breakthrough was realized by Li and coworkers with the introduction of EBX reagents in the realm of radical C-alkynylation (Scheme 10).34 They developed a general decarboxylative alkynylation of aliphatic carboxylic acids using catalytic silver nitrate and stoichiometric amounts of potassium persulfate under aqueous conditions (products 15a–15e). A classical addition–elimination mechanism was proposed for this reaction. Using similar reaction conditions developed by Li,34 Qi and coworkers reported a double decarboxylative radical alkynylation of arylpropiolic acids with α-ketoacids to access ynones.35
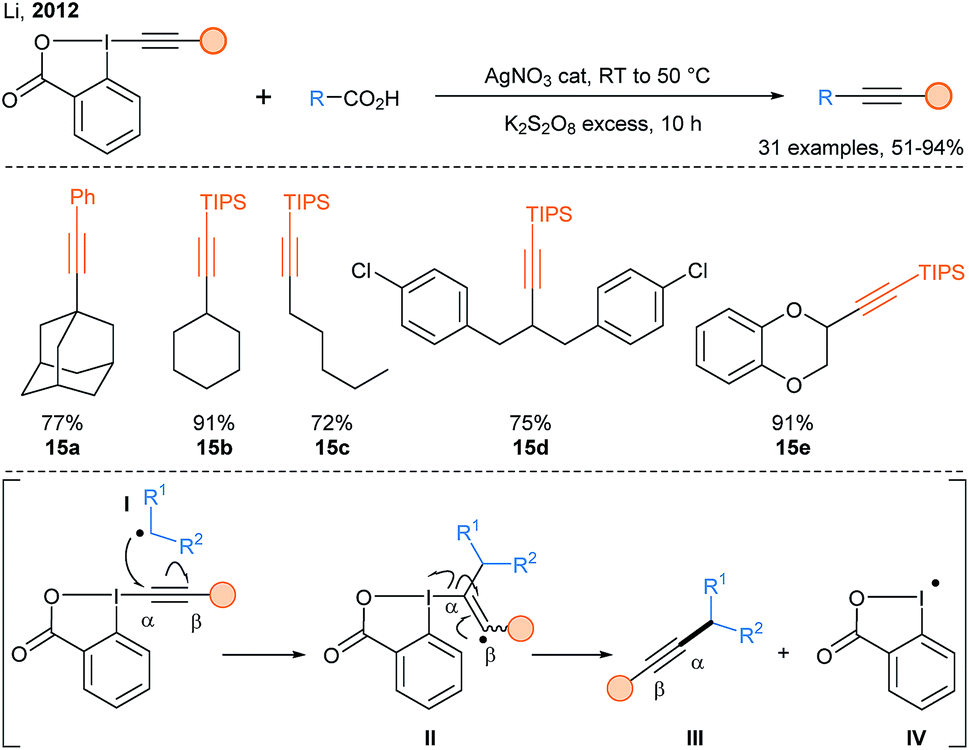 | ||
| Scheme 10 Silver catalyzed decarboxylative Csp3-alkynylation developed by Li and putative mechanism proposed by the authors. | ||
In 2014, Yu and coworkers documented the metal free Cα–H alkynylation of protected amines and ethers using tert-butylhydroperoxide (TBHP) as oxidant (Scheme 11, eqn (1)).36 In 2016, this transformation was extended by Xu and Feng using di-tert-butylperoxide (eqn (2)).37
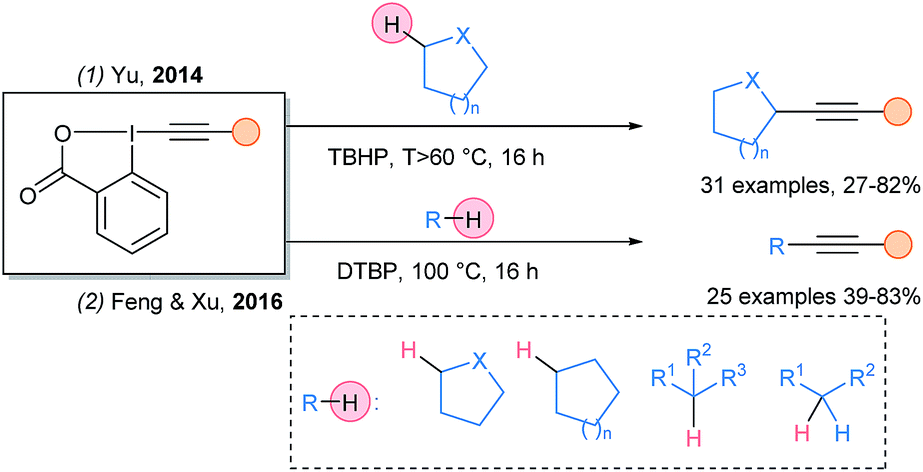 | ||
| Scheme 11 Alkynylation of unactivated C–H bonds using peroxides as radical initiators and EBX reagents. | ||
Since 2015, an increasing number of reports highlighted the exceptional reactivity of EBXs. First, the alkynylation of acyl radicals starting directly from aldehydes was achieved independently by several research groups using peroxides such as TBHP or DTBP at high temperatures (Scheme 12, eqn (1)).38 Similar valuable ynones were synthesized by Duan and coworkers upon the facile decarboxylation of α-keto acids under Li's oxidative conditions (eqn (2)).39 Almost simultaneously, Xu and Feng documented the same transformation without silver catalyst.40 The decarboxylative alkynylation of aryl- and thio-difluoroacetic acids was also successful.41
 | ||
| Scheme 12 Radical alkynylation using EBX for the synthesis of ynones and ynamides. | ||
In 2018, Cheng and Li introduced Hantzsch esters (Z = ester) and Meyer nitriles (Z = CN) as radical precursors for SOMOphilic alkynylations under oxidative conditions (Scheme 13, eqn (1)).42 Such starting materials are easily obtained from the corresponding aldehydes. These oxidative conditions were also successful for the oxyalkynylation of alkenes, as reported by Liu and coworkers (eqn (2)).43 Water and alcohols can be introduced, thus generating attractive scaffolds in only one step.
 | ||
| Scheme 13 (1) Use of DHP as radical precursors for SOMOphilic alkynylation and (2) oxyalkynylation of alkenes using EBX reagents. | ||
Heteroatom-centered radicals initiated fragmentation is a powerful strategy to access alkyl radicals. In 2015, Duan and coworkers used persulfate radicals in a HAT process with cyclopropanols and cyclobutanols to generate O-centered radicals (intermediate I, Scheme 14, eqn (1)).44 Fragmentation to radical II followed by alkynylation led to β- or γ-alkynyl ketones. In 2016, α-alkenyl and α-ethynyl cyclobutanols were used in this cascade transformation by Chen and Yu (eqn (2)).45 In this case, an extra cyclization step occurs from II to III, before the final alkynylation.
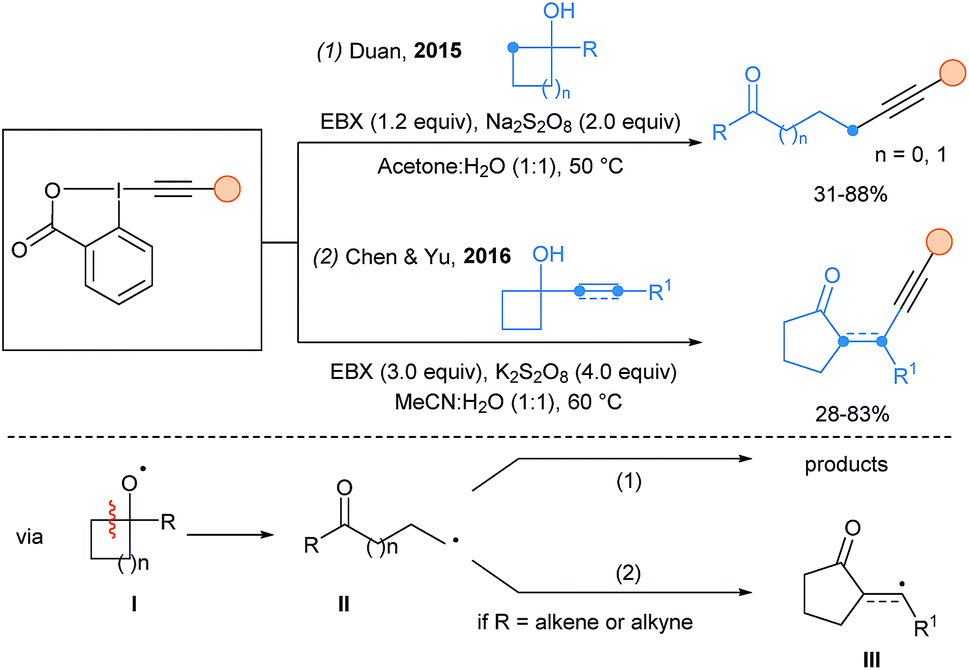 | ||
| Scheme 14 Ring-fragmentation-alkynylation cascades. | ||
The use of EBX reagents has open new possibilities in radical alkynylation, mainly based on oxidative conditions using peroxides and persulfates. These classical methods still have severe drawbacks: neat conditions for peroxide reactions, elevated temperatures, often large excess of both oxidant and reagent. With the ideal goal of having more broadly applicable alkynylation methods, milder conditions based on photoredox catalysis were developed.
3. The photoredox catalysis revolution
Photoredox catalysis harnesses the energy of visible light to promote single electron transfer (SET).46 The excited state of the photocatalyst can act both as an oxidant or reductant, depending on the other redox partners. This allows to use a broad variety of radical precursors, and to adapt the alkynylating reagent to close the catalytic cycle. The application of photoredox catalysis for the alkynylation of radicals has just started within the last seven years, yet a tremendous number of reports already showcased its importance.In 2007, Osawa and Akita described the first alkynylation assisted by Ru(bpy)3(PF6)2.47 Under light irradiation, they observed that the copper free Sonogashira coupling between aryl bromides and terminal alkynes led to higher yields in shorter time when Ru(bpy)3(PF6)2 is used as co-catalyst. In 2012, independent studies from Stephenson, Rueping and Fu on the photoredox catalyzed alkynylation of tetrahydroquinolines (THQ) were published (Scheme 15).48 A copper(I) catalyst is necessary for the alkynylation step. While Stephenson and Rueping chose Ru(bpy)3(PF6)2 as best photocatalyst, Fu found that Rose Bengal is also efficient. Stephenson used BrCCl3 as oxidant, whereas Fu and Rueping reported the use of oxygen as a greener oxidant. Later, Zeitler and coworkers reinvestigated the transformation using BrCCl3 and discovered that the photocatalyst was not necessary.49 In 2015, the enantioselective variant of this transformation was successfully achieved by Li and coworkers. The catalytic system involved a copper catalyst with the P–N chiral ligand QUINAP, the common Ir(ppy)2dtbbpyPF6 photocatalyst and either benzoyl peroxide or oxygen as oxidant.50 Concerning the mechanism, all the authors envisaged an iminium formation after oxidation of the electron rich tertiary amine by the excited state of the photocatalyst, and regeneration of the ground state photocatalyst by the external oxidant. In situ generation of copper acetylides followed by nucleophilic addition to the iminium delivered the alkyne products.
 | ||
| Scheme 15 Photocatalytic alkynylation of THQ derivatives using terminal alkynes and copper catalysis. | ||
In 2012, the Hwang group developed the cross coupling between terminal alkynes and aryl bromides (and aryl iodides) under light irradiation and oxygen atmosphere (Scheme 16, eqn (1)).51 They discovered that copper(I) acetylides can be excited using blue light (λmax = 460 nm). Using similar photocatalytic conditions, they also described the synthesis of symmetrical52 and unsymmetrical diynes (eqn (2) and (3)).53
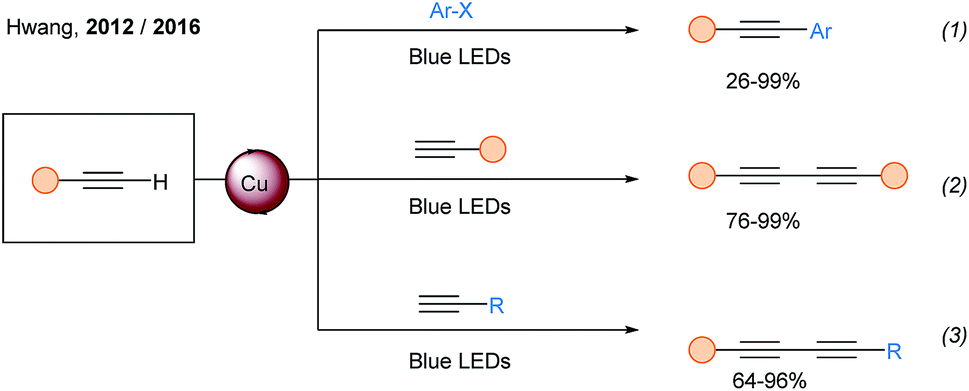 | ||
| Scheme 16 Metal free Sonogashira and alkyne dimerization mediated by visible light. | ||
In 2017, a new synthesis of propargylic amines starting from α-amino N-acyloxyphthalimides was reported by Fu and coworkers using a photocatalytic system similar to the one of Rueping for THQ (Scheme 17, eqn (1)).54 However, after oxidative quenching, an α-amino radical is generated upon decarboxylation and an additional SET is proposed to oxidize it to the iminium. In 2018, Lalic and coworkers developed an alkyl-Sonogashira coupling catalyzed by a copper–tripyridine complex (eqn (2)).55
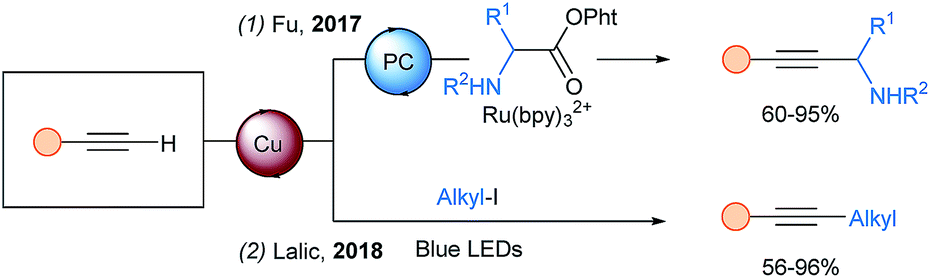 | ||
| Scheme 17 Visible light mediated alkyl–alkyne couplings using terminal alkynes. | ||
Terminal alkynes were also used in a Sonogashira coupling with diazonium salts based on dual gold photoredox catalysis by the Glorius group (Scheme 18, eqn (1)).56 They proposed that a low valent gold(I) species could trap the formed aryl radical to give a gold(II) complex. Upon oxidation to gold(III), the latter would regenerate the ground state photocatalyst. Then, the gold(III) species reacts with the alkyne to form an aryl acetylide gold(III) complex, which readily undergoes reductive elimination. Using TMS protected alkynes and similar reaction conditions, the Toste group independently reported the same transformation (eqn (2)).57
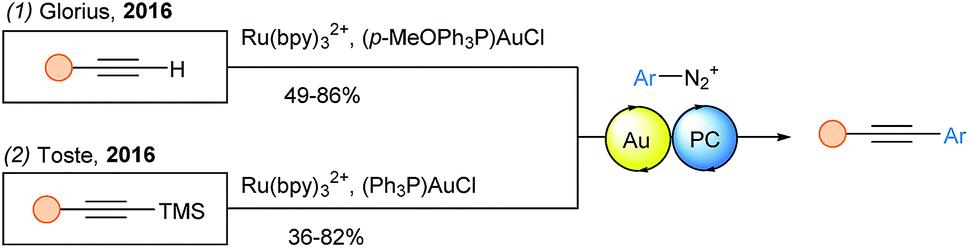 | ||
| Scheme 18 Dual gold-photoredox catalyzed Sonogashira type coupling. | ||
In 2015, Hashmi and coworkers documented the use of the gold photocatalyst [Au2(μ-dppm)2](OTf)216 for the photoredox catalyzed alkynylation of α-amino C–H bonds with iodoalkynes (Scheme 19, eqn (1)).58 The excited state of the gold species is strongly reducing (−1.5 to −1.7 V). Therefore, the authors proposed an oxidative quenching by the 1-iodoalkyne (−1.29 V vs. Fc in MeCN), followed by regeneration of the ground state of the photocatalyst upon reduction by the tertiary amine. Finally, radical coupling (eqn (2)) between the α-amino radical (long lifetime) and the alkynyl radical (short lifetime) is envisioned. In 2019, Chan and coworkers reported the alkynylation of THQs derivatives using alkynylbromides and the same gold catalyst as reported by Hashmi.59
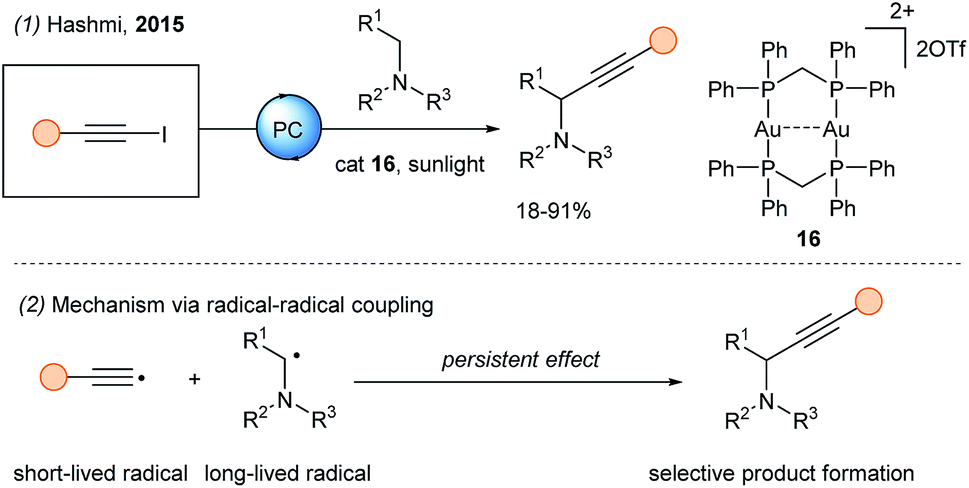 | ||
| Scheme 19 Gold photoredox catalyzed alkynylation using 1-iodo alkynes. | ||
An alternative mechanism in which an α-addition/β-elimination process is operative has also been proposed for other alkynylations using halogenoalkynes reagents, such as the alkynylation of (per)fluoroalkyl radicals or Hantzsch esters.60
Alkynylsulfone reagents were not used before 2015 under photoredox conditions. Since then, they have been particularly successful in oxidative quenching transformations, which start with the oxidation of the excited state of the photocatalyst (Scheme 20). Chen and coworkers first used alkynyl sulfones for the alkynylation of alkyl radicals starting from N-(acyloxy)phthalimides (eqn (1)).61 The reaction is fast (30 min) and a broad variety of carboxylic acids are tolerated. Interestingly, this reaction is compatible with biomolecules. Using the same approach, Fu reported in 2016 the functionalization of side chains for the synthesis of chiral unnatural amino acids.62 Later König and coworkers improved the reaction of Chen by using Eosin Y as a replacement of Ru(bpy)32+.63 In 2016, Fu and coworkers harnessed the radical fragmentation of N-phthalimidoyl oxalates, first introduced by Overman,64 for the preparation of alkynylated quaternary centers (eqn (2)).65 In 2018, the Gryko lab developed a metal free deaminative alkynylation using reaction conditions similar to those of König (eqn (3)).66 The use of alcohols and primary amines greatly expanded the scope of radical precursors that can be used in SOMOphilic alkynylation.
 | ||
| Scheme 20 Photoredox catalyzed alkynylation using alkynylsulfones. | ||
Arylalkynylsulfones were also used in a radical cyclization/alkynylation cascade by Rueping and coworkers (Scheme 21).67 Upon a PCET event realized by fine tuning the photocatalyst and base ({Ir(dFCF3ppy)2bpy}PF6 and NBu4(OMe)2PO2 is the best combination), a nitrogen-centered radical is generated and can easily add onto alkenes to form a 5-membered ring along with an alkyl radical, which reacts with alkynylsulfones. Another example of photoredox catalyzed difunctionalization of alkenes using arylalkynylsulfones is the three components alkynyl difluoroalkylation reported by Zhu.68
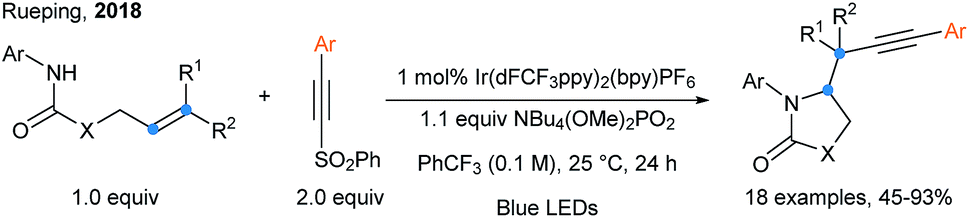 | ||
| Scheme 21 Intramolecular aminoalkynylation of alkenes using alkynylsulfones. | ||
Finally, a major impact of the use of photoredox catalysis for radical alkynylation was realized using EBX reagents. In 2014, a breakthrough was made by Chen and coworkers: they introduced EBX as suitable reagent for the photoredox catalyzed alkynylation of organotrifluoroborate salts using Ru(bpy)3(PF6)2 (Scheme 22).69 An excess of trifluoroborates (3.0 equiv.) was needed for some specific alkynylations, for example with alkyl or TIPS substituents. The method developed was very robust and could be even run in phosphate buffers in presence of biomolecules such as proteins and DNA.
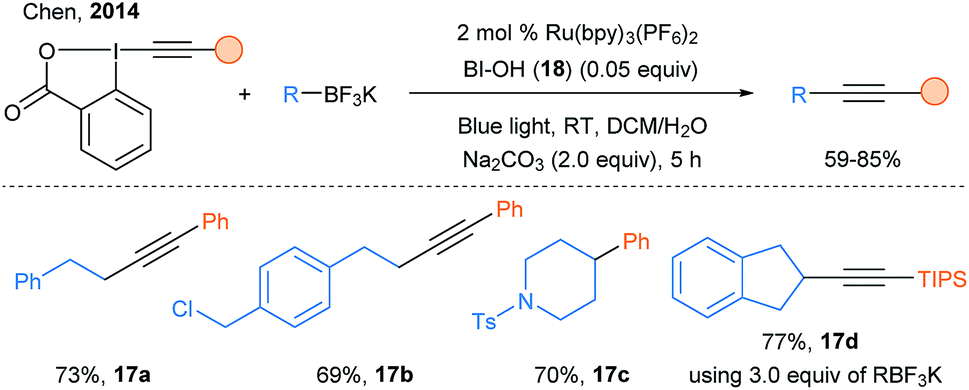 | ||
| Scheme 22 Chen's photoredox catalyzed deboronative alkynylation. | ||
The reaction starts upon homolytic cleavage of a catalytic amount of hypervalent iodine reagent 18 (BIOH, 0.05 equiv.), and then turnover is possible due to the generation of a benziodoxolonyl radical I from the EBX reagent (Scheme 23). Based on Stern–Volmer studies, and the fact that I should be easily reduced, oxidative quenching of Ru(II)*, would give 2-iodobenzoate 19 along with Ru(III). Oxidation of potassium organotrifluoroborate salts by the strongly oxidizing Ru(III) generates alkyl radicals, which can then be alkynylated. An α-addition – β-elimination pathway was proposed by the authors and labelling experiments with a 13C reagent confirmed the regioselectivity of the radical addition.
 | ||
| Scheme 23 Proposed mechanism for the deboronative alkynylation using photoredox catalysis and EBXs. | ||
After this first work, the visible light driven decarboxylative alkynylation of α-keto acids was documented independently by two groups (Scheme 24). A cooperative system using photoredox catalysis, EBX reagents and the stoichiometric oxidant BIOAc 20 was described by Chen (eqn (1));70 while catalytic amount of BIOH 18 in combination with bromoalkynes under sunlight irradiation was used by Wang (eqn (2)).71 The authors proposed an in situ generation of EBX reagents.
 | ||
| Scheme 24 Photoredox catalyzed decarboxylative alkynylation of oxamic and keto-acids using hypervalent iodine reagents. | ||
In 2015, our group72 and Xiao group73 reported independently the decarboxylative alkynylation of free carboxylic acids under visible light irradiation with an iridium photocatalyst using EBX reagents (Scheme 25). This transformation was very general allowing good yields for the transfer of silyl-, aryl- and alkyl-substituted alkynes. α-amino and α-oxy acids were the best substrates, while synthetically useful yields could still be achieved with less reactive aliphatic carboxylic acids. Xiao and coworkers also reported a decarboxylative carbonylative alkynylation under high pressure of CO (60 bars) to access ynone products. Cheng and coworkers described a metal free variation of this transformation using DCA as organic dye.74
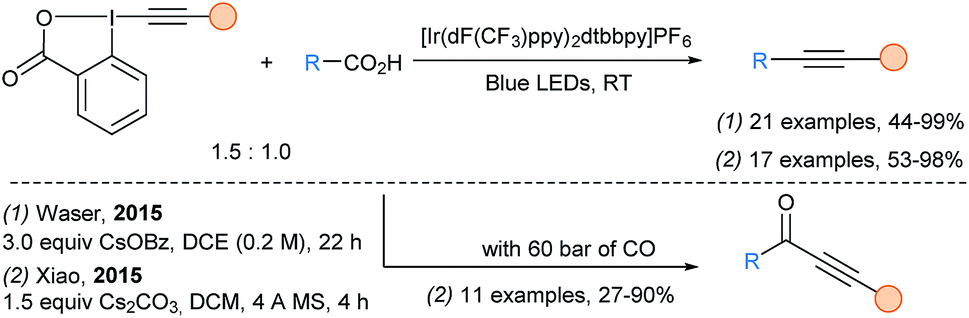 | ||
| Scheme 25 Photoredox catalyzed decarboxylative (carbonylative) alkynylation using EBX reagents. | ||
Using photoredox catalyzed decarboxylative alkynylation, our group developed in 2019 the C-terminal selective bioconjugation of peptides (Scheme 26).75 The use of tetra(carbazolyl)isophthalonitrile (4CzIPN) as dye allowed a metal free, fast (30 min) and selective C-terminus alkynylation on peptides up to hexamers. Introduction of alkynes bearing bioorthogonal groups such as azides or terminal alkynes was possible, and a broad functional group tolerance was observed.
 | ||
| Scheme 26 C-terminal bioconjugation of peptides using photoredox catalyzed decarboxylative alkynylation. | ||
In 2016, Chen and coworkers described the remote alkynylation of tertiary cyclopropanols and cyclobutanols using hypervalent iodine activation in combination with photoredox catalysis (Scheme 27, eqn (1)).76 The formation of a highly reactive O-centered radical triggered a β-scission and released the β- or γ- alkylketone radical. The β-scission was also successful in the case of non cyclic alcohols with two aryl substituents in α position. Soon after, the same strategy was employed to access acyl radical en route to ynones using reagent 21 (eqn (2)).77 In 2018, phosphorous-centered radicals were generated using a similar β-scission of O-centered radical to access phosphonoalkynes.78
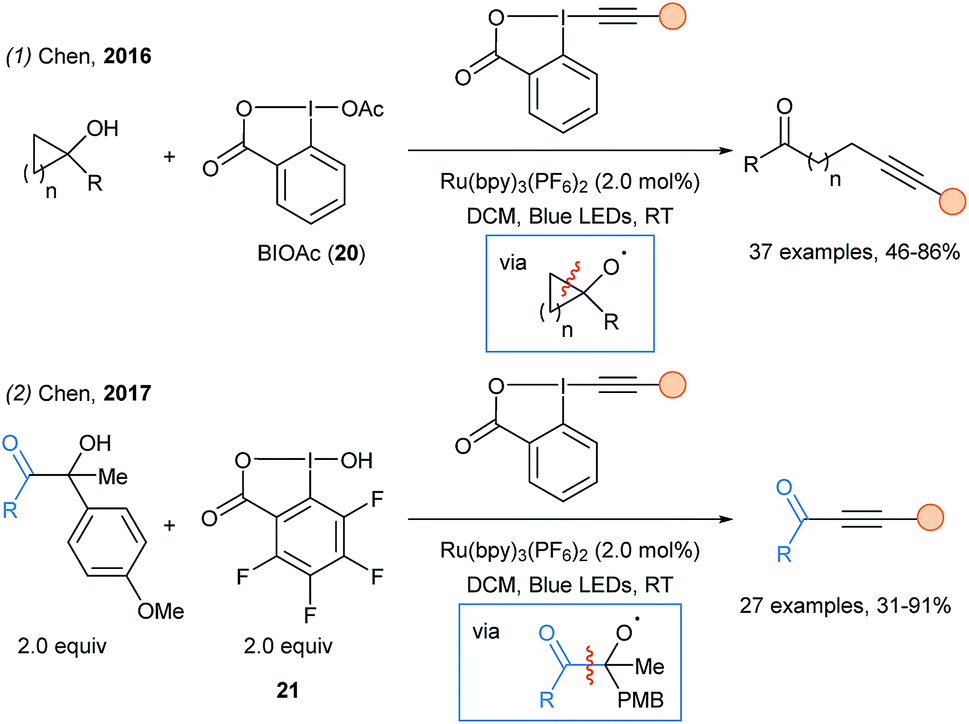 | ||
| Scheme 27 Photoredox catalyzed alkynylation via β-scission of O radicals. | ||
In 2017, Glorius and coworkers harnessed the potential of the (iodo)benzocarboxyl radical as HAT reagent in the photoredox catalyzed C–H alkynylation of aldehydes and formamides using EBX reagents (Scheme 28).79
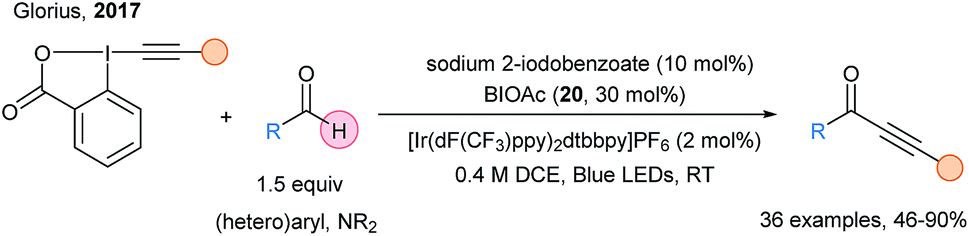 | ||
| Scheme 28 Ynones synthesis via photoredox alkynylation of aldehydes. | ||
In 2017, new transformations based on the reactivity of nitrogen-centered radicals were developed. First, Leonori and coworkers reported iminyl radical driven cascades under photoredox catalysis (Scheme 29).80 Photoredox oxidation of the carboxylic acid activating group gives radical I. Decarboxylation, acetone extrusion, and cyclization of iminyl radical II forms a alkyl radical III. The latter is nucleophilic and therefore well suited for further functionalization with various somophilic reagents, especially EBXs for the efficient transfer of alkynes.
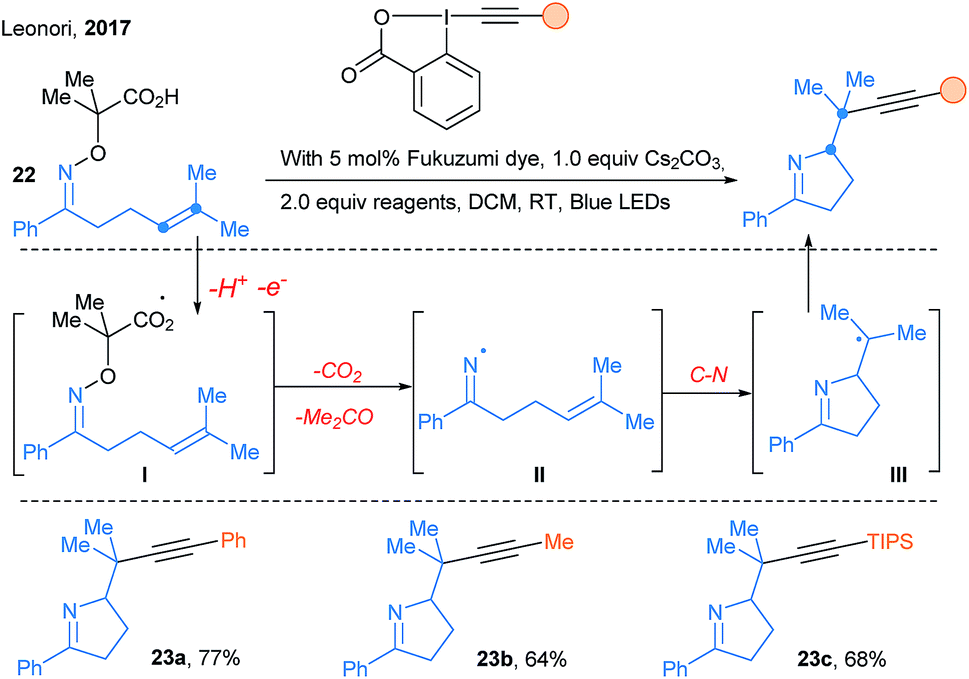 | ||
| Scheme 29 Use of EBX reagents in photoredox catalyzed cascades by Leonori. | ||
Soon after, the remote alkynylation of oxime ethers upon photoredox catalyzed cascade (decarboxylation/fragmentation/alkynylation) was successfully reported by our group using a similar activation strategy (Scheme 30, eqn (1)).81 The best yield of alkynylnitrile was obtained with the new chlorinated 4ClCzIPN photocatalyst (24). Simultaneously, Castle and coworkers have developed the microwave-assisted fragmentation of oximes ethers (eqn (2)).82 Only one example was reported using EBX, delivering δ-ethynylnitrite 26 in 54% yield.
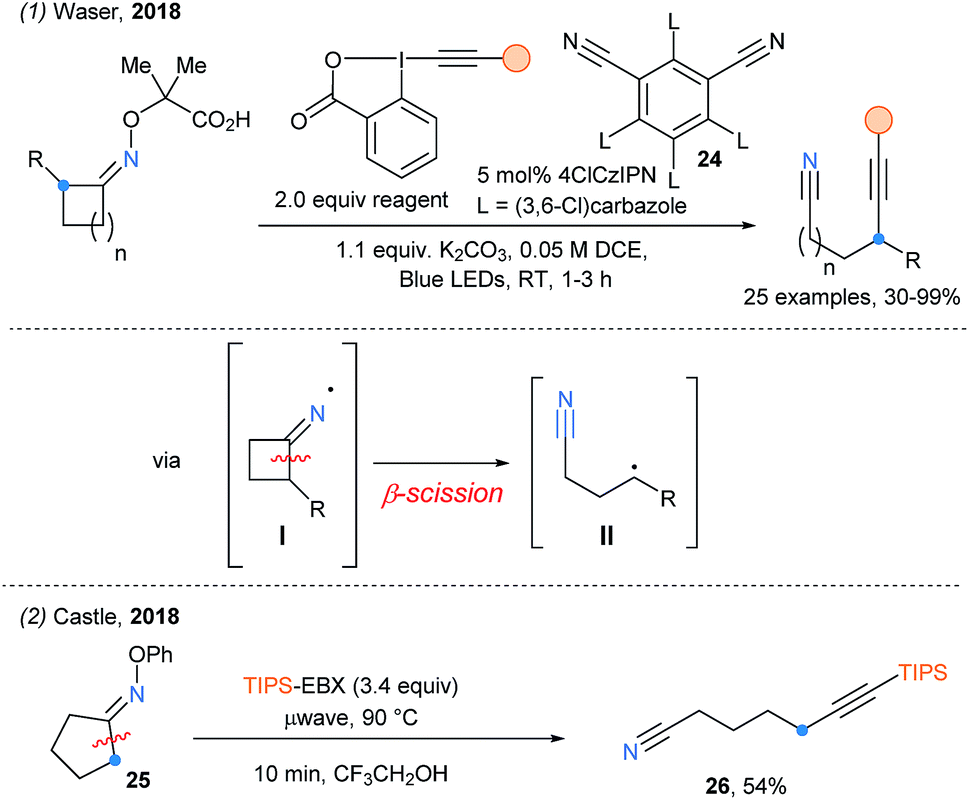 | ||
| Scheme 30 Iminyl radical initiated fragmentation for remote alkynylation. | ||
Leonori and coworkers successfully developed a remote alkynylation of aliphatic amides (Scheme 31, eqn (1)).83 The generated amidyl radical is used to abstract the hydrogen at the delta position (1,5-HAT). The resulting nucleophilic δ-amidoalkyl radical can finally be alkynylated using EBX reagents. A MCR aminoalkynylation of alkenes was achieved by Studer and coworkers using the same activating group in 2019 (eqn (2)).84
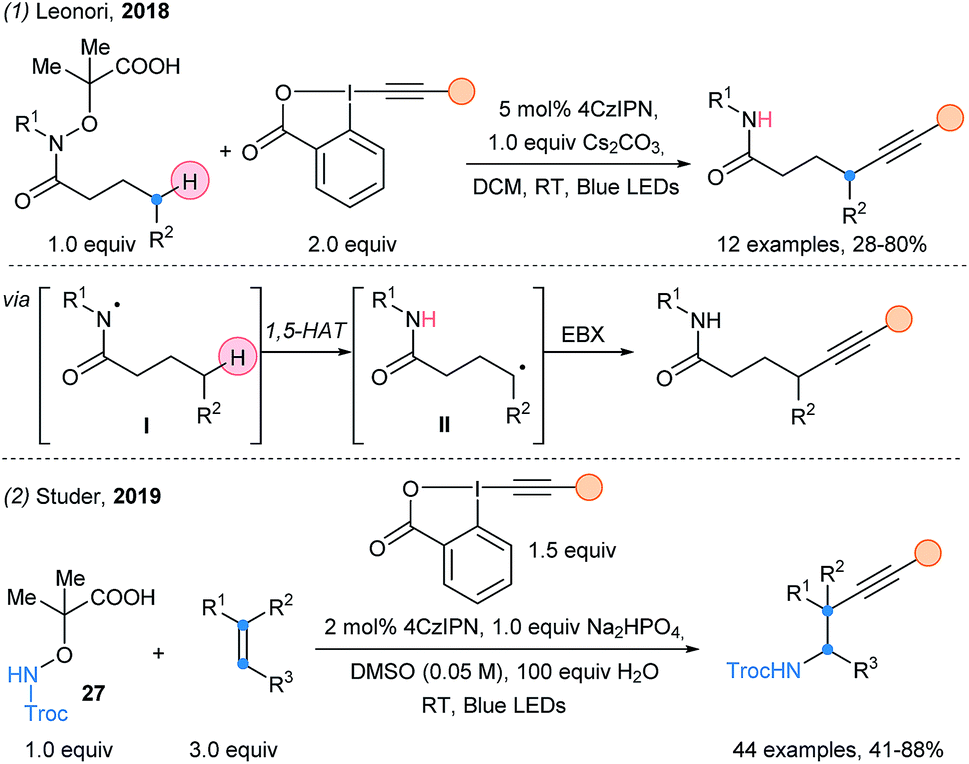 | ||
| Scheme 31 Use of EBX reagents in photoredox catalyzed cascades by Leonori and Studer. | ||
The mechanism of the reaction of alkyl radicals with EBX reagents was studied by our group in 2017 using DFT calculations (Scheme 32).85 Depending on the substituent of the alkyne, two possible transition states involving the iodine atom and different carbons of the alkyne could be operative: (1) TS-I with the C-α atom of the alkyne (a); direct product formation is favored, without a vinyl radical intermediate I as proposed previously; (2) TS-II with the C-β atom of the alkyne (b); an addition/elimination sequence viaII would form a carbene III, which upon subsequent 1,2-shift would lead to the alkyne product. An additional SET event base on the oxidative properties of hypervalent iodine reagents was also calculated (TS-III). However, the collapse of radical anion IV into the iodanyl radical and the acetylide V is too high in energy to be competitive. Sterics and electronics of both the radical and the substituent on the EBX reagents are crucial to determine which pathway is operative. For example, Chen and coworkers did a C13 labelling experiment in their photoredox catalyzed deboronative alkynylation using Ph-EBX showing that the reaction proceeds via α-addition.69 They then developed modified reagents in order to increase the radical transfer ability of EBX reagents, with the best result obtained when introducing two methoxy groups on the benzene ring.86
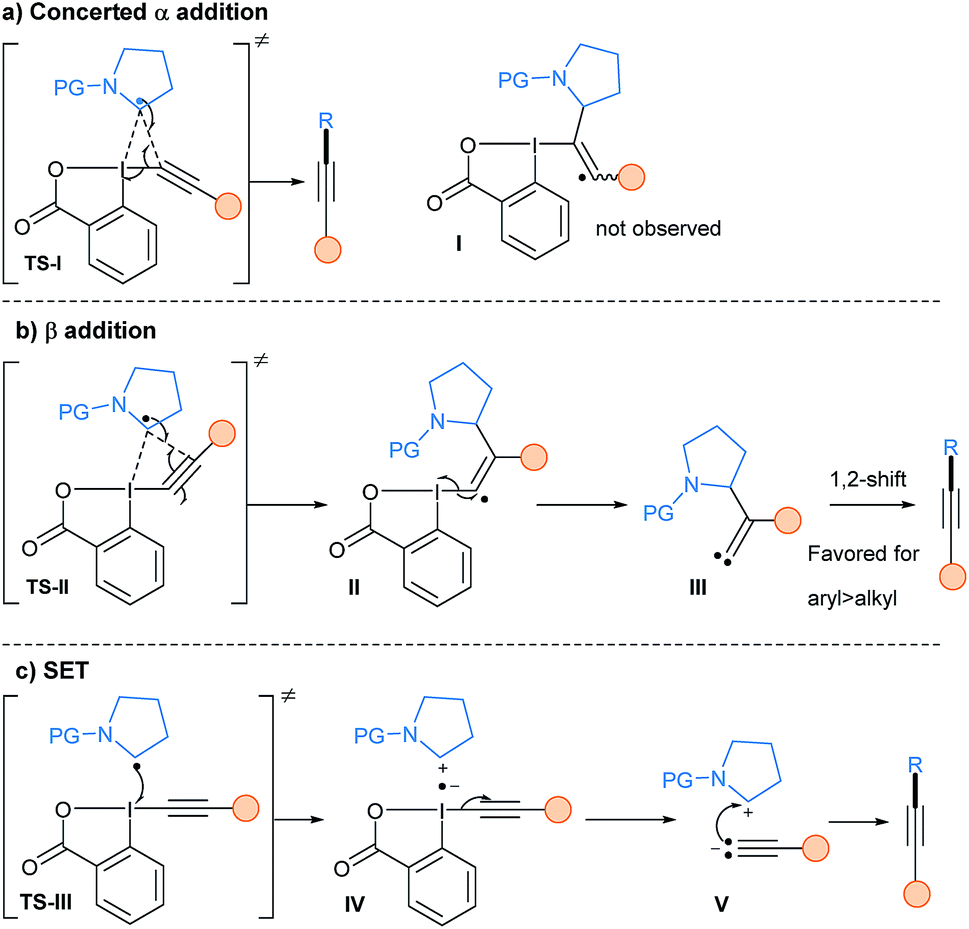 | ||
| Scheme 32 Mechanistic details of the alkynylation step of alkyl radicals using EBX reagents involving either a concerted α-addition (a), a β-addition (b) or a SET pathway (c). | ||
4. Transition metal catalyzed alkynylation of alkyl radicals
During the last few years, the use of transition metals that can promote SET events has known a growing interest and constitutes an interesting alternative to photoredox chemistry. Transition metals have been used for radical generation only, or in a dual function of radical generation followed by radical capture and alkynylation upon reductive elimination. Oshima and coworkers reported the radical alkynylation of alkyl iodides using a cobalt catalyst and ethynylmagnesium bromide reagents in 2006 (Scheme 33).87 A SET event between the iodides and the Co catalyst releases the alkyl radical, which can then be trapped by the low valent Co species. Finally, transmetallation with the Grignard reagents followed by reductive elimination affords the alkyne products.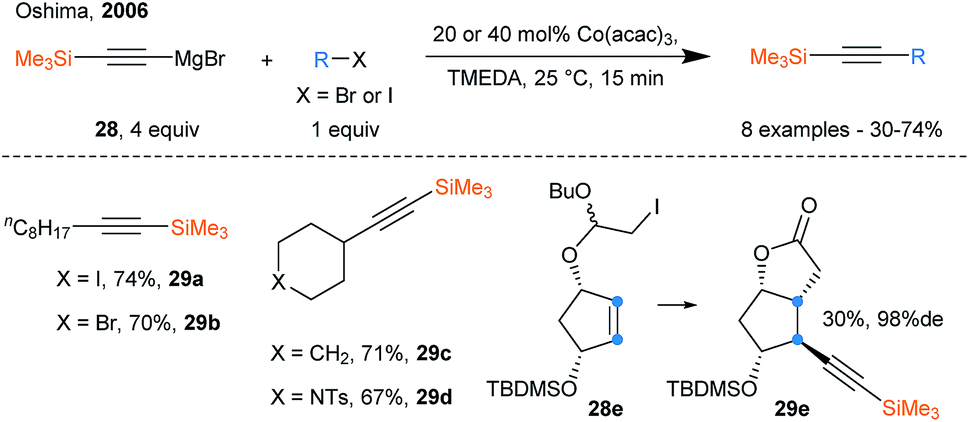 | ||
| Scheme 33 SOMOphilic alkynylation using ethynylmagnesium bromide reagents and Co catalyst. | ||
A decade later, nickel has emerged as one of the most promising metal for such strategy. By fine tuning the ligands, the redox properties of the Ni complexes can be optimized, thus allowing efficient SET events. Radical capture can then occur, giving access to Ni(III) species prone to reductive elimination. Weix and coworkers reported a cross coupling of two electrophiles: N-acyloxyphthalimides and bromoalkynes (Scheme 34, eqn (1)).88 Regeneration of the catalyst was realized by reduction with Mn. Simultaneously, Baran and coworkers used nucleophilic sources of alkynes to alkynylate alkyl radicals generated after reduction of N-acyloxyphthalimides by a transition metal (eqn (2)).89 Two sets of conditions were reported: (1) a nickel catalyst and alkynylzincates were found to be efficient for the preparation of terminal alkynes; (2) the combination of an iron catalyst and Grignard reagents at −15 °C allowed the synthesis of internal alkynes.
 | ||
| Scheme 34 Nickel- and iron-catalyzed alkynylation of N-acyloxyphthalimides. | ||
Lei and coworkers reported in 2016 a Ni/Cu/Ag system for Csp3–H/Csp–H coupling with a broad scope of terminal alkynes (Scheme 35, eqn (1)).90 However, the alkyl substrates were used as cosolvent to ensure good yields. The same group reported latter the alkynylation of various alkyl radicals using a similar multimetallic system (eqn (2)).91 The use of copper and silver salts is crucial for the generation of the alkyl radicals and acetylides. Then, an iron or nickel catalyst is required to promote coupling, affording the alkynylated products.
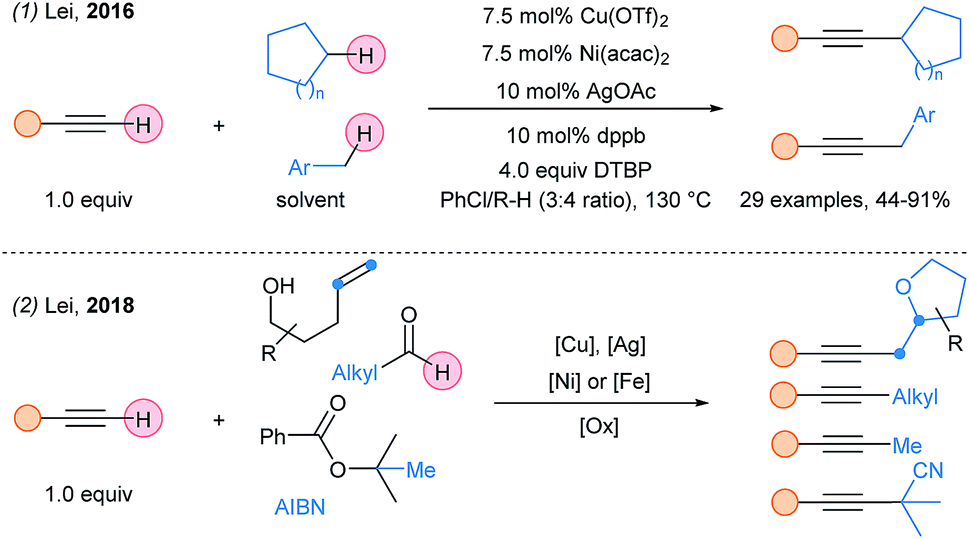 | ||
| Scheme 35 Multimetallic system for alkyl–alkynes cross coupling using terminal alkynes. | ||
In 2017, a hydroalkynylation of alkenes was reported by Cui and Liu using Fe(acac)3 with silanes and bromoalkynes (Scheme 36, eqn (1)).92 An α-addition/β-elimination sequence was proposed for the alkynylation step. In 2018, Liu and coworkers described a remote alkynylation method starting from alkylhydroperoxides and acetylenic triflones (eqn (2)).93 Finally, Shi and coworkers developed the iron-catalyzed Cα radical alkynylation of tertiary amines (eqn (3)).94
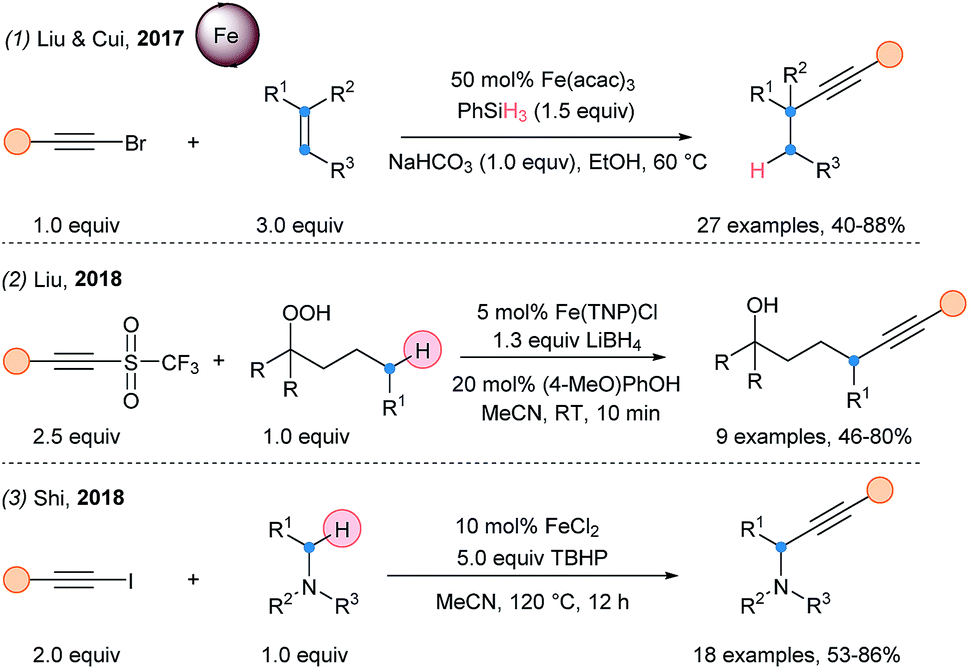 | ||
| Scheme 36 Recent iron-catalyzed SOMOphilic alkynylations. | ||
Wang and Shen described the aminoalkynylation of alkenes based on the copper catalyzed cyclization of tethered alkenyl Weinreb amides (Scheme 37, eqn (1)).95 Radical intermediates could be generated through the thermal homolytic cleavage of alkyl–Cu(II) species. The radical nature of the reaction was supported by TEMPO-radical trap experiments as well as radical clock experiments. In 2018, an extension of this work was reported for the radical cyclization alkynylation of unsaturated ketoximes (eqn (2) & (3)).96 Depending of the position of the oxime (β–γ or γ–δ), either C–O bond or C–N bond formation was obtained.
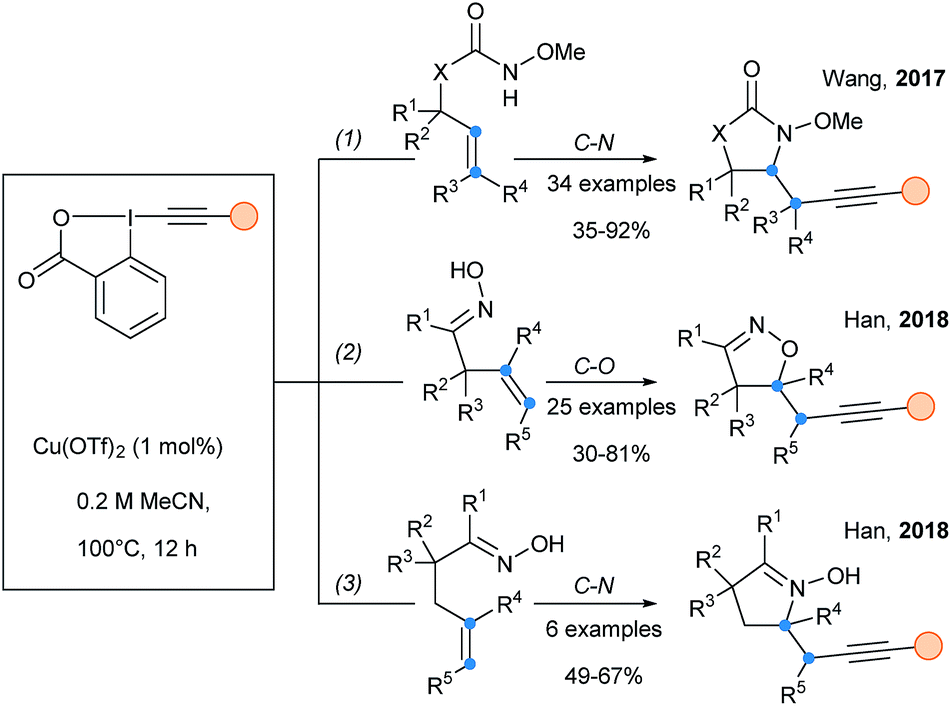 | ||
| Scheme 37 Copper-catalyzed radical cyclization–alkynylation of alkenes. | ||
In 2016, Zhu and coworkers reported the use of a Mn(III) catalyst with Dess–Martin Periodinane or PIDA as oxidant for the cleavage of cyclobutanols at room temperature yielding nucleophilic γ-alkylketone radicals (Scheme 38, eqn (1)).97 In presence of acetylenic sulfones, efficient alkynylation was observed. In 2018, Maruoka and coworkers developed a fragmentation/alkynylation cascade of larger cycloalkanols using a copper catalyst and preoxidized starting materials (cycloalkylsilylperoxides) (eqn (2)). Interestingly, terminal alkynes could be use in this transformation, and one example of non-cyclic substrate is reported.98
 | ||
| Scheme 38 Manganese- and copper-catalyzed radical fragmentation-alkynylation cascade. | ||
5. Asymmetric methodologies
Inoue and coworkers reported in 2015 an extension of their radical alkynylation using alkynylsulfones and a photosensitizer with chiral reagents leading to propargyl amines with enantiomeric ratio up to 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11.99 However, this approach requires stoichiometric amount of the chiral sulfoximine reagents, which are synthesized in several steps. Concerning catalytic approaches, Li and coworkers described in 2015 the copper-catalyzed addition of acetylides on iminiums generated in situ from THQ compounds using photoredox catalysis (Scheme 15, p. 5).50 In 2017, Zhang and Luo reported the asymmetric alkynylation of activated carbonyl compounds using dual organophotoredox catalysis with hypervalent iodine reagents in combination with a chiral amine catalyst (Scheme 39).100 Condensation of chiral amine catalyst 30 with a β-keto ester forms first an enamine, which upon oxidation and deprotonation can delivered a chiral α-iminyl radical II. Simultaneously, in situ formation of highly reactive carboxylate hypervalent iodine reagents I from ynoic acids and hydroxybenziodoxolone 18 is important: radical alkynylation may be assisted by H-bonding, thus delivering the alkynes with high enantiomeric excess, albeit in low to medium yields.
:11.99 However, this approach requires stoichiometric amount of the chiral sulfoximine reagents, which are synthesized in several steps. Concerning catalytic approaches, Li and coworkers described in 2015 the copper-catalyzed addition of acetylides on iminiums generated in situ from THQ compounds using photoredox catalysis (Scheme 15, p. 5).50 In 2017, Zhang and Luo reported the asymmetric alkynylation of activated carbonyl compounds using dual organophotoredox catalysis with hypervalent iodine reagents in combination with a chiral amine catalyst (Scheme 39).100 Condensation of chiral amine catalyst 30 with a β-keto ester forms first an enamine, which upon oxidation and deprotonation can delivered a chiral α-iminyl radical II. Simultaneously, in situ formation of highly reactive carboxylate hypervalent iodine reagents I from ynoic acids and hydroxybenziodoxolone 18 is important: radical alkynylation may be assisted by H-bonding, thus delivering the alkynes with high enantiomeric excess, albeit in low to medium yields.
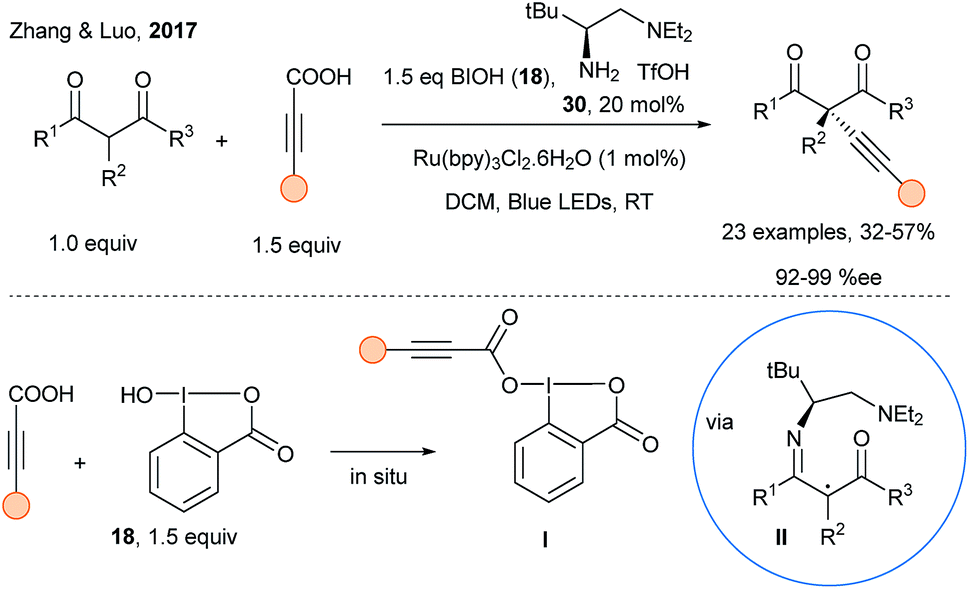 | ||
| Scheme 39 Asymmetric alkynylation using organocatalysis, photoredox catalysis and hypervalent iodine reagents. | ||
Finally, Liu and coworkers reported an enantioselective trifluoromethylalkynylation of alkenes using Togni I reagent (12), alkynyl–Si(OMe)3, Cu(CH3CN)4BF4 and Box ligand 31 (Scheme 40).101 Upon a SET event, a CF3 radical is generated and adds onto the styrene derivative, affording a benzylic radical. The chiral Cu(II) acetylide complex is then able to capture the benzylic radical, thus leading to a highly reactive Cu(III) intermediate I. Finally, fast reductive elimination allows the formation of chiral benzylic alkynes in good yield and high ee.
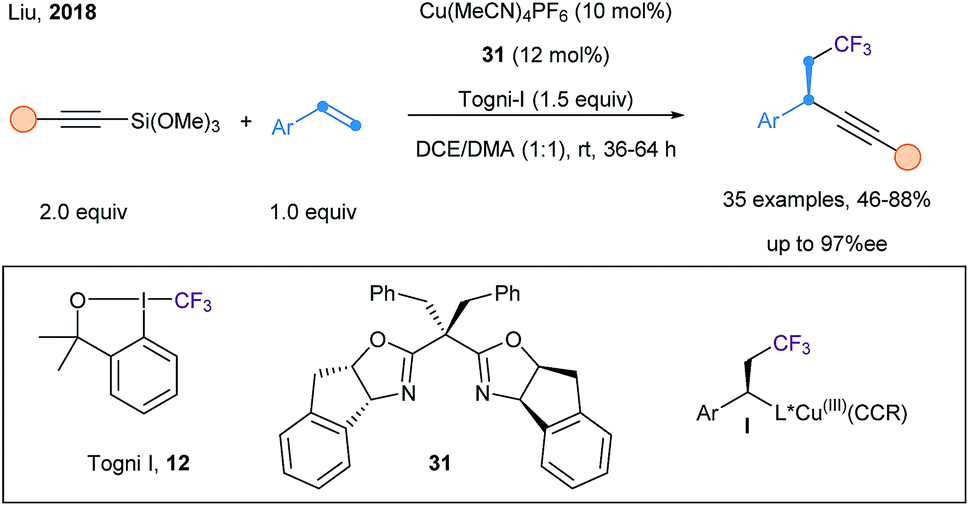 | ||
| Scheme 40 Catalytic asymmetric trifluoromethylalkynylation of alkenes using a copper catalyst and a Box ligand 30. | ||
6. Migration
A very recent strategy for radical alkynylation is based on alkynyl migration. It has been first introduced independently by Zhu102 and Studer,103 in a visible light driven alkynyl migration starting from tertiary propargylic alcohols (Scheme 41). The trifluoromethylalkynylation of alkenes can be achieved using the photoredox catalyst fac-Ir(ppy)3 and Umemoto's reagent (32) (eqn (1)), while the perfluoroalkyl alkynylation of alkenes is realized using perfluoroalkyliodides and DABCO, via a radical chain propagation (eqn (2)). Later, using the same strategy, Xie and Zhu extended their work to the difluoroalkylalkynylation of alkenes using various bromine reagents.104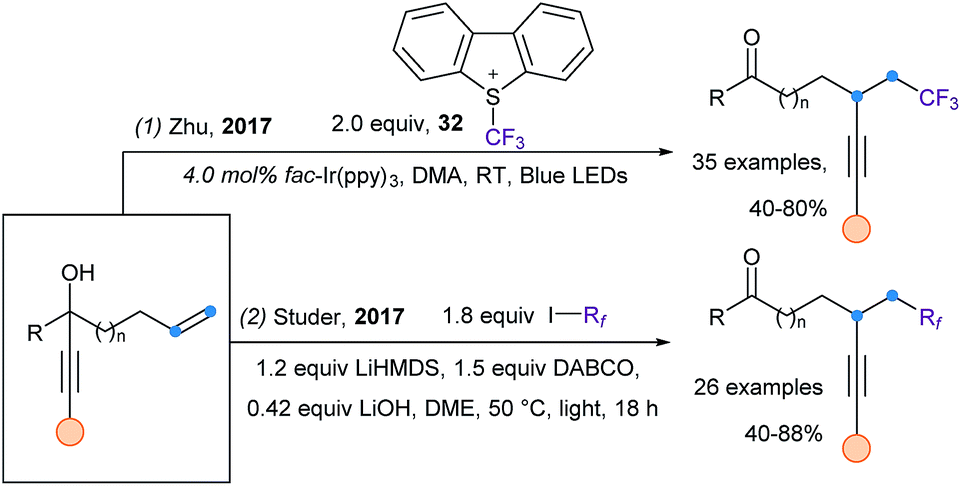 | ||
| Scheme 41 Visible light mediated 1,4- and 1,5-alkynyl migration. | ||
In 2018, Jiang and coworkers reported a 1,2 alkynyl migration using 1,4-enynes and cycloalkanes (Scheme 42, eqn (1)).105 The proposed mechanism included an “anti-Baldwin” 3-exo-dig cyclization for the alkynyl migration. A catalytic system composed of iron(II) chloride and DTPB at elevated temperature allowed the generation of alkyl radicals upon a HAT process. A 1,3-alkynyl migration was then reported for the stereospecific synthesis of (Z)-2-amino conjugated enynals/enynones (eqn (2)).106 This transformation is promoted by a copper catalyst and NFSI derivatives. A radical aminoalkynylation–oxidation cascade was proposed by the authors.
 | ||
| Scheme 42 1,2 and 1,3-alkynyl migration of propargyl alcohols. | ||
Promising preliminary results have been recently obtained using electrochemical methods as an alternative to photoredox catalysis. Pan and coworkers used it for the sulfonylation/alkynylation of unactivated alkenes via 1,4-alkynyl migration (Scheme 43).107
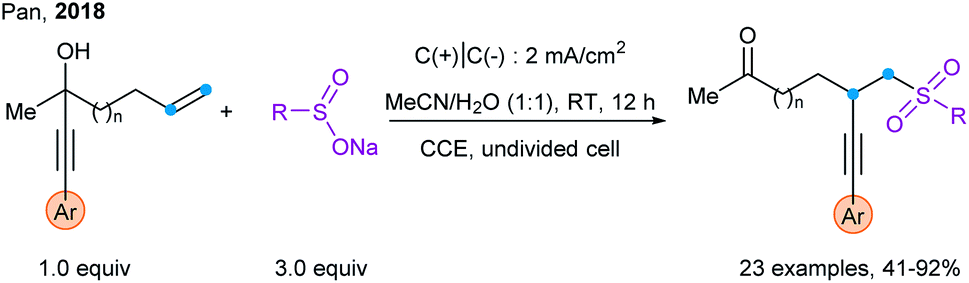 | ||
| Scheme 43 Electrochemical method for the sulfonylation–alkynylation of unactivated alkenes via 1,4-alkynyl migration. | ||
7. Conclusion/outlook
In Fig. 3, a chronological representation of our personal choice of milestones achieved in radical alkynylation is presented. The first alkynylation of radical was reported in 1986 by Russel using sulfone reagents and UV light. In the 90's, Fuchs and coworkers did important developments for the alkynylation of either C–H or C–I bond, including mechanistic studies and applications in total synthesis. In 2002, Oshima and coworkers developed organo–gallium and –indium reagents for the alkynylation of electrophilic radicals. In 2012, EBX reagents were introduced as radical alkynylating reagents, together with broadly applicable oxidative decarboxylative conditions. Since then, the number of reports of alkynylations has known an exponential growth. This is also due to two other important breakthroughs in radical generation based on photoredox and non-redox innocent transition metal catalysis (Cu, Co, Ni, Fe). These novel modes of activation allowed new applications such as the asymmetric alkynylation of radicals and the functionalization of peptides. | ||
| Fig. 3 Personal choice of important breakthroughs in SOMOphilic alkynylation over the last three decades. | ||
With this impressive progress, the field of radical alkynylation is not anymore in its infancy. Nevertheless, breakthroughs are still needed, especially for:
(i) Broadly applicable enantioselective transformations.
(ii) Applications on complex (bio)molecules such as natural products and peptides/proteins.
(iii) Atom-economical transformations directly from terminal alkynes in cases currently limited to the use of pre-formed reagents.
(iv) Multi-bond forming processes, such as cascade and multi-component reactions, giving access to more complex, natural-product- or drug-like scaffolds.
(v) More in-depth mechanistic studies.
(vi) The use of other techniques to generate and control the reactivity of radicals, such as electro- and flow-chemistry, the latter especially in the case of photoredox-based methods.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the ERC (European Research Council, Starting Grant iTools4MC, number 334840) and EPFL for financial support.Notes and references
- (a) B. I. Morinaka, C. K. Skepper and T. F. Molinski, Org. Lett., 2007, 9, 1975 CrossRef CAS PubMed; (b) S. Nickel, R. A. Serwa, F. Kaschani, S. Ninck, S. Zweerink, E. W. Tate and M. Kaiser, Chem.–Eur. J., 2015, 21, 10721 CrossRef CAS PubMed.
- (a) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS PubMed; (b) J. Lehmann, M. H. Wright and S. A. Sieber, Chem.–Eur. J., 2016, 22, 4666 CrossRef CAS PubMed.
- D. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry: Chemistry, Biology and Material Science, ed, D. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH, 2005 Search PubMed.
- (a) H. H. Inhoffen and W. Hohlweg, Naturwissenschaften, 1938, 26, 96 CrossRef CAS; (b) J. W. Corbett, S. S. Ko, J. D. Rodgers, L. A. Gearhart, N. A. Magnus, L. T. Bacheler, S. Diamond, S. Jeffrey, R. M. Klabe, B. C. Cordova, S. Garber, K. Logue, G. Trainor, P. S. Anderson and S. K. Erickson-Vittanen, J. Med. Chem., 2000, 43, 2019 CrossRef CAS PubMed; (c) A. Bianchetti, A. Lavezzo and P. J. Carminati, J. Pharm. Pharmacol., 1982, 34, 51 CrossRef CAS PubMed; (d) Z. Fisar, J. Hroudová and J. Raboch, Neuroendocrinol. Lett., 2010, 31, 645 CAS.
- B. M. Trost and C.-J. Li, Modern Alkynes Chemistry: Catalytic and Atom-Economic Transformations, ed, B. M. Trost and C.-J. Li, Wiley-VCH, Weinheim, 2014, p. 424 Search PubMed.
- (a) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2004, 4095 CrossRef CAS; (b) B. M. Trost and A. H. Weiss, Adv. Synth. Catal., 2009, 351, 963 CrossRef CAS PubMed.
- (a) Metal-Catalyzed Cross-Coupling Reactions, ed. A. De Meijere and F. Diederich, Wiley-VCH, 2nd edn, 2004 Search PubMed; (b) K. Sonogashira, Chapter 2.5 – Coupling Reactions Between sp Carbon Centers in Comprehensive Organic Synthesis, Elsevier, 1991, vol. 3, p. 551 Search PubMed.
- D. Seebach, Angew. Chem., Int. Ed., 1979, 18, 239 CrossRef.
- Selected examples: (a) F. M. Beringer and S. A. Galto, J. Org. Chem., 1965, 30, 1930 CrossRef CAS; (b) M. Ochiai, M. Kunishima, Y. Nagao, K. Fuji, M. Shiro and E. Fujita, J. Am. Chem. Soc., 1986, 108, 8281 CrossRef CAS; (c) D. Fernández González, J. P. Brand and J. Waser, Chem.–Eur. J., 2010, 16, 9457 CrossRef PubMed.
- For reviews, see: (a) J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165 RSC; (b) Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436 CrossRef CAS PubMed; (c) J. Waser, Synlett, 2016, 27, 2761 CrossRef CAS; (d) D. P. Hari, P. Caramenti and J. Waser, Acc. Chem. Res., 2018, 51, 3212 CrossRef CAS PubMed.
- (a) G. Martelli, P. Spagnolo and M. Tiecco, J. Chem. Soc. D, 1969, 282 RSC; (b) G. Martelli, P. Spagnolo and M. Tiecco, J. Chem. Soc. B, 1970, 1413 RSC; (c) K. M. Erwin, S. Gronert, S. E. Barlow, M. K. Gilles, A. G. Harrison, V. M. Bierbaum, C. H. DuPuy, W. C. Lineberger and G. B. Ellison, J. Am. Chem. Soc., 1990, 112, 5750 CrossRef; (d) M. S. Robinson, M. L. Polek, V. M. Bierbaum, C. H. DuPuy and W. C. Lineberger, J. Am. Chem. Soc., 1995, 117, 6766 CrossRef CAS; (e) P. Boutillier and S. Z. Zard, Chem. Commun., 2001, 1304 RSC.
- (a) G. A. Russel and P. Ngoviwatchai, Tetrahedron Lett., 1986, 27, 3479 CrossRef; (b) G. A. Russel, P. Ngoviwatchai, H. I. Tashtoush, A. Dalmau and R. K. Khanna, J. Am. Chem. Soc., 1988, 110, 3530 CrossRef; (c) G. A. Russel and P. Ngoviwatchai, J. Org. Chem., 1989, 54, 1836 CrossRef.
- (a) J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1996, 118, 4486 CrossRef CAS; (b) J. Xiang, W. Jiang and P. L. Fuchs, Tetrahedron Lett., 1997, 38, 6635 CrossRef CAS.
- J. S. Xiang and P. L. Fuchs, Tetrahedron Lett., 1996, 37, 5269 CrossRef CAS.
- J. Gong and P. L. Fuchs, Tetrahedron Lett., 1997, 38, 787 CrossRef CAS.
- J. Xiang and P. L. Fuchs, Tetrahedron Lett., 1998, 39, 8597 CrossRef CAS.
- J. Bian, M. Van Wingerden and J. M. Ready, J. Am. Chem. Soc., 2006, 128, 7428 CrossRef CAS PubMed.
- T. M. Brütsch, P. Bucher and K. -H. Altmann, Chem.–Eur. J., 2016, 22, 1292 CrossRef PubMed.
- A.-P. Schaffner, V. Darmency and P. Renaud, Angew. Chem., Int. Ed., 2006, 45, 5847 CrossRef CAS PubMed.
- V. Liautard, F. Robert and Y. Landais, Org. Lett., 2011, 13, 2658 CrossRef CAS PubMed.
- T. Hoshikawa, S. Kamijo and M. Inoue, Org. Biomol. Chem., 2013, 11, 164 RSC.
- S. Yoshioka, M. Nagatomo and M. Inoue, Org. Lett., 2015, 17, 90 CrossRef CAS PubMed.
- S. Paul and J. Guin, Green Chem., 2017, 19, 2530 RSC.
- S. Zhou, T. Song, H. Chen, Z. Liu, H. Shen and C. Li, Org. Lett., 2017, 19, 698 CrossRef CAS PubMed.
- H. Jiang, Y. He, Y. Cheng and S. Yu, Org. Lett., 2017, 19, 1240 CrossRef CAS PubMed.
- L. Wang, Y. Xia, K. Bergander and A. Studer, Org. Lett., 2018, 20, 5817 CrossRef CAS.
- Y. Xia and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 9836 CrossRef CAS PubMed.
- S.-I. Usugi, H. Yorimitsu, H. Shinokubo and K. Oshima, Bull. Chem. Soc. Jpn., 2002, 75, 2687 CrossRef CAS.
- For a review see: K. Takami, S.-I. Usugi, H. Yorimitsu and K. Oshima, Synthesis, 2005, 5, 824 Search PubMed.
- (a) T. Hirashita, A. Hayashi, M. Tsuji, J. Tanaka and S. Araki, Tetrahedron, 2008, 64, 2642 CrossRef CAS; (b) I. Suzuki, K. Kiyokawa, M. Yasuda and A. Baba, Org. Lett., 2013, 15, 1728 CrossRef CAS PubMed; (c) I. Suzuki, N. Esumi, M. Yasuda and A. Baba, Chem. Lett., 2015, 44, 38 CrossRef.
- W. Lu, L. Li and C.-J. Li, Nat. Commun., 2015, 6, 6526 CrossRef PubMed.
- L. Wang, W. Wei, D. Yang, H. Cui, H. Yue and H. Wang, Tetrahedron Lett., 2017, 58, 4799 CrossRef CAS.
- H.-Y. Huang, L. Cheng, J.-J. Liu, D. Wang, L. Liu and C.-J. Li, J. Org. Chem., 2017, 82, 2656 CrossRef CAS PubMed.
- X. Liu, Z. Wang, X. Cheng and C. Li, J. Am. Chem. Soc., 2012, 134, 14330 CrossRef CAS PubMed.
- M. Meng, G. Wang, L. Yang, K. Cheng and C. Qi, Adv. Synth. Catal., 2018, 360, 1218 CrossRef CAS.
- R.-Y. Zhang, L.-Y. Xi, L. Zhang, S. Liang, S.-Y. Chen and X.-Q. Yu, RSC Adv., 2014, 4, 54349 RSC.
- Z.-F. Cheng, Y.-S. Feng, C. Rong, T. Xu, P.-F. Wang, J. Xu, J.-J. Daia and H.-J. Xu, Green Chem., 2016, 18, 4185 RSC.
- (a) X. Liu, L. Yu, M. Luo, J. Zhu and W. Wei, Chem.–Eur. J., 2015, 21, 8745 CrossRef CAS PubMed; (b) R.-Y. Zhang, L.-Y. Xi, L. Zhang, S.-Y. Chen and X.-Q. Yu, Tetrahedron, 2015, 71, 6176 CrossRef CAS; (c) X.-H. Ouyang, R.-J. Song, C.-Y. Wang, Y. Yanga and J.-H. Li, Chem. Commun., 2015, 51, 14497 RSC.
- H. Wang, L.-N. Guo, S. Wang and X.-H. Duan, Org. Lett., 2015, 17, 3054 CrossRef CAS PubMed.
- P.-F. Wang, Y.-S. Feng, Z.-F. Cheng, Q.-M. Wu, G.-Y. Wang, L.-L. Liu, J.-J. Dai, J. Xu and H.-J. Xu, J. Org. Chem., 2015, 80, 9314 CrossRef CAS PubMed.
- (a) F. Chen and A. S. K. Hashmi, Org. Lett., 2016, 18, 2880 CrossRef CAS; (b) X. Li, S. Li, S. Sun, F. Yang, W. Zhu, Y. Zhu, Y. Wu and Y. Wu, Adv. Synth. Catal., 2016, 358, 1699 CrossRef CAS; (c) E. Ismalaj, Q. Glenadel and T. Billard, Eur. J. Org. Chem., 2017, 14, 1911 CrossRef; (d) F. Shen, P. Zhang, L. Lu and Q. Shen, Org. Lett., 2017, 19, 1032 CrossRef CAS PubMed.
- X. Liu, Ru. Liu, J. Dai, X. Cheng and G. Li, Org. Lett., 2018, 20, 6906 CrossRef CAS PubMed.
- Y. Li, R. Lu, S. Sun and L. Liu, Org. Lett., 2018, 20, 6836 CrossRef CAS PubMed.
- S. Wang, L.-N. Guo, H. Wang and X.-H. Duan, Org. Lett., 2015, 17, 4798 CrossRef CAS PubMed.
- R.-Y. Zhang, L.-Y. Xi, L. Shi, X.-Z. Zhang, S.-Y. Chen and X.-Q. Yu, Org. Lett., 2016, 18, 4024 CrossRef CAS PubMed.
- For selected reviews, see: (a) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785 CrossRef CAS PubMed; (b) T. P. Yoon, M. A. Ischay and J. N. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (d) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (e) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97 RSC; (f) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- M. Osawa, H. Nagai and M. Akita, Dalton Trans., 2007, 827 RSC.
- (a) D. B. Freeman, L. Furst, A. G. Condie and C. R. J. Stephenson, Org. Lett., 2012, 14, 94 CrossRef CAS PubMed; (b) M. Rueping, R. M. Koenigs, K. Poscharny, D. C. Fabry, D. Leonori and C. Vila, Chem.–Eur. J., 2012, 18, 5170 CrossRef CAS PubMed; (c) W. Fu, W. Guo, G. Zou and C. Xu, J. Fluorine Chem., 2012, 140, 88 CrossRef CAS.
- J. F. Franz, W. B. Kraus and K. Zeitler, Chem. Commun., 2015, 51, 8280 RSC.
- I. Perepichka, S. Kundu, Z. Hearne and C.-J. Li, Org. Biomol. Chem., 2015, 13, 447 RSC.
- A. Sagadevan and K. C. Hwang, Adv. Synth. Catal., 2012, 354, 3421 CrossRef CAS.
- A. Sagadevan, V. P. Charpe and K. C. Hwang, Catal. Sci. Technol., 2016, 6, 7688 RSC.
- A. Sagadevan, P.-C. Lyub and K. C. Hwang, Green Chem., 2016, 18, 4526 RSC.
- H. Zhang, P. Zhang, M. Jiang, H. Yang and H. Fu, Org. Lett., 2017, 19, 1016 CrossRef CAS PubMed.
- A. Hazra, M. T. Lee, J. F. Chiu and G. Lalic, Angew. Chem., Int. Ed., 2018, 57, 5492 CrossRef CAS PubMed.
- A. Tlahuext-Aca, M. N. Hopkinson, B. Sahoo and F. Glorius, Chem. Sci., 2016, 7, 89 RSC.
- S. Kim, J. Rojas-Martin and F. D. Toste, Chem. Sci., 2016, 7, 85 RSC.
- J. Xie, S. Shi, T. Zhang, N. Mehrkens, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2015, 54, 6046 CrossRef CAS PubMed.
- Y. Zhao, J. Jin and P. W. H. Chan, Adv. Synth. Catal., 2019, 361, 1313 CrossRef CAS.
- (a) N. Iqbal, N. Iqbal, S. S. Han and E. J. Cho, Org. Biomol. Chem., 2019, 17, 1758 RSC; (b) Z.-Y. Song, C.-L. Zhang and S. Ye, Org. Biomol. Chem., 2019, 17, 181 RSC.
- J. Yang, J. Zhang, L. Qi and Y. Chen, Chem. Commun., 2015, 51, 5275 RSC.
- M. Jiang, Y. Jin, H. Yang and H. Fu, Sci. Rep., 2016, 6, 26161 CrossRef CAS PubMed.
- J. Schwarz and B. König, ChemPhotoChem, 2017, 1, 237 CrossRef CAS.
- G. L. Lackner, K. W. Quasdorf and L. E. Overman, J. Am. Chem. Soc., 2013, 135, 15342 CrossRef CAS PubMed.
- C. Gao, J. Li, J. Yu, H. Yang and H. Fu, Chem. Commun., 2016, 52, 7292 RSC.
- M. Ociepa, J. Turkowska and D. Gryko, ACS Catal., 2018, 8, 11362 CrossRef CAS.
- J. Jia, Y. A. Ho, R. F. Bülow and M. Rueping, Chem.–Eur. J., 2018, 24, 14054 CrossRef CAS PubMed.
- W. Jin, M. Wu, Z. Xiong and G. Zhu, Chem. Commun., 2018, 54, 7924 RSC.
- H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280 CrossRef CAS PubMed.
- H. Huang, G. Zhang and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 7872 CrossRef CAS PubMed.
- H. Tan, H. Li, W. Ji and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 8374 CrossRef CAS PubMed.
- F. Le Vaillant, T. Courant and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 11200 CrossRef CAS PubMed.
- Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196 CrossRef CAS PubMed.
- C. Yang, J.-D. Yang, Y.-H. Li, X. Li and J.-P. Cheng, J. Org. Chem., 2016, 81, 12357 CrossRef CAS PubMed.
- M. Garreau, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 8182 CrossRef CAS PubMed.
- K. Jia, F. Zhang, H. Huang and Y. Chen, J. Am. Chem. Soc., 2016, 138, 1514 CrossRef CAS PubMed.
- K. Jia, Y. Pan and Y. Chen, Angew. Chem., Int. Ed., 2017, 56, 2478 CrossRef CAS PubMed.
- K. Jia, J. Li and Y. Chen, Chem.–Eur. J., 2018, 24, 3174 CrossRef CAS PubMed.
- S. Mukherjee, R. A. Garza-Sanchez, A. Tlahuext-Aca and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 14723 CrossRef CAS PubMed.
- J. Davies, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 13361 CrossRef CAS PubMed.
- F. Le Vaillant, M. Garreau, S. Nicolai, G. Gryn'ova, C. Corminboeuf and J. Waser, Chem. Sci., 2018, 9, 5883 RSC.
- M. M. Jackman, S. Im, S. R. Bohman, C. C. L. Lo, A. L. Garrity and L. S. Castle, Chem.–Eur. J., 2018, 24, 594 CrossRef CAS PubMed.
- S. P. Morcillo, E. M. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 12945 CrossRef CAS PubMed.
- H. Jiang and A. Studer, Chem.–Eur. J., 2019, 25, 516 CAS.
- (a) F. Le Vaillant, M. D. Wodrich and J. Waser, Chem. Sci., 2017, 8, 1790 RSC; (b) F. Le Vaillant and J. Waser, Chimia, 2017, 71, 226 CrossRef CAS PubMed.
- Y. Pan, K. Jia, Y. Chen and Y. Chen, Beilstein J. Org. Chem., 2018, 14, 1215 CrossRef CAS PubMed.
- H. Ohmiya, H. Yorimitsu and K. Oshima, Org. Lett., 2006, 8, 3093 CrossRef CAS.
- L. Huang, A. M. Olivares and D. J. Weix, Angew. Chem., Int. Ed., 2017, 56, 11901 CrossRef CAS.
- J. M. Smith, T. Qin, R. R. Merchant, J. T. Edwards, L. R. Malins, Z. Liu, G. Che, Z. Shen, S. A. Shaw, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 11906 CrossRef CAS PubMed.
- S. Tang, P. Wang, H. Li and A. Lei, Nat. Commun., 2016, 7, 11676 CrossRef PubMed.
- S. Tang, Y. Liu, X. Gao, P. Wang, P. Huang and A. Lei, J. Am. Chem. Soc., 2018, 140, 6006 CrossRef CAS PubMed.
- Y. Shen, B. Huang, J. Zheng, C. Lin, Y. Liu and S. Cui, Org. Lett., 2017, 19, 1744 CrossRef CAS PubMed.
- H. Guan, S. Sun, Y. Mao, L. Chen, R. Lu, J. Huang and L. Liu, Angew. Chem., Int. Ed., 2018, 57, 11413 CrossRef CAS PubMed.
- L. Ma, X. Shi, X. Li and D. Shi, Org. Chem. Front., 2018, 5, 3515 RSC.
- K. Shen and Q. Wang, Chem. Sci., 2017, 8, 8265 RSC.
- W.-J. Han, Y.-R. Wang, J.-W. Zhang, F. Chen, B. Zhou and B. Han, Org. Lett., 2018, 20, 2960 CrossRef CAS PubMed.
- R. Ren, Z. Wu, Y. Xu and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2866 CrossRef CAS.
- R. Sakamoto, T. Kato, S. Sakurai and K. Maruoka, Org. Lett., 2018, 20, 1400 CrossRef CAS PubMed.
- M. Nagatomo, S. Yoshioka and M. Inoue, Chem.–Asian. J., 2015, 10, 120 CrossRef CAS PubMed.
- D. Wang, L. Zhang and S. Luo, Org. Lett., 2017, 19, 4924 CrossRef CAS PubMed.
- L. Fu, S. Zhou, X. Wan, P. Chen and G. Liu, J. Am. Chem. Soc., 2018, 140, 10965 CrossRef CAS PubMed.
- Y. Xu, Z. Wu, J. Jiang, Z. Ke and C. Zhu, Angew. Chem., Int. Ed., 2017, 56, 4545 CrossRef CAS PubMed.
- X. Tang and A. Studer, Chem. Sci., 2017, 8, 6888 RSC.
- (a) J. Yu, D. Wang, Y. Xu, Z. Wu and C. Zhu, Adv. Synth. Catal., 2018, 360, 744–750 CrossRef CAS; (b) J. Liu, W. Li, J. Xie and C. Zhu, Org. Chem. Front., 2018, 5, 797 RSC.
- Q. Zhao, X.-S. Ji, Y.-Y. Gao, W.-J. Hao, K.-Y. Zhang, S.-J. Tu and B. Jiang, Org. Lett., 2018, 20, 3596 CrossRef CAS PubMed.
- J. Sun, G. Zheng, Y. Fu, L. Wang, Y. Li and Q. Zhang, Org. Lett., 2018, 20, 5597 CrossRef CAS PubMed.
- Y. Gao, H. Mei, J. Han and Y. Pan, Chem.–Eur. J., 2018, 24, 17205 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |