
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ROMPI-CDSA: ring-opening metathesis polymerization-induced crystallization-driven self-assembly of metallo-block copolymers†
Ye
Sha
 ,
Md Anisur
Rahman
,
Tianyu
Zhu
,
Yujin
Cha
,
C. Wayne
McAlister
and
Chuanbing
Tang
*
,
Md Anisur
Rahman
,
Tianyu
Zhu
,
Yujin
Cha
,
C. Wayne
McAlister
and
Chuanbing
Tang
*
Department of Chemistry and Biochemistry, University of South Carolina, Columbia, South Carolina 29208, USA. E-mail: tang4@mailbox.sc.edu
First published on 4th September 2019
Abstract
Polymerization-induced self-assembly (PISA) and crystallization-driven self-assembly (CDSA) are among the most prevailing methods for block copolymer self-assembly. Taking the merits of scalability of PISA and dimension control of CDSA, we report one-pot synchronous PISA and CDSA via ring-opening metathesis polymerization (ROMP) to prepare nano-objects based on a crystalline poly(ruthenocene) motif. We denote this self-assembly methodology as ROMPI-CDSA to enable a simple, yet robust approach for the preparation of functional nanomaterials.
Introduction
Block copolymer (BCP) self-assembly in solution provides a versatile tool to build a wide variety of micellar morphologies, ranging from spheres to vesicles to many other complex structures.1–8 Notwithstanding significant advances, the preparation of BCP micelles is usually a protracted multi-step process, which typically includes precise sequential polymerization, tedious purification and sophisticated assembly steps, making it formidable for scalability.9–12Scalable processing (under concentrated conditions, typically 5–50% w/w solids) of well-defined micelles can be realized by an approach termed polymerization-induced self-assembly (PISA) where the polymerization of BCPs and self-assembly take place in situ.10,13–21 However, the preparation of fiber-like or platelet micelles with uniformity is challenging to access from the self-assembly of non-crystalline BCPs.22 This has led to the development of crystallization-driven self-assembly (CDSA) of BCPs where the crystalline core largely dictates the formation of 1D and 2D nanostructures.1,23–28 Nevertheless, multiple steps of synthesis and purification together with very low concentration solution processing (typically <0.5% w/w solids) severely limit the scalability of CDSA.29
Taking advantage of the scalability and dimensional control respectively from PISA and CDSA, a new process called polymerization-induced CDSA (or PI-CDSA) would be promising, but challenging.22,29 PISA is predominantly conducted via living radical polymerization technologies, where reversible addition–fragmentation chain transfer (RAFT) polymerization prevails.10 However, radical polymerization usually leads to atactic polymers, which are not preferred for crystallization. Recently, Manners and his co-workers reported the first and so far the only example of PI-CDSA by using unique properties of poly(ferrocenyldimethylsilane) via living anionic polymerization under stringent conditions.22,29 This unparalleled paradigm has inspired us to question whether this approach can be applicable to more mild polymerization methods and other crystalline polymers. Ring-opening metathesis polymerization (ROMP) has shown the potential in PISA due to its access to different classes of monomers.12,30,31 Additionally, highly conjugated crystalline polymer species can be also prepared via ROMP.32,33
In this work, we report ROMP as a new polymerization method to achieve the PI-CDSA with ruthenocene-containing polymers as new crystallizable species. It is termed as ring-opening metathesis polymerization-induced crystallization-driven self-assembly (ROMPI-CDSA). Platelet hybrid nanostructures were obtained using this one-pot process without any post-treatment. The crystalline polyruthenocene core segment is an exceptional driving force to dictate the self-assembly. The work represents new efforts toward emerging directions of metallopolymers for advanced materials by us and others.34–45
Results and discussion
The synchronous process of PI-CDSA requires the incipient production of a corona-forming block that is carried out under living polymerization in a good solvent medium. The same solvent system becomes non-selective for the second core-forming block as the degree of polymerization (DP) of the second segment proceeds to a threshold that induces crystallization.22To meet this criteria, we first targeted a block copolymer poly(5-methoxycyclooctene)-b-polyferrocene (PMCOE-b-PFc) in tetrahydrofuran (THF) (Scheme S1†), as THF is a good solvent for PMCOE but poor for PFc, and the CDSA behavior of this BCP in THF was recently demonstrated by us.46 However, this seemingly straightforward process proved nontrivial. After the complete consumption of 5-methoxycyclooctene monomer to form the corona block, cyclic ferrocenyl olefin monomer was added into the polymerization system to achieve a final concentration of 10% w/w solids, which is within the typical concentration regime of PISA but well above the concentration for CDSA (<0.5% wt).22 It was anticipated that the PFc block would become insoluble and precipitate from the solution when its DP reaches a threshold where ROMPI-CDSA might occur. As shown in Table S1,† initial experiments with a wide range of block ratios from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1 to 1:1 in the polymerization system were carried out. When the conversion of second monomer reached the upper limit, ethyl vinyl ether (EVE) was added to quench the polymerization, and small aliquots of the solutions were diluted by THF and then cast onto copper grids for transmission electron microscopy (TEM) analysis. Only non-crystalline spheres or irregular aggregations were observed for these compositions (Fig. S9†). One possible reason is that the target molecular weight might be too high (over 100000 Da) to form non-spherical morphologies,47 and the rate of crystallization for the PFc core-forming block would decrease when its chain length increases.48 We then decreased the target molecular weight to 45000 Da with a wide range of block ratios (Table S2†). However, spheres or irregular aggregations were still observed, as shown in Fig. S10.† The ROMP process to prepare polycyclooctene is generally not a living process,49 thus it may lead to ineffective initiation and growth of the core-forming block, which could make the architecture of diblock copolymers in an ill-defined manner.
:0.1 to 1:1 in the polymerization system were carried out. When the conversion of second monomer reached the upper limit, ethyl vinyl ether (EVE) was added to quench the polymerization, and small aliquots of the solutions were diluted by THF and then cast onto copper grids for transmission electron microscopy (TEM) analysis. Only non-crystalline spheres or irregular aggregations were observed for these compositions (Fig. S9†). One possible reason is that the target molecular weight might be too high (over 100000 Da) to form non-spherical morphologies,47 and the rate of crystallization for the PFc core-forming block would decrease when its chain length increases.48 We then decreased the target molecular weight to 45000 Da with a wide range of block ratios (Table S2†). However, spheres or irregular aggregations were still observed, as shown in Fig. S10.† The ROMP process to prepare polycyclooctene is generally not a living process,49 thus it may lead to ineffective initiation and growth of the core-forming block, which could make the architecture of diblock copolymers in an ill-defined manner.
It is well established that norbornene-based strained olefins undergo living ROMP efficiently.50–53 Since pristine polynorbornene can be crystalline,54 we grafted a bulky tricyclic group (dehydroabietic) as the side-chain moiety onto exo-5-norbornenecarboxylic acid to not only increase the solubility of the corona-forming block but also elevate its glass transition temperature (∼110 °C) to facilitate assembled morphologies unaffected under room temperature.52 As shown in Fig. 1, main-chain ferrocene-containing diblock copolymers can be prepared by sequential ROMP. Narrow molecular weight distribution (Fig. S6†) and predicted molecular weight indicated the living manner of the first block of polynorbornene (PNR), the yielded BCPs with a series of segment compositions are given in Table 1. TEM measurements showed the formation of fiber-like morphologies, demonstrating that self-assembly took place during the polymerization process (Fig. S11†). However, these micelles are surrounded by pronounced background, which can be attributed to the low crystallinity of PFc prepared by ROMP.46 It has been demonstrated that higher crystallinity is required to achieve successful CDSA.46,55,56 Based on these results, it was suggested that PFc-based BCPs are not sufficient to produce well-defined nanostructures via the ROMPI-CDSA process, but inspire further exploration on other metallocene-based BCPs with higher crystallinity.
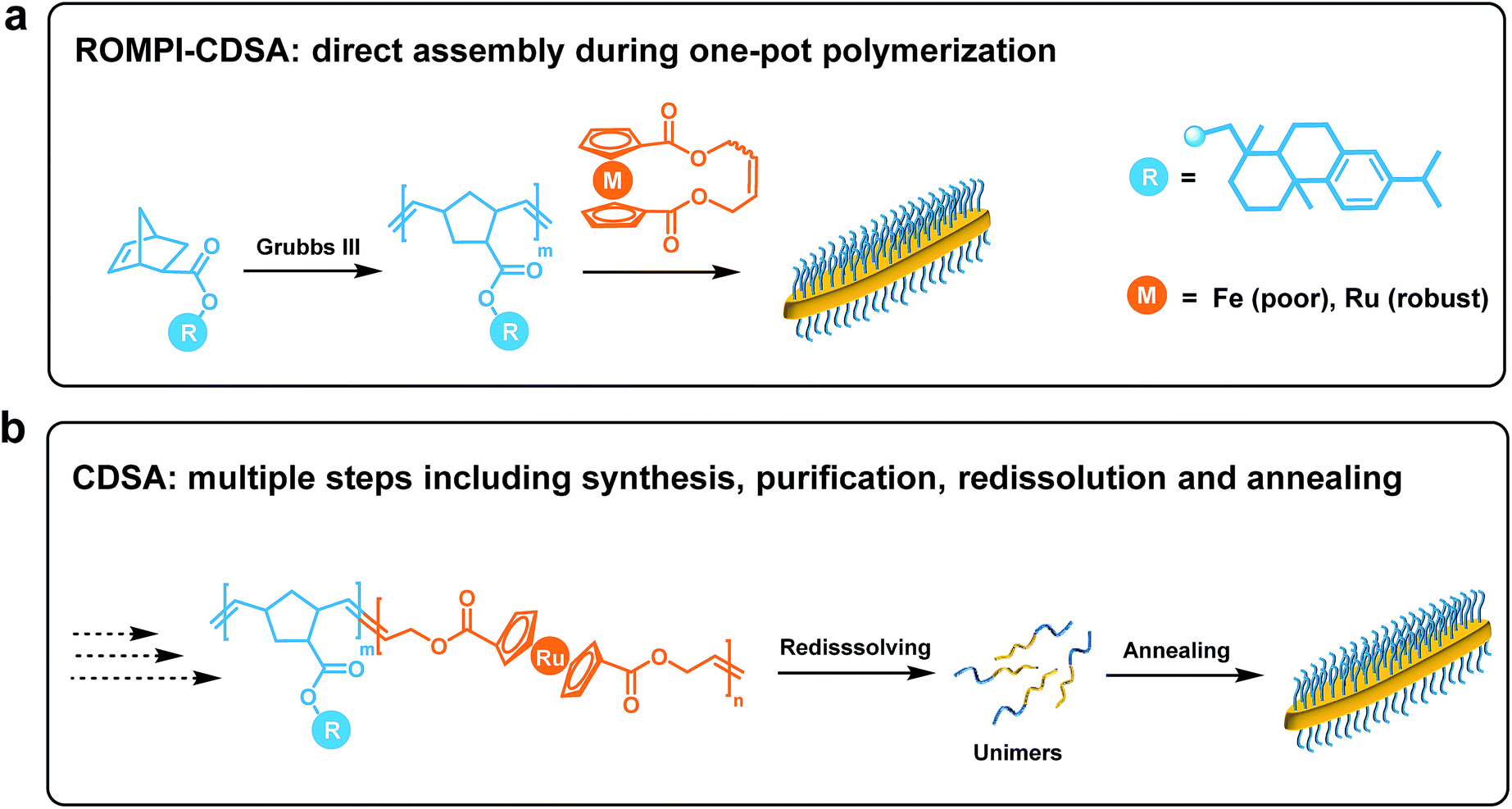 | ||
| Fig. 1 Schematic comparison between (a) ROMPI-CDSA process that allows simple, direct assembly of block copolymers during one-pot synthesis, and (b) CDSA process that involves multiple steps including synthesis, purification, redissolution and annealing. | ||
| Sample | Block copolymera | M n (Da) |
|---|---|---|
| a The DP of the first block was determined from GPC, the DP of the second block was determined from 1H NMR via compositional analysis. b The number-average molecular weight was calculated based on the DP of the diblock copolymer. | ||
| 1 | PNR123-b-PFc93 | 80300 |
| 2 | PNR123-b-PFc36 | 61800 |
| 3 | PNR52-b-PFc71 | 44500 |
| 4 | PNR52-b-PFc43 | 35400 |
| 5 | PNR31-b-PRc49 | 30800 |
| 6 | PNR31-b-PRc21 | 20400 |
| 7 | PNR48-b-PRc56 | 40300 |
As an analogy to ferrocene, ruthenocene-based polymers are somehow much less explored.57–64 Using a similar methodology,46,64,65 a cyclic ruthenocenyl olefin monomer was prepared (Fig. 2a). Interestingly, polyruthenocene (PRc) is crystalline as confirmed by X-ray Diffraction (XRD, Fig. 2b and S1†) and Differential Scanning Calorimetry (DSC, a melting transition observed during the heating scan, Fig. 2c and S3†). Surprisingly, it has much higher crystallinity (∼38.3%) than PFc (∼17.4%). It might be due to the presence of the trans-dominated conformation of carbon–carbon double bonds along the backbone (as indicated by solid-state 13C NMR spectra, Fig. S4†), which induces higher crystallization.66,67 It is thus intriguing to explore ROMPI-CDSA for ruthenocene-containing polymers.
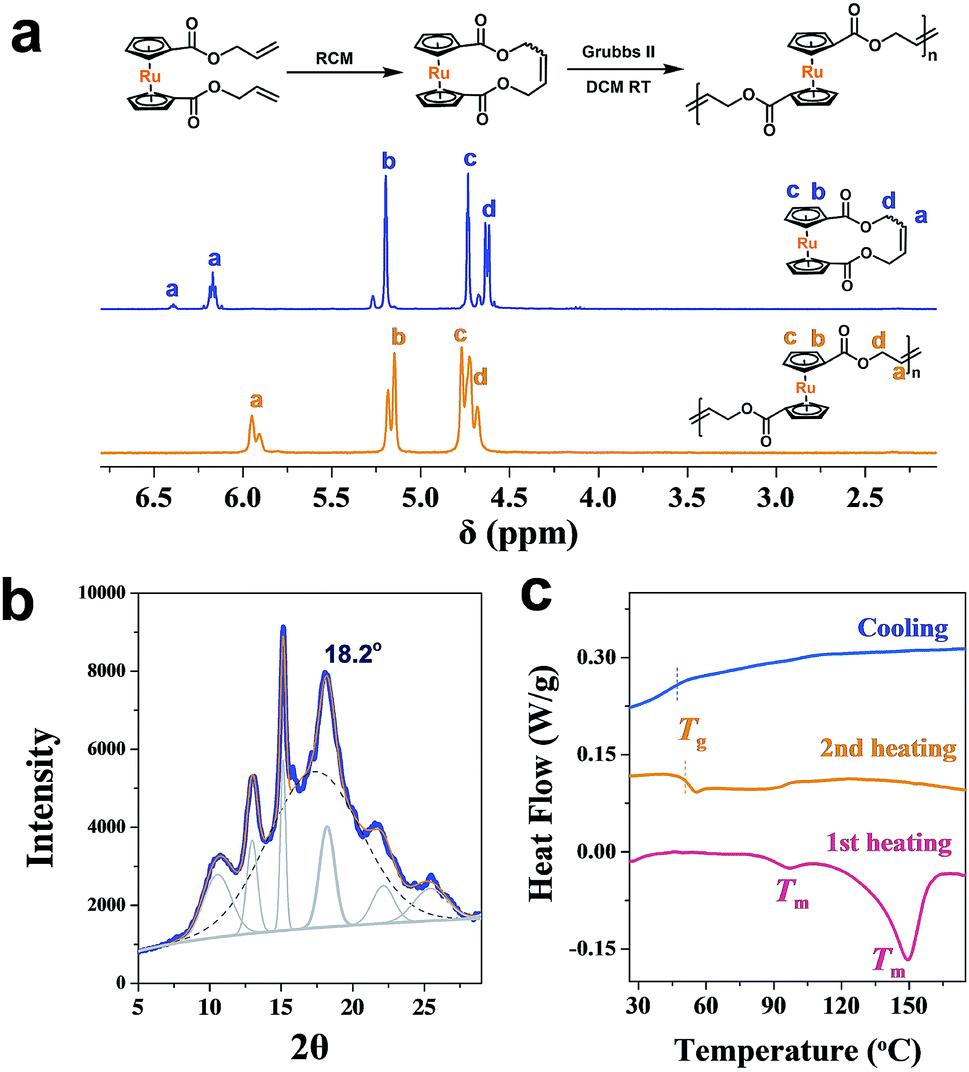 | ||
| Fig. 2 (a) 1H NMR spectra (CDCl3) of cyclic ruthenocene olefin and main-chain ruthenocene-based homopolymer; (b) XRD powder spectrum of PRc homopolymer; (c) DSC curves of PRc. | ||
Following the same procedure as ROMPI-CDSA of the ferrocene monomer, ruthenocene-containing block copolymer PNR-b-PRc was prepared with different block ratios (Table 1). TEM measurements of diluted PNR31-b-PRc49 samples showed exclusive lenticular platelet structures with a number-average length of Ln = 645 nm and a low dispersity of 1.10 (Fig. 3a). The aspect ratio (Ln/Wn, where Wn is the number-average width) was statistically analyzed as 9.5 with a low dispersity of 1.09 (Fig. 3a). Selected-area electron diffraction (SAED) pattern of TEM (Fig. 3b) showed a well-defined diffraction ring with d = 4.9 Å, indicating the crystalline nature of PRc core. This result confirms that the self-assembly process was driven by the crystallization of PRc core, as suggested in related to the distance calculated from Fig. 2b. AFM height image indicated that the height of platelets is approx. 8 nm (Fig. 3c). The crystalline core of PRc is expected to be sandwiched between the PNR coronas (Fig. 3d). The thickness of crystalline domains was estimated to be 4 nm (see ESI† for analysis).68,69 Together with the estimated contour length of crystalline chain, the PRc segment (DP = 49) is folded ca. 16 times (nf = 16, Fig. 3d), considering the proximity of chain packing in the direction normal to the fold surface.70
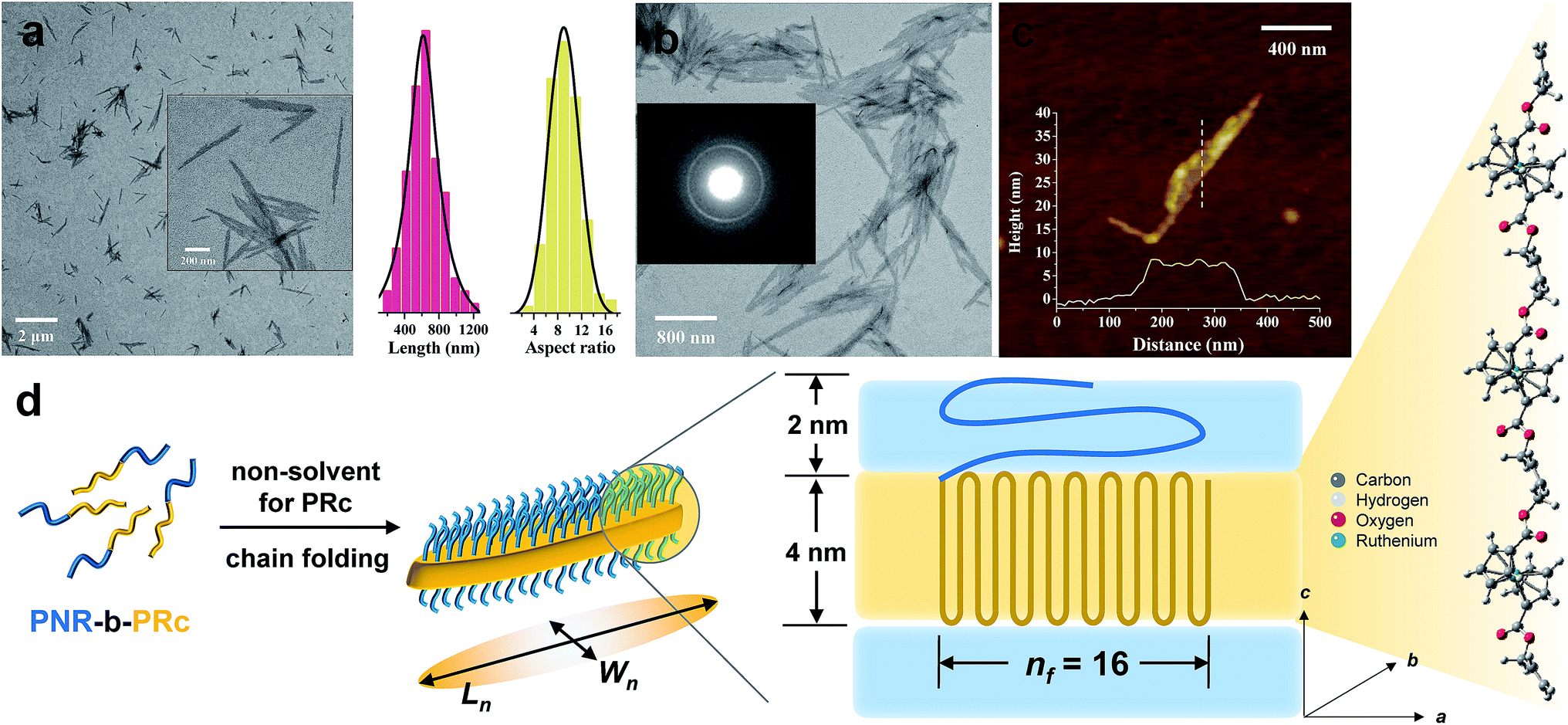 | ||
| Fig. 3 (a) Low resolution TEM image of PNR31-b-PRc49 micelles via ROMPI-CDSA; the inset shows the high resolution image; the statistical plots show data of lengths and aspect ratios; (b) high-resolution TEM image of PNR31-b-PRc49 micelles formed after polymerization, the inset image shows the SAED pattern; (c) AFM height image and profile of PNR31-b-PRc49 micelles; the height profile is from the indicated dash line; (d) a schematic diagram illustrating the chain folding of the crystalline PRc core. The stick-ball structure indicates the possible alignment of PRc segment along the perpendicular direction. | ||
In a separate experiment, this BCP was isolated and purified for studying the CDSA process as illustrated in Fig. 1b. This standalone process resulted in the formation of similar platelet structures (Fig. S13†). This additional experiment demonstrated that the one-pot synthesis of nanostructures was largely driven by crystallization of the PRc core. ROMPI-CDSA of other BCPs (PNR31-b-PRc21 and PNR48-b-PRc56) also produced lenticular platelets, as shown in Fig. S14 and S15.† The decrease in platelet width of PNR31-b-PRc21 compared to PNR31-b-PRc49 is in a good agreement with the decreased chain length of the crystalline PRc segment.
In addition to the CDSA process, it is equally important to determine if the polymerization and self-assembly took place simultaneously, i.e., whether is it only a PISA process? During the ROMPI-CDSA process to target a BCP of PNR48-b-PRc56, aliquots were taken out at specified time intervals for 1H NMR and TEM characterization to provide mechanistic insights into the self-assembly. The diblock copolymer composition was summarized in Table S3.† As shown in Fig. 4 (higher resolution) and Fig. S16† (lower resolution), no micelles were observed during the first minute with a DP = 9 of PRc. At 5 min, ill-defined fiber-like features were observed. At 20 min, large quantities of aggregated fibers appeared, and platelet structures started to form. When polymerization went for 60 min with DP = 33, platelet structures became more pronounced. At time 120 min with a DP = 38 of PRc, a morphological transition occurred with dominated platelet micelles present and almost without fiber-like micelles. When time reached over 240 min, exclusive platelet micelles were observed. A test on the Tyndall effect of reaction solutions could provide additional evidence on the PISA process, simply by projecting a laser beam through the reaction vials. It clearly showed the increasing Tyndall effect with evolution of reaction time, until the formation of visibly turbid solutions (Fig. S17†). It was then concluded that ROMP and self-assembly occurred simultaneously through the ROMPI-CDSA process, corresponding to a morphological transformation from fibers at low DP to platelets at high DP.22,71–73
 | ||
| Fig. 4 Representative higher resolution TEM images of PNR48-b-PRcn micelles by ROMPI-CDSA with aliquots taken out at specific time intervals. The scale bar in each image is 400 nm. n is the DP of PRc. | ||
Conclusions
In summary, we presented a new self-assembly process termed as ROMPI-CDSA. This one-pot synthesis combined synchronous ROMP, PISA and CDSA to yield uniform crystalline nano-objects in a mild, fast, scalable and controlled manner. The mechanistic insights by exploring ferrocene-containing BCPs towards ROMPI-CDSA unveiled the interplay of various parameters including molecular weight, chain extension and crystallinity. It was discovered that higher crystalline ruthenocene-containing BCPs self-assembled via the ROMPI-CDSA process to allow the scalable production of lenticular platelet nanomaterials at concentrations ∼10% w/w solids. The ROMPI-CDSA could be viewed as a new paradigm that is applicable to other BCP systems with a crystalline core-forming block, which can be prepared by mild polymerization techniques.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is partially supported by the National Science Foundation EPSCoR Program under NSF Award # OIA-1655740.References
- U. Tritschler, S. Pearce, J. Gwyther, G. R. Whittell and I. Manners, Macromolecules, 2017, 50, 3439–3463 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS PubMed.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- Z. B. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98–101 CrossRef CAS PubMed.
- A. H. Groschel, A. Walther, T. I. Lobling, F. H. Schacher, H. Schmalz and A. H. E. Muller, Nature, 2013, 503, 247 CrossRef PubMed.
- J. Rodriguez-Hernandez, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS.
- J. C. Brendel and F. H. Schacher, Chem.–Asian J., 2018, 13, 230–239 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- K. Y. Yoon, I. H. Lee, K. O. Kim, J. Jang, E. Lee and T. L. Choi, J. Am. Chem. Soc., 2012, 134, 14291–14294 CrossRef CAS PubMed.
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS.
- J. Rieger, F. Stoffelbach, C. Bui, D. Alaimo, C. Jerome and B. Charleux, Macromolecules, 2008, 41, 4065–4068 CrossRef CAS.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef PubMed.
- G. Wang, M. Schmitt, Z. Wang, B. Lee, X. Pan, L. Fu, J. Yan, S. Li, G. Xie, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2016, 49, 8605–8615 CrossRef CAS.
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS.
- W. Wan and C. Pan, Polym. Chem., 2010, 1, 1475–1484 RSC.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- S. Varlas, J. C. Foster and R. K. O'Reilly, Chem. Commun., 2019, 55, 9066–9071 RSC.
- C. E. Boott, J. Gwyther, R. L. Harniman, D. W. Hayward and I. Manners, Nat. Chem., 2017, 9, 785–792 CrossRef CAS PubMed.
- J. Massey, K. N. Power, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 1998, 120, 9533–9540 CrossRef CAS.
- S. Ganda, M. Dulle, M. Drechsler, B. Foerster, S. Foerster and M. H. Stenzel, Macromolecules, 2017, 50, 8544–8553 CrossRef CAS.
- Z. X. Du, J. T. Xu and Z. Q. Fan, Macromolecules, 2007, 40, 7633–7637 CrossRef CAS.
- J. Schobel, M. Karg, D. Rosenbach, G. Krauss, A. Greiner and H. Schmalz, Macromolecules, 2016, 49, 2761–2771 CrossRef.
- A. Pitto-Barry, N. Kirby, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2014, 5, 1427–1436 RSC.
- Z. M. Hudson, C. E. Boott, M. E. Robinson, P. A. Rupar, M. A. Winnik and I. Manners, Nat. Chem., 2014, 6, 893–898 CrossRef CAS PubMed.
- A. M. Oliver, J. Gwyther, C. E. Boott, S. Davis, S. Pearce and I. Manners, J. Am. Chem. Soc., 2018, 140, 18104–18114 CrossRef CAS PubMed.
- D. B. Wright, M. A. Touve, L. Adamiak and N. C. Gianneschi, ACS Macro Lett., 2017, 6, 925–929 CrossRef CAS.
- S. Varlas, J. C. Foster, L. A. Arkinstall, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O'Reilly, ACS Macro Lett., 2019, 8, 466–472 CrossRef CAS PubMed.
- I. Choi, S. Yang and T. L. Choi, J. Am. Chem. Soc., 2018, 140, 17218–17225 CrossRef CAS PubMed.
- M. A. Buretea and T. D. Tilley, Organometallics, 1997, 16, 1507–1510 CrossRef CAS.
- Y. Yan, J. Zhang, L. Ren and C. Tang, Chem. Soc. Rev., 2016, 45, 5232–5263 RSC.
- J. Zhang, Y. Chen, K. P. Miller, M. S. Ganewatta, M. Bam, Y. Yan, M. Nagarkatti, A. W. Decho and C. Tang, J. Am. Chem. Soc., 2014, 136, 4873–4876 CrossRef CAS PubMed.
- J. Zhang, Y. Yan, M. W. Chance, J. Chen, J. Hayat, S. Ma and C. Tang, Angew. Chem., Int. Ed., 2013, 52, 13387–13391 CrossRef CAS PubMed.
- T. Zhu, Y. Sha, J. Yan, P. Pageni, M. A. Rahman, Y. Yan and C. Tang, Nat. Commun., 2018, 9, 4329 CrossRef PubMed.
- T. Zhu, S. Xu, A. Rahman, E. Dogdibegovic, P. Yang, P. Pageni, M. P. Kabir, X. D. Zhou and C. Tang, Angew. Chem., Int. Ed., 2018, 57, 2388–2392 CrossRef CAS PubMed.
- P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC.
- Y. Wang, D. Astruc and A. S. Abd-El-Aziz, Chem. Soc. Rev., 2019, 48, 558–636 RSC.
- R. L. N. Hailes, A. M. Oliver, J. Gwyther, G. R. Whittell and I. Manners, Chem. Soc. Rev., 2016, 45, 5358–5407 RSC.
- A. Voevodin, L. M. Campos and X. Roy, J. Am. Chem. Soc., 2018, 140, 5607–5611 CrossRef CAS PubMed.
- B. V. K. J. Schmidt, J. Elbert, D. Scheid, C. J. Hawker, D. Klinger and M. Gallei, ACS Macro Lett., 2015, 4, 731–735 CrossRef CAS.
- M. Karayilan, W. P. Brezinski, K. E. Clary, D. L. Lichtenberger, R. S. Glass and J. Pyun, Angew. Chem., Int. Ed., 2019, 58, 7537–7550 CrossRef CAS PubMed.
- A. Rabiee Kenaree, B. M. Berven, P. J. Ragogna and J. B. Gilroy, Chem. Commun., 2014, 50, 10714–10717 RSC.
- Y. Sha, Y. Zhang, T. Zhu, S. Tan, Y. Cha, S. L. Craig and C. Tang, Macromolecules, 2018, 51, 9131–9139 CrossRef CAS.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- M. S. Hsiao, S. F. M. Yusoff, M. A. Winnik and I. Manners, Macromolecules, 2014, 47, 2361–2372 CrossRef CAS.
- H. Martinez, N. Ren, M. E. Matta and M. A. Hillmyer, Polym. Chem., 2014, 5, 3507–3532 RSC.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- M. A. Rahman, H. N. Lokupitiya, M. S. Ganewatta, L. Yuan, M. Stefik and C. Tang, Macromolecules, 2017, 50, 2069–2077 CrossRef CAS.
- M. S. Ganewatta, W. Ding, M. A. Rahman, L. Yuan, Z. Wang, N. Hamidi, M. L. Robertson and C. Tang, Macromolecules, 2016, 49, 7155–7164 CrossRef CAS.
- J. K. Su, J. D. Feist, J. H. Yang, J. A. M. Mercer, J. A. H. Romaniuk, Z. X. Chen, L. Cegelski, N. Z. Burns and Y. Xia, J. Am. Chem. Soc., 2018, 140, 12388–12391 CrossRef CAS PubMed.
- K. Sakurai, T. Kashiwagi and T. Takahashi, J. Appl. Polym. Sci., 1993, 47, 937–940 CrossRef CAS.
- J. A. Massey, K. Temple, L. Cao, Y. Rharbi, J. Raez, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2000, 122, 11577–11584 CrossRef CAS.
- N. Petzetakis, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2011, 2, 955–960 RSC.
- C. G. Hardy, J. Zhang, Y. Yan, L. Ren and C. Tang, Prog. Polym. Sci., 2014, 39, 1742–1796 CrossRef CAS.
- Y. Yan, J. Zhang, Y. Qiao, M. Ganewatta and C. Tang, Macromolecules, 2013, 46, 8816–8823 CrossRef CAS.
- A. Alkan, T. Gleede and F. R. Wurm, Organometallics, 2017, 36, 3023–3028 CrossRef CAS.
- U. Vogel, A. J. Lough and I. Manners, Angew. Chem., Int. Ed., 2004, 43, 3321–3325 CrossRef CAS PubMed.
- T. C. Willis and J. E. Sheats, J. Polym. Sci., Polym. Chem. Ed., 1984, 22, 1077–1084 CrossRef CAS.
- M. Erhard, K. Lam, M. Haddow, G. R. Whittell, W. E. Geiger and I. Manners, Polym. Chem., 2014, 5, 1264–1274 RSC.
- A. R. Kenaree and J. B. Gilroy, Organometallics, 2017, 36, 2483–2486 CrossRef.
- Y. Sha, Y. Zhang, E. Xu, C. W. McAlister, T. Zhu, S. L. Craig and C. Tang, Chem. Sci., 2019, 10, 4959–4965 RSC.
- Y. Sha, Y. Zhang, E. Xu, Z. Wang, T. Zhu, S. L. Craig and C. Tang, ACS Macro Lett., 2018, 7, 1174–1179 CrossRef CAS PubMed.
- B. K. Keitz, A. Fedorov and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 2040–2043 CrossRef CAS PubMed.
- P. Dounis, W. J. Feast and A. M. Kenwright, Polymer, 1995, 36, 2787–2796 CrossRef CAS.
- W. Chen, J. Zheng, S. Z. D. Cheng, C. Li, P. Huang, L. Zhu, H. Xiong, Q. Ge, Y. Guo, R. P. Quirk, B. Lotz, L. Deng, C. Wu and E. L. Thomas, Phys. Rev. Lett., 2004, 93, 028301 CrossRef PubMed.
- W. Zhu, B. Peng, J. Wang, K. Zhang, L. Liu and Y. Chen, Macromol. Biosci., 2014, 14, 1764–1770 CrossRef CAS PubMed.
- T. Vilgis and A. Halperin, Macromolecules, 1991, 24, 2090–2095 CrossRef CAS.
- M. E. Robinson, A. Nazemi, D. J. Lunn, D. W. Hayward, C. E. Boott, M.-S. Hsiao, R. L. Harniman, S. A. Davis, G. R. Whittell, R. M. Richardson, L. De Cola and I. Manners, ACS Nano, 2017, 11, 9162–9175 CrossRef CAS PubMed.
- Y. Wei, J. Tian, Z. Zhang, C. Zhu, J. Sun and Z. Li, Macromolecules, 2019, 52, 1546–1556 CrossRef CAS.
- G. Rizis, T. G. M. van de Ven and A. Eisenberg, Angew. Chem., Int. Ed., 2014, 53, 9000–9003 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03056e |
| This journal is © The Royal Society of Chemistry 2019 |